
Abstract
Severely birth-asphyxiated human infants develop delayed(“secondary”) cerebral energy failure, which carries a poor prognosis, during the first few days of life. This study tested the hypothesis that i.v. magnesium sulfate (MgSO4) after severe transient cerebral hypoxia-ischemia decreases the severity of delayed energy failure in the newborn piglet. Twelve piglets underwent temporary occlusion of the common carotid arteries and hypoxemia. Resuscitation was started when cerebral[phosphocreatine (PCr)]/[inorganic phosphate (Pi)], as determined by phosphorus magnetic resonance spectroscopy, had fallen virtually to zero, and nucleotide triphosphate (NTP) had fallen below a third of baseline. The piglets were randomized to receive, blind, either: 1) three i.v. infusions of 12.5% MgSO4 heptahydrate solution: 400 mg·kg-1 MgSO4·7H2O starting 1 h after resuscitation, and 200 mg·kg-1 12 and 24 h later (n = 6); or 2) three infusions of placebo, 0.9% NaCl (n = 6). Phosphorus and proton spectroscopy were continued until 48 h after resuscitation, and values were compared between the two groups. Mean plasma magnesium levels, 1 h after each of the three doses of MgSO4, were 2.1, 2.0, and 1.9 mmol·L-1, respectively. The severity of the primary insult, determined by the time-integral of depletion of cerebral[NTP]/[exchangeable phosphate pool (EPP)], was similar in the MgSO4-treated and placebo groups. After resuscitation, there was no difference in the progression or severity of delayed energy failure between the two groups, as judged by cerebral [PCr]/[Pi], [NTP]/[EPP], or lactate/creatine and N-acetylaspartate/creatine peak-area ratios. We conclude that MgSO4 did not decrease the severity of delayed cerebral energy failure.
Similar content being viewed by others

Main
Perinatal hypoxia-ischemia, often due to “birth-asphyxia”(critically impaired intrapartum gas exchange), remains an important cause of neonatal mortality and permanent neurodevelopmental disability worldwide(1), and yet clinical management continues to rely on supportive and symptomatic treatments only. Studies of birth-asphyxiated infants using 31P MRS have shown that the development of cerebral energy failure, which carries a bad prognosis, commences some 8-24 h after birth(2, 3). This has suggested the potential for early treatment to prevent or ameliorate the evolution of cerebral injury.
In an animal model of perinatal hypoxia-ischemia, which was developed in the newborn piglet(4, 5), we have demonstrated that delayed or “secondary” energy failure, with the same characteristics as seen in birth-asphyxiated human infants, can be initiated by a preceding “primary” episode of energy failure caused by severe hypoxia-ischemia, and reversed by resuscitation. Sham-operated control piglets did not develop these changes. We have argued that this model provides a good opportunity for testing cerebroprotective strategies and have used it to show that mild postinsult hypothermia markedly reduced delayed cerebral energy failure(6).
Magnesium ions gate the NMDA channel and might therefore prevent glutamate-induced calcium entry, which is thought to be a major cause of cell damage due to hypoxia-ischemia(7, 8). Clinical trials of MgSO4 for neuronal rescue therapy are being initiated in birth-asphyxiated infants(7). The aim of this study was to test, in the newborn piglet, the hypothesis that i.v. MgSO4, given after an acute hypoxic-ischemic insult, would prevent or reduce the severity of delayed cerebral energy failure, determined by [PCr]/[Pi] and[NTP]/[EPP] between 24 and 48 h after resuscitation.
METHODS
These experiments were performed with Home Office approval and according to UK guidelines.
Subjects. Twelve healthy Large White piglets, born at term(mean gestation 116 ± SD 2 d) and weighing 1.75 ± 0.33 kg, were studied on the 1st d of life. Full details of the maintenance and monitoring of the piglets have been given previously(4, 6). Briefly, after sedation with intramuscular midazolam (0.2 mg·kg-1), anesthesia was induced with 5% isoflurane followed by tracheostomy and ventilation with nitrous oxide, oxygen, and isoflurane(<1.5%). Umbilical catheters were sited, and inflatable occluders positioned around both common carotid arteries. Rectal and tympanic temperatures were kept normal at 38.5-39.0°C. The piglets were maintained inside the magnet with full intensive care, continuous monitoring of vital functions, intermittent observations of a range of biochemical variables by standard techniques, and attention to fluid balance and plasma volume. Dopamine was commenced if MABP fell <5.3 kPa.
MRS. Spectra were acquired with a 7 tesla Bruker Biospec spectrometer and a 25-mm diameter surface coil centered on the intact scalp over the parietal lobes. The acquisition and analysis of fully relaxed31 P spectra were as previously described(4). NTP(mainly ATP), was quantified using the β-triplet. [NTP] was expressed as a fraction of [EPP], defined as [Pi] + [PCr] + [(γ + α +β) NTP]. The cerebral intracellular free Mg2+ concentration was estimated from the chemical shifts of the α- and β-NTP resonances and intracellular pH (from the Pi chemical shift)(9), using MAGPAC software(10).
For 1H MRS, a 11-22 spin-echo sequence(11) was optimized for Naa with echo time 270 ms, spectral width 4 kHz, 2048 quadrature data points, and 32 summed echoes. The repetition time was 5 s for baseline measurements, 1 s for the acute insult, and 5 s thereafter. 1H echoes were baseline-corrected and zero-filled to 2048 points, followed by convolution difference to reduce spectrum baseline roll. Data were then Fourier-transformed, followed by manual phasing and subtraction of a cubic-spline baseline. Peak areas were measured as for 31P MRS, and peak-area ratios calculated. The Lac methyl resonance was fitted as a doublet; total Cr and Naa were fitted as singlets.
Acute cerebral hypoxia-ischemia. After stable baseline observations, each piglet underwent a cerebral hypoxic-ischemic insult achieved by reducing the inspired oxygen fraction to 0.12 and inflating the carotid occluders. This was continued until severe cerebral energy failure developed, as shown by almost total depletion of PCr, and a fall in [NTP] to below a third of baseline. The animals were then resuscitated by releasing the carotid occluders and increasing the inspired oxygen fraction to normalize arterial Po2. The duration of the insult was 53 ± SD 32 min.
MgSO4 administration. The 12 piglets were randomized, blind to the investigators, into two groups of 6, one of which received MgSO4 and the other placebo (0.9% NaCl). A power calculation showed that this would give an 80% chance of detecting, at the 5% level, a reduction of the delayed fall in [PCr]/[Pi] of similar magnitude to that previously produced in this model using mild hypothermia(6). Three doses of 12.5% MgSO4·7H2O solution (0.5 mmol Mg2+/mL) were administered: 400 mg·kg-1 MgSO4·7H2O 1 h after resuscitation, and 200 mg·kg-1 12 and 24 h later. Doses were infused slowly via the umbilical venous catheter, the first over 20 min, and the latter two over 10 min. Arterial samples for the measurement of plasma Mg2+ levels were taken at baseline, then 1, 3, 6, and 11 h after the first dose, and 1 and 11 h after the two subsequent doses. The plasma was immediately separated from the cells and stored at 4°C. Observations by MRS and of systemic variables continued until 48 h after resuscitation.
Neuropathology. At the end of the study, the animals were killed by overdose of anesthetic. The brains were perfusion-fixed using 1% paraformaldehyde in PBS. The results of the histologic examinations will be reported separately.
Data analysis. Data were examined for normality and equality of variance, and parametric or nonparametric tests used as appropriate. For systemic variables only values of p < 0.01 were considered significant, as multiple tests were performed. The severity of the hypoxic-ischemic insult was assessed as previously described, by calculating the time-integral of the depletion of [NTP]/[EPP] from the onset of hypoxia-ischemia until 1 h after resuscitation(4). For MRS outcome variables, values of p < 0.05 were considered significant.
RESULTS
Physiologic variables. There were no significant differences between the two groups of piglets for observations of a wide range of systemic and biochemical variables (including arterial Po2, Pco2, pH, base excess, hematocrit, blood glucose, MABP, heart rate, rectal and tympanic temperatures, and plasma Na+, K+, urea, creatinine, and Lac), either at baseline, the end of the insult, or 2, 24, and 48 h after resuscitation. These variables remained stable, except that arterial Po2 fell during the insult (from 13.6 ± SD 3.5 to 3.3 ± 0.1 kPa in the MgSO4 group, and from 14.9 ± 2.0 to 3.3 ± 0.4 kPa in the placebo group). Tympanic temperature also fell slightly (by a mean of 0.3°C) in both groups during the insult.
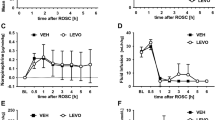
Figure 1 shows the plasma Mg2+ levels from both groups of piglets. When the acute cardiovascular effects of MgSO4 infusion were assessed, a trend toward a fall in MABP was found after the first dose of MgSO4, but the only significant difference between the two groups was 1 h after the second dose when MABP was lower after MgSO4 (p < 0.01, unpaired t test). This difference had resolved 1 h later; the mean value in both groups was 7.6 kPa. The fall in MABP was accompanied by a trend toward a reduction in heart rate. Only one piglet had a MABP which fell <5.3 kPa. This piglet was from the MgSO4 group and received dopamine at 5μg·kg-1·min-1 for 8 h with a good response.

Plasma Mg2+ levels in the MgSO4 (•) and placebo (○) groups. Values are mean and SEM. In the MgSO4 group, peak levels 1 h after each of the three doses were 2.12, 2.04 and 1.91 mmol·L-1, respectively, and after treatment, levels remained significantly above those of the placebo group (p < 0.01 unpaired t test) throughout the experiment (n = 5 in the MgSO4 group after 24 h).
MRS. The severity of the acute cerebral insult, as judged by the time-integral of depletion of [NTP]/[EPP], was similar in the two groups; for the MgSO4 group it was 0.098 ± 0.039 h, and for the placebo group, 0.092 ± 0.032 h. The cerebral intracellular free [Mg2+] was 0.69 ± 0.06 mM at baseline in both groups. Two hours after resuscitation (1 h after the first dose of MgSO4) it had risen significantly to 0.82 ± 0.07 mM (p < 0.02) in the MgSO4 group, and 0.89 ± 0.12 mM (p < 0.005) in the placebo group. Mean values for cerebral intracellular free [Mg2+] then remained between 0.7 and 0.9 mM throughout the rest of the experiment, with no significant differences between the two groups. As in previous studies(4), a cerebral intracellular acidosis developed during the hypoxic-ischemic insult, but not during delayed energy failure; after resuscitation, mean values for intracellular pH remained between 6.91 and 7.11 in both groups.
Figure 2 illustrates changes in [PCr]/[Pi],[NTP]/[EPP], Lac/Cr and Naa/Cr throughout the duration of the experiment. After recovery, evidence of delayed energy failure developed similarly in both groups. There were no significant differences between the two groups in minimum [PCr]/[Pi], [NTP]/[EPP], and Naa/Cr, or maximum Lac/Cr, between 24 and 48 h after resuscitation (t test and Mann-Whitney). Intensive care was withdrawn 24 h after resuscitation from one piglet in the MgSO4 group, which had developed early onset, profound, delayed energy failure ([PCr]/[Pi] and [NTP]/[EPP] = 0.00) after a moderate insult. The final values from this piglet were used in the calculation of subsequent points in Fig. 2. Excluding these values made no difference to the conclusions reached.

(a) [PCr]/[Pi] and (b)[NTP]/[EPP] concentration ratios; (c) Lac/Cr, and (d) Naa/Cr peak-area ratios, in the two groups of piglets. Values are mean and SEM. Symbols: •, MgSO4 group; ○, placebo group. The fall in Naa/Cr during the acute insult reflects a transient increase in the transverse relaxation time of Cr(23).
DISCUSSION
Several lines of reasoning have suggested that systemic MgSO4 might prevent brain damage in birth-asphyxiated human infants. Nelson and Grether(12) observed that the incidence of cerebral palsy was reduced in preterm infants whose mothers received MgSO4, although the cause of the association remains uncertain(13). Studies of newborn animals have also suggested that MgSO4 is neuroprotective. When given 15 min after intracerebral injection of NMDA, MgSO4 reduced brain injury in 7-d-old rat pups(14); and MgSO4 reduced damage in 5-d-old mice when given before or 10 min after the injection of ibotenate, a glutamatergic agonist, into the brain(8). A combination of MgSO4 and oxygen free radical scavengers given immediately after unilateral carotid artery ligation and hypoxia reduced cerebral damage in 7-d-old rat pups(15). In 2-4 d-old piglets, infusion of MgSO4 before and during hypoxia, to produce high plasma levels of ≈7 mmol·L-1, prevented the hypoxia-induced modification of the NMDA receptor, and attenuated the reduction in Na+,K+-ATPase activity(16). The piglets were not resuscitated after hypoxia in that study.
No previous study had reported the effects of MgSO4 given as a single agent after severe transient cerebral hypoxia-ischemia, and the results of the present study provide no evidence of a cerebroprotective effect of MgSO4 in these circumstances. The severity of the insult in the treated and placebo groups was closely similar. As in previous studies(4, 5), cerebral metabolism apparently recovered on resuscitation, but subsequently delayed or secondary energy failure developed in spite of normal values for a wide range of systemic variables. No differences were found in the severity of delayed energy failure between the two groups, as judged by falls in [PCr]/[Pi] and [NTP]/[EPP]. Concomitant with these falls, the Lac/Cr peak-area ratio rose, indicating increased glycolysis relative to oxidative phosphorylation, and Naa/Cr fell, probably indicating neuronal loss(17). No support was therefore found for our hypothesis that MgSO4 would decrease the severity of delayed cerebral energy failure.
In the light of previous studies, this result was disappointing. One explanation for the lack of a cerebroprotective effect may be that the MgSO4 was given too late. Tan et al.(18) have shown in fetal sheep that acute reversed cerebral ischemia caused transient glutamate release with a return to baseline by 2 h. Later a secondary rise took place. Our study was designed to resemble the situation in asphyxiated infants, in whom treatment could be difficult to justify or achieve within 1 h of birth. If the timing of glutamate release in the piglets was similar to that in the fetal sheep(18), then the primary rise may already have initiated a cascade of damaging biochemical reactions before MgSO4 was given; there was apparently no beneficial effect of treatment before and during the secondary rise.
Another explanation for our failure to demonstrate any effect of MgSO4 may have been that Mg2+ did not reach the synaptic cleft in sufficient concentration to gate the NMDA receptors and prevent calcium influx. Normally, cerebrospinal fluid Mg2+ is maintained ≈30% above plasma levels, but after MgSO4 treatment in two asphyxiated infants, cerebrospinal fluid levels were lower than plasma levels(7), suggesting a limited transfer across the blood brain barrier of Mg2+ administered into the circulation. Mg2+ within cells is mainly bound to nucleic acids and proteins: if ATP generation is impaired then free [Mg2+] should increase due to dephosphorylation of MgATP. This makes interpretation of intracellular free Mg2+ levels difficult, but it is notable that these did not differ between the two groups in the present study, despite plasma Mg2+ levels within the range of those found in a pilot study for the use of MgSO4 in clinical trials(7). Doses that achieved higher levels were considered unsafe for infants because of their adverse systemic effects, and in our study, MABP and heart rate tended to fall with MgSO4 administration. It is possible that in both infants(19), and piglets(16), continuous infusion of MgSO4, rather than bolus doses, might have reduced adverse cardiovascular effects.
A final explanation for the failure of MgSO4 is that glutamate release is only one of a range of damaging biochemical reactions initiated by acute hypoxia-ischemia(20). Any beneficial effect of NMDA blockade after reoxygenation and reperfusion could have been overwhelmed by the adverse effects of other reactions. In contrast, protection against an insult provoked by intracerebral injection of a glutamatergic agonist(8, 14) is clearly specific for just one route to neuronal injury.
Delayed cerebral energy failure, as determined by 31P MRS, carries a very poor prognosis in birth-asphyxiated infants. Many infants die, and the survivors often suffer from severe neurodevelopmental disabilities and microcephaly(2, 3). Elevated cerebral Lac(21, 22) and reduced Naa(21) after birth asphyxia also convey an adverse prognosis. The failure of MgSO4 to prevent or ameliorate delayed energy failure in the piglet, as found in this study, does not support the wisdom of setting up clinical trials in human infants, at least until more information is available.
Abbreviations
- Cr:
-
creatine
- EPP:
-
exchangeable phosphate pool
- Lac:
-
lactate
- MABP:
-
mean arterial blood pressure
- MRS:
-
magnetic resonance spectroscopy
- Naa:
-
N-acetylaspartate
- NMDA:
-
N-methyl-D-aspartate
- PCr:
-
phosphocreatine
- Pi:
-
inorganic phosphate
References
Volpe JJ 1995 Neurology of the Newborn, 3rd Ed. WB Saunders, Philadelphia, pp 260–313
Azzopardi D, Wyatt JS, Cady EB, Delpy DT, Baudin J, Stewart AL, Hope PL, Hamilton PA, Reynolds EOR 1989 Prognosis of newborn infants with hypoxicischemic brain injury assessed by phosphorus magnetic resonance spectroscopy. Pediatr Res 25: 445–451
Roth SC, Edwards AD, Cady EB, Delpy DT, Wyatt JS, Azzopardi D, Baudin J, Townsend J, Stewart AL, Reynolds EOR 1992 Relation between cerebral oxidative metabolism following birth asphyxia and neurodevelopmental outcome and brain growth at one year. Dev Med Child Neurol 34: 285–295
Lorek A, Takei Y, Cady EB, Wyatt JS, Penrice J, Edwards AD, Peebles D, Wylezinska M, Owen-Reece H, Kirkbride V, Cooper CE, Aldridge RF, Roth S, Brown G, Delpy DT, Reynolds EOR 1994 Delayed(“secondary”) cerebral energy failure after acute hypoxia-ischemia in the newborn piglet: continuous 48-hour studies by phosphorus magnetic resonance spectroscopy. Pediatr Res 36: 699–706
Penrice J, Cady EB, Lorek A, Amess PN, Wylezinska M, D'Souza P, Cooper CE, Edwards AD, Wyatt JS, Reynolds EOR 1995 Proton MRS of the brain during acute hypoxia-ischaemia and delayed cerebral energy failure in the newborn piglet. Neuropediatrics 26: 341–342
Thoresen M, Penrice J, Lorek A, Cady EB, Wylezinska M, Kirkbride V, Cooper CE, Brown GC, Edwards AD, Wyatt JS, Reynolds EOR 1995 Mild hypothermia after severe transient cerebral hypoxia-ischemia ameliorates delayed cerebral energy failure in the newborn piglet. Pediatr Res 37: 667–670
Levene M, Blennow M, Whitelaw A, Hanko E, Fellman V, Hartley R 1995 Acute effects of two different doses of magnesium sulphate in infants with birth-asphyxia. Arch Dis Child 73:F174–F177
Marret S, Gressens P, Gadisseux J-F, Evrard P 1995 Prevention by magnesium of excitotoxic neuronal death in the developing brain: an animal model for clinical intervention studies. Dev Med Child Neurol 37: 473–484
Mosher TJ, Williams GD, Doumen C, La Nove KF, Smith MB 1992 Error in the calibration of the MgATP chemical-shift limit: effects on the determination of free magnesium by 31P NMR spectroscopy. Magn Reson Med 24: 163–169
Williams GD, Mosher TJ, Smith MB 1993 Simultaneous determination of intracellular magnesium and pH from the three 31P NMR chemical shifts of ATP. Anal Biochem 214: 458–467
Hore PJ 1983 Solvent suppression in Fourier transform nuclear magnetic resonance spectroscopy. J Magn Reson 55: 283–300
Nelson KB, Grether JK 1995 Can magnesium sulphate reduce the risk of cerebral palsy in very low birthweight infants?. Pediatrics 95: 263–269
Blair E, Palmer L, Stanley F 1996 Cerebral palsy in very low birth weight infants, pre-eclampsia and magnesium sulphate. Pediatrics 97: 780–781
McDonald JW, Sliverstein FS, Johnston MV 1990 Magnesium reduces N-methyl-D-aspartate (NMDA)-mediated brain injury in perinatal rats. Neurosci Lett 109: 234–238
Thordstein M, Bågenholm R, Thiringer K, Kjellmar I 1993 Scavengers of free oxygen radicals with magnesium ameliorate perinatal hypoxic-ischemic brain damage in the rat. Pediatr Res 34: 23–26
Hoffman DJ, Marro PJ, McGowan JE, Mishra OP, Delivoria-Papadopoulos M 1994 Protective effect of MgSO4 infusion on NMDA receptor binding characteristics during cerebral cortical hypoxia in the newborn piglet. Brain Res 644: 144–149
Miller BL 1991 A review of chemical issues in 1H NMR spectroscopy: N-acetyl-L-aspartate, creatine and choline. NMR Biomed 4: 47–52
Tan WKM, Williams CE, During MJ, Mallard CE, Gunning MI, Gunn AJ, Gluckman PD 1996 Accumulation of cytotoxins during the development of seizures and edema after hypoxic-ischemic injury in late gestation fetal sheep. Pediatr Res 39: 791–797
Wu TJ, Teng RJ, Yau K-IT 1995 Persistent pulmonary hypertension of the newborn treated with magnesium sulfate in premature neonates. Pediatrics 96: 472–474
Volpe JJ 1995 Neurology of the Newborn, 3rd Ed. WB Saunders, Philadelphia, pp 228–237
Groenendaal F, Veenhoven RH, Van Der Grond J, Jansen GH, Witkamp TD, De Vries LS 1994 Cerebral lactate and N-acetylaspartate/choline ratios in asphyxiated full-term neonates demonstrated in vivo using proton magnetic resonance spectroscopy. Pediatr Res 35: 148–151
Penrice J, Cady EB, Lorek A, Wylezinska M, Amess PN, Aldridge RF, Stewart A, Wyatt JS, Reynolds EOR 1996 Proton magnetic resonance spectroscopy of the brain in normal preterm and term infants, and early changes following perinatal hypoxia-ischemia. Pediatr Res 40: 6–14
Cady EB, Lorek A, Penrice J, Wylezinska M, Cooper CE, Brown GC, Owen-Reece H, Kirkbride V, Wyatt JS, Reynolds EOR 1994 Brain-metabolite transverse relaxation times in magnetic resonance spectroscopy increase as adenosine triphosphate depletes during secondary energy failure following acute hypoxia-ischaemia in the newborn piglet. Neurosci Lett 182: 201–204
Author information
Authors and Affiliations
Additional information
Supported by the Medical Research Council, UK, the Wellcome Trust, and University College London Hospitals Trust.
Rights and permissions
About this article
Cite this article
Penrice, J., Amess, P., Punwani, S. et al. Magnesium Sulfate after Transient Hypoxia-Ischemia Fails to Prevent Delayed Cerebral Energy Failure in the Newborn Piglet. Pediatr Res 41, 443–447 (1997). https://doi.org/10.1203/00006450-199703000-00024
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199703000-00024
This article is cited by
-
Short-term effects of early initiation of magnesium infusion combined with cooling after hypoxia–ischemia in term piglets
Pediatric Research (2019)
-
Magnesium sulfate: a last roll of the dice for anti-excitotoxicity?
Pediatric Research (2019)
-
Magnesium treatment for neuroprotection in ischemic diseases of the brain
Experimental & Translational Stroke Medicine (2013)
-
Hypothermia for the treatment of infants with hypoxic–ischemic encephalopathy
Journal of Perinatology (2010)
