
Abstract
The aim of this study was to produce a neonatal piglet model which, avoiding vessel ligation, exposed the whole animal to hypoxia and produced dose-dependent clinical encephalopathy and neuropathologic damage similar to that seen after birth asphyxia. Twenty-three piglets were halothane-anesthetized. Hypoxia was induced in 19 piglets by reducing the fractional concentration of inspired oxygen (Fio2) to the maximum concentration at which the EEG amplitude was below 7 μV (low amplitude) for 17-55 min. There were transient increases in Fio2 to correct bradycardia or hypotension. Posthypoxia, the piglets were extubated when breathing was stable. Four were shamtreated controls. We aimed at 72-h survival; seven died prematurely due to posthypoxic complications. EEG and a videotaped itemized neurologic assessment were recorded regularly. We found that 95% of the animals showed neuropathologic damage. The duration of low amplitude EEG during the insult and the arterial pH at the end of the insult correlated with cortical/white matter damage; r = 0.75 and 0.81, respectively. Early postinsult EEG background amplitude (r = 0.86 at 3 h) and neurologic score (r = 0.79 at 8 h) correlated with neuropathology. Epileptic seizures in seven animals were always associated with severe neuropathologic damage. We conclude that EEG-controlled hypoxia and subsequent intensive care enabled the animals to survive with an encephalopathy which correlated with the cerebral hypoxic insult. The encephalopathy was clinically, electrophysiologically, and neuropathologically similar to that in the asphyxiated term infant. This model is suitable for examining mechanisms of damage and evaluation of potential protective therapies after birth asphyxia.
Similar content being viewed by others

Main
No intervention after human birth asphyxia has been shown to reduce hypoxic ischemic encephalopathy and subsequent brain damage(1, 2). However, from recent work on both newborn and adult animals there is convincing evidence that neuronal rescue therapy is effective even if applied after the insult(3, 4).
Current understanding of the pathophysiology of hypoxic ischemic encephalopathy has evolved mainly from animal studies using a range of models. We aimed at developing a survival animal model which produced the clinical insults and pathologic outcome experienced by newborn babies, without the need for surgical incisions which might complicate postinsult handling of the animals requiring pain relief and so forth. Unlike other cerebral insult models, the whole body should be exposed to the same hypoxic stress in a model of newborn asphyxia, because the effects of therapeutic interventions are also relevant on the posthypoxic heart, kidney, and gut. Some newborn pigs tolerate only 10-15 min of a preset degree of hypoxia (i.e. Fio2 = 0.08) before cardiac arrest, whereas others tolerate 180 min(5). The newborn piglet heart appears to be more vulnerable to hypoxia than is the brain(6, 7).
In contrast to previous cerebral hypoxia models, we therefore individualized the insult according to the cerebral response (EEG) of each piglet rather than using a set Fio2 values. This enabled animal survival with significant neurologic and neuropathologic findings. The animals were followed by EEG recordings and neurologic examinations, and these measurers were related to the neuropathologic examination.
METHODS
Animal preparation. Norwegian Animal Research Committee approval was obtained. Twenty three Landrace piglets (median age, 24 h; range, 10-72 h) were supplied on the morning of the study by local breeders. Four animals were used as controls and went through the same protocol except for exposure to low Fio2. Before instrumentation the animals were weighed, observed, and video filmed during normal activity.
Anesthesia and ventilation. Anesthesia was initiated by passing 1.5-2% halothane in oxygen through the closed box where the piglet was kept. The piglet was intubated, moved to an incubator, and mechanically ventilated with halothane in a mixture of 30-40% oxygen and 60-70% nitrogen using a Neovent 90 ventilator (Vickers Medical, Basingstoke, England). The snout and mouth were covered with plastic to avoid air entering the trachea around the uncuffed endotracheal tube. The inspired halothane concentration was adjusted to keep the end-tidal halothane concentration at 1.0% as monitored 3 cm down the endotracheal tube by a sidestream multigas analyzer (Capnomac Ultima, Datex, Helsinki, Finland), and the ventilation was adjusted to keep end-tidal CO2 (PAco2) at 4.0-5.0 kPa during baseline. The ventilator setting was not adjusted further during the insult. Transcutaneous arterial oxygen saturation (Sao2) was registered from a pulsoxymeter probe (501+, Criticare Systems Inc., Pewakee, WI) positioned on a hind leg hoof and kept above 96% by adjusting the Fio2 before and after the hypoxia.
Temperature and cardiovascular monitoring. Temperature was monitored rectally and at the tympanic membrane (which is similar to intracerebral temperature in this model)(8) with cromel-alumel temperature sensors. During the insult and for 3-5 h while anesthetized, the temperature at the tympanic membrane was kept stable at 39°C (range 38.7-39.2 °C) as small changes in intraischemic temperature alter outcome(9). After discontinuation of anesthesia, rectal temperature was kept around 39 °C.
No surgical incisions were made, the only procedures being intubation and umbilical vessel catheterization. MABP of 5.3 kPa was the minimum acceptable pressure during baseline recordings as well as in the posthypoxic period. Before the insult, hypotension (MABP < 5.3 kPa) was treated with 10 mL-1·kg of 10% human albumin. During the insult no lower MABP limit was set, and the degree of hypotension allowed was dependent on the individual tolerance to hypoxia. Three-lead ECG was continuously recorded.
Monitoring of EEG activity and storing of data. Two-channel(interelectrode distance of 3 cm) EEG from each hemisphere in the parasagittal direction was recorded. Skin impedance was lowered by shaving and washing, and impedance of the surface electrodes was always <2kΩ during recordings. The raw EEG signal was continuously recorded on tape (Oxford Medilog system 9000) before, during, and for the first 6 h after the insult and then intermittently. The EEG signal was continuously printed out simultaneously with ECG, MABP, PAco2, and end-expiratory oxygen tension(PAo2) on a chart recorder. A high frequency filter of 35 Hz but no low frequency filter was used on the EEG paper printout at either 1 mm/s or 25 mm/s paper speed. MABP, PAco2, PAo2, and tympanic and rectal temperature were also logged into a computer (Apple Macintosh SE, Workbench program).
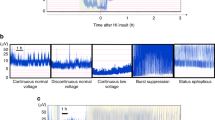
Experimental protocol. The piglet was in the prone position, supported with pads in both axilla and inguinal regions for the thorax to move freely. After stabilization, a 30-min baseline period was allowed before commencing the insult by abruptly reducing Fio2 to around 6% (seeFig. 1). LA EEG, defined as a peak-to-peak amplitude of less than 7 μV for more than 5 s, was achieved after 1.7 min (median, range 0.7-4.0 min). The oxygen fraction was regulated to the highest value which gave LA EEG. The ability of the cardiovascular system to tolerate the reduced Fio2 varied. When severe bradycardia and/or severe hypotension occurred, the Fio2 was transiently increased until the heart rate and blood pressure recovered, whereupon the Fio2 was again reduced according to the tolerance of the animal. The response to bradycardia had to be fast, within about 20 s, and the step increase in Fio2 depended on the severity of bradycardia, usually 0.5-1.5%. With cardiac arrest 100% O2 and cardiac compressions were given. The duration of the increased Fio2 lasted until heart rate, MABP, and PAco2 started to rise which varied between 15 s and several min. The increased Fio2 would sometimes transiently increase the EEG amplitude to above 7 μV, thus interrupting the LA EEG period. The individual average MABP obtained at the Fio2 value which gave a LA EEG varied between 23 and 40 mm Hg (3.1 and 5.3 kPa) (see Table 3).

The upper panels show intermittent periods from a continuous EEG from an animal who suffered a mild insult with summated duration of LA EEG of 17 min (total duration of insult, 45 min) and who recovered completely and showed no brain pathology. To the left, after Fio2 was reduced to 6.0%, the EEG amplitude decreased to below 7 μV within 1.5 min. The EEG had fully recovered by 1 h after reoxygenation. After 12 h a pattern of normal sleep stage cycles in the sleeping unsedated animal was seen. The lower panels show selected EEGs from an animal with total insult duration of 45 min, including a 40-min LA EEG at reoxygenation time; 0 and 2 min, and 1, 3, and 5 h after the insult. There was a burst suppression pattern at 1 h and electroconvulsive activity at varying frequencies from 3 h, which was accompanied by clinical seizures at 5 h. This animal had maximal damage in most brain regions.
The insult was terminated by reoxygenating with 100% oxygen for 30 min. One hundred percent O2 was chosen because this was current clinical practice when the study started. Thereafter Fio2 was reduced to 30% or to the minimum level giving Sao2 > 96%. The metabolic acidosis at the end of the insult was half corrected with NaHCO3 (base deficit× 0.4 × weight in kg × 0.5). After the insult, MABP was kept at 40 mm Hg or more by infusing dopamine at a rate of 5-20μg-1·kg-1·min as required. Dopamine was never given during the hypoxic insult. The pigs were anesthetized for 3-5 h after the insult, after which they were weaned from the ventilator when breathing was adequate.
Blood chemistry sampling. Arterial samples were analyzed for blood gases (AVL 945, Biomedical Instruments, Schaffhausen, Switzerland), Hb(IL 482 CO-Oximeter, Instrumentation Laboratory, Lexington, MA), lactate(Technicon RA-1000 Chemistry Analysator, Tarrytown, NY), and glucose(Haemo-Glucotest 1-44, Boehringer Mannheim, Mannheim, Germany) taken at baseline, end of hypoxia, and 3 h after hypoxia. Blood gases and glucose were later analyzed according to clinical need.
Drugs, fluids, and nutrition. Antibiotics (gentamicin 2.5 mg-1·kg and cephalothin 20 mg-1·kg) were given i.v. twice daily. Before and during the insult glucose 35 g-1·L in NaCl 4.5 g-1·L was given at 7.5 mL-1·kg-1·h. At the end of the insult and for the next 24 h, glucose at 5 mL-1·kg-1·h was given at varying concentrations (35 to 50 g-1·L) to keep the blood glucose between 2 and 10 mmol-1·L. From the 2nd d, according to their clinical state, the piglets were bottle-fed 10 mL-1·kg-1·h (pig milk formula Baby Lactal, Peter Möller a/s, Oslo, Norway) at 2-3-h intervals. The piglets were weighed daily. The weight increased (mean (SD)) from 1.7 (0.2) kg to 2.1 (0.4) kg for those who survived to 3 d.
EEG evaluation of the insult and the posthypoxic period. The following EEG values during the total duration of the insult were noted: the total period of LA EEG (<7.0 μV) from summating all individual LA EEG periods, the summated duration of EEG with amplitude ≤3.5 μV (very LA EEG) and the longest single period of LA EEG. The delay until electrical activity above 7 μV appeared after resuscitation was noted as well as the delay until onset of and duration of any electroconvulsive activity (seeTable 2). EEG recordings were continuous until extubation and examined with respect to background activity and presence of pathology at 1, 3, 6, 12, 24, and 48 h after the insult.
Seizures and anticonvulsive treatment. Electroconvulsive activity was defined as spike or sharp wave activity with an amplitude more than double the background activity (amplitude of background activity was taken from compressed EEG, as in Figure 1, ignoring peaks) at a regular frequency and lasting longer than 20 s. Clinical seizures were defined as rhythmic pathologic movements accompanied by electroconvulsive activity.
Electroconvulsive activity being continuous for more than 10 min with or without clinical seizures was treated with one, two, or more (depending on clinical effect) of the following four drugs used in clinical practice (listed in order of administration): lidocaine bolus (4 mg-1·kg) followed by continuous infusion (2-8μg-1·kg-1·min), diazepam bolus injection 0.5-0.75 mg-1·kg, phenobarbital 20 mg-1·kg, and phenytoin 20 mg-1·kg. When the first drug chosen had no effect after 30 min, the first dose was repeated or a second drug was tried.
Neurologic assessment. Each piglet was filmed before hypoxia and then approximately every 8 h for the first 24 h. Daily filming until death or the end of the study was performed in 14 animals. The piglet was filmed in an active state if possible, walking around, feeding, being held by an examiner, and being laid supine to see how quickly the piglet could right itself. If the piglet still needed intensive care, gentle stimulation and manipulation were used according to the piglet's tolerance. Pathologic movements were filmed whenever possible. After many observations of newborn piglets under experimental conditions, it was possible to make a list of nine neurologic items which could be scored as: 2, normal; 1, moderately abnormal; or 0, definitely pathologic. The maximum score for a normal piglet was 18. Neurologic items were: 1) Normal respiration, without apnea, retractions, or need for oxygen; 2) consciousness; 3) orientation. Looking at and investigating the surroundings; 4) ability to walk on all four limbs in one direction without falling;5) ability to control the forelimbs using them to raise quickly from a lying position; 6) ability to control the hind limbs using them to raise quickly from a lying position and keeping them together in the upright position; 7) maintenance of steady and equal tone in forelimbs and hind limbs; 8) almost continuous activity when awake; 9) absence of pathologic movements were scored as 2. Sustained clonic movements or persistent tonic postures were scored as 0. Occasional cycling movements or jerks were scored as 1.
Having drawn up this scoring system, a neonatologist with experience in neurologic evaluation (A.W.) who was unaware of the treatment, EEG, or neuropathological results, assessed each videotape of each piglet.
At the conclusion of the experiment the piglets were again anesthetized with halothane, and the brains were perfusion fixed through the transcardiac route with 4% phosphate-buffered formaldehyde running for 40 min. Nineteen of the 23 animals were also autopsied.
Histologic evaluation. Coronal blocks through the brain were embedded in paraffin, subserially sectioned at 5 μm, and stained with hematoxylin and eosin. At regular intervals from each brain 15 sections, including frontal and parietal cortex, hippocampus, basal ganglia, thalamus, and one sagittal section through the right cerebellar hemisphere were examined by E.M.L., who was blinded to the mode of treatment and all clinical information. The extent of damage for each of the five regions was graded with 0.5-intervals on a 9-step scale from 0.0 to 4.0 as described inTable 1. The term “incomplete infarct” means in gray matter a localized area where the neurons are mainly necrotic whereas the other tissue elements (i.e. glia and vessels) are preserved. In a completely infarcted area all tissue elements are damaged.
The minimum time for visible histologic damage to develop is shorter the more severe the injury, and an increasing number of damaged neurons are visible from 6 to 24 h(10, 11). Early evaluation might therefore underestimate the damage. In one animal who survived 13 h, the damage score was already maximal (4.0) in cortex and hippocampus, and the regional distribution was similar to that of those surviving longer after seizures. One animal with the most severe clinical course (early seizures followed by cardiovascular collapse and death at 4 h) had no visible damage by light microscopy. In calculations, an estimate of damage, equal to the maximum found in surviving piglets with seizures, was used.
Data analysis. Based on the summated duration of LA EEG(Table 2) and the presence or absence of posthypoxic seizures, the piglets were post hoc divided into four subgroups for further analysis:
Group A. Four sham-treated control piglets.
Group B. Six piglets with short 27 (6.9)-min (mean, SD) duration of summated LA EEG where five developed brain damage and none developed seizures.
Group C. Six piglets with long 40 (3.1)-min duration of summated LA EEG where all developed brain damage and none developed seizures.
Group D. Seven piglets with long 45 (8.7)-min duration of summated LA EEG where all developed seizures.
Between group comparisons were performed using one-way analysis of variance with Bonferroni correction for repeated testing. Within group comparison between different experimental conditions was performed using one-way analysis of variance for repeated measures. The data were also analyzed using nonparametric (Kruskall-Wallis, Wilcoxon signed rank) tests, and similar results were obtained (not given). Simple regression was used to describe the relationships between the duration of LA EEG, EEG background activity, and neurology score with the main outcome variable brain pathology. Comparisons of brain damage between different brain regions were performed using pairedt tests with Bonferroni corrections [Statview 4.0, Abacus Concepts Inc., Berkeley, CA; level of significance was set to 0.05 (two-sided)]. For descriptive statistics, mean with SD or median with total range was used.
RESULTS
Cardiovascular effects of the hypoxic insult. The average MABP was significantly reduced by 30% during the insult and did not differ between the three groups; group B (short duration of LA EEG without seizures), C (long duration of LA EEG without seizures), and D (long duration of LA EEG with seizures). To achieve a MABP of >5.3 kPa after hypoxia, six piglets (two from group D and four from group C) received dopamine for a mean duration of 3 h (range 1-5 h). The mean delay before the start of dopamine infusion was 1 h after reoxygenation (range 15 min-2h). At the end of the insult, arterial pH was significantly lower in piglets who subsequently developed seizures than in those who did not. Mean blood glucose for experimental animals was 7.8 (3.8) mmol·L-1 at the end of the insult with no relation to the pathologic outcome nor experimental subgroup. Changes in heart rate, Paco2, Pao2, and lactate were similar in all subgroups(see Table 3).
Transient increases in Fio2 successfully counteracted hypotension and bradycardia during the insult. Cardiac massage with or without i.v. adrenaline was needed in five animals. Three of these animals had severe bradycardia (heart rate 0-50 beats·min-1) and hypotension (MABP< 2 kPa) for 3-4 min and received adrenaline (0.2-0.5 mg). Two of these three developed seizures and died within 13 h, the third did not develop seizures but became severely damaged. Two animals received brief cardiac massage (0.3 and 1.3 min) and transient increase in Fio2 before recovery from bradycardia. They did not develop seizures and had minor brain damage. Four shamtreated controls achieved spontaneous ventilation 60 min after anesthesia (median, range 45-90 min), 16 experimental animals after 100 min (30 min to 12 h). Three pigs were never extubated.
Early mortality. We aimed at a minimum of 72-h survival, but seven piglets had shorter survival. Five early deaths were due to complications secondary to the hypoxic-ischemic insult: one had cessation of cerebral circulation with evidence of raised intracranial pressure, two had cardiac depression with hypotension and subsequent arrest. Mechanical ventilation was stopped at 24 h in two piglets who were persistently unresponsive with EEG amplitude <3.5 μV. Four of these five developed seizures. In addition, a sixth pig died from aspiration pneumonia and multiple lung emboli, and a seventh pig died from Escherichia coli septicemia and peritonitis.
EEG changes after hypoxia. Figure 1, upper tracing, shows intermittent periods of EEG obtained before, during, and after the insult in the animal with the shortest duration (17 min) of LA EEG, the only animal which did not develop any brain damage. In this piglet the EEG amplitude was normalized within 1 h after the insult with normal EEG sleep stage variation after 12 h. The lower tracing shows examples of postinsult seizure activity in a piglet who developed severe brain damage.
Table 2 show the EEG characteristics at different times in each subgroup of the animals. There was a consistent trend for the seizure group (D) to be subjected to more severe insults. Although not significant, they tended to have longer periods with very LA EEG (<3.5μV) than group C, and longer duration of the longest single period of LA EEG. The median delay from the end of the insult until the EEG activity had reached 7 μV or more was significantly longer among those who developed seizures (one animal with seizures never restored EEG activity with amplitude above 7 μV). The EEG background activity for the first 12 h was also significantly more depressed in this group than in the others. The time delay after the insult until EEG amplitude reached 7 μV was highly predictive of whether seizures would occur or not. Median delay before resumption of threshold activity was 60 min in the seizure group (D), compared with 8.0 min in group C.
Posthypoxic seizures. In group D, electroconvulsive activity and clinical seizures were first observed at a median of 167 (range 2-330) min after the end of the insult. The duration of seizures varied from 0.5 to 20 h. Electrical and clinical seizures started simultaneously in four animals. In three animals electrical seizures preceded clinical seizures by 1-15 min. Two animals did not receive any anticonvulsive treatment due to short lasting seizures followed by very LA EEG and later cardiovascular collapse. Five animals received anticonvulsive treatment after a median duration of seizures of 30 min (range 15 min to 6 h). In general, the response to anticonvulsive treatment was poor with only one animal responding within minutes and four animals continuing to have seizures despite multiple anticonvulsant drugs.
Clinical encephalopathy and subsequent neuropathology. All animals who developed seizures had neurology scores less than 6 for the first 24 h, and neither of those who did not develop seizures had such low scores(Fig. 2). One animal with seizures recovered to near normal neurology score at 3 d; this animal had a pH of 7.1 at the end of insult, regained EEG activity readily after 8 min, received only one anticonvulsant before seizure control, and had the lowest cortical pathology score (3.0) among those with seizures. The neurology score predicted altered pathologic damage better if performed early rather than late.

Time course of neurology score in the three subgroups of animals. Not all of the animals were examined regularly and seven died before 72 h (marked with †). •, long insults with seizures; ○, long insults without seizures; ▵, short insults without seizures.
Posthypoxic damage in other organs. The four shamtreated controls were normal. Eleven of the 15 experimental animals autopsied had pathologic findings in one or more organs other than the brain. We saw mild bronchopneumonia, thrombi in the pulmonary arteries, a subpleural pulmonary hemorrhage, small areas of liver necrosis, subcapsular liver hematomas, a macroscopic adrenal hemorrhage, a kidney infarct, mild acute changes in the superficial intestinal mucosa, and a small (<1 mm) cell infiltrate in the heart, probably of antenatal origin.
Distribution of neuropathologic damage. Eighteen of the 19 experimental animals had neuropathologic damage in one or more regions of the brain. Seventeen were damaged in both gray and white matter; one had minor damage in white matter only. Cerebellar lesions were seen in 17 animals, 15 had hippocampal damage, 13 basal ganglia damage, whereas only 9 had thalamic damage. The degree of damage varied from localized areas with selective nerve cell necrosis to large infarcts seen as pale areas with vacuolization of the neuropil and necrotic neurons with eosinophilic cytoplasm. A typical localization in mild to moderately damaged animals was necrosis most marked in the depths of the gyri as seen in infants(12)(Fig. 3a). White matter lesions were first seen in the subcortical regions, varying from small areas with vacuolization of neurophil and retraction balls (incomplete infarcts) (Fig. 3b), to large complete infarcts. In severely damaged brains, large confluent infarcts affecting both cortex and white matter were located mainly in the watershed areas between the large cerebral arteries. Typically, the parasagittal regions of the hemispheres were the first to be affected. Cerebellar damage varied from selective Purkinje cell necrosis (Fig. 3d), to confluent infarcts mainly in watershed areas (Fig. 3c).

(a) A small infarct (three curved arrows) in the depth of the cortical gyrus (ulegyria). Beneath the infarct (one arrow) there is a white matter lesion with vacuolization of neuropil. Hematoxylin and eosin (H&E) stain, magnification × 25. (b) White matter lesion with retraction balls (curved arrows). H&E stain, × 250.(c) A cerebellar infarct in the upper left part of the figure is delineated by curved arrows. Healthy tissue with selective Purkinje cell necrosis (large arrow) to the lower right, H&E stain, × 25.(d) Purkinje cell necrosis (eosinophilic cells) in a noninfarcted area (curved arrows. H&E stain, × 250.
The mean (SD) damage score (defined in Table 1) for cortex/white matter was 2.6 (1.2), hippocampus 2.6 (1.5), cerebellum 2.4(1.2), basal ganglia 1.5 (1.5), and thalamus 0.6 (0.8). The thalamus was significantly less damaged than all other regions, and basal ganglia were significantly less damaged than cortex/white matter, hippocampus, and cerebellum. The order of the degree of damage for each region of the brain was always group B < C < D (Fig. 4). Comparisons of groups of similar duration both with (D) and without (C) seizures showed significantly more damage in the seizure group in cortex and white matter, hippocampus and the basal ganglia. Within each subgroup cortex and white matter, hippocampus and cerebellum were the most damaged areas.

Mean (SD) for pathology score from five regions of the brain as indicated for each of the groups B, C, and D. Damage in cortex and white matter, hippocampus, and basal ganglia was significantly (p< 0.05) greater in animals with seizures (group D) than in those without(groups B and C). Cortex, hippocampus, and cerebellum had damage of the same magnitude and significantly more than the basal ganglia and thalamus.
Physiologic variables during the insult which correlated with pathologic outcome. There was a significant correlation between the duration of LA EEG and the pathology score in cortex/white matter (r= 0.75) (Fig. 5A). The corresponding correlation was 0.71, 0.60, 0.58, and 0.45 for the hippocampus, cerebellum, basal ganglia, and thalamus, respectively. The longest single period of LA EEG(Table 3) predicted pathologic damage in each region equally well, r being 0.80, 0.69, 0.64, 0.73, and 0.56, respectively.

(A) On the x axis is the summated period of LA EEG (<7 μV) in min. On the y axis is the corresponding pathology score from cortex/white matter. ♦ = Animals who developed seizures after asphyxia; • = animals without seizures. Sham-treated control animals had no insult and were not damaged (not shown in the figure). The damage in one animal (*) was estimated (see“Methods”). (B) Relationship between arterial pH at the end of the insult and later pathology score. (C) Relationship between background EEG amplitude at 1 h after the insult and later pathology score. (D) Relationship between the neurology score obtained 8 h after the insult and the later pathology score.
In one animal LA EEG was less than 20 min, and there was no brain damage. If LA EEG was >40 min, most animals developed posthypoxic seizures.Figure 5B shows a linear relationship between arterial pH and pathology score (r = 0.77).
Physiologic variables after the insult which correlated with pathologic outcome. EEG background activity at 1 h postinsult correlated with pathology score for cortical/white matter at death (r = 0.86)(Fig. 5C). At 3, 6, 12, 24, and 48 h postinsult, this correlation was 0.86 (n = 19), 0.58 (n = 18), 0.76(n = 15), 0.81 (n = 10), and 0.31 (n = 9), respectively. All animals were not examined at all times, and seven died before 72 h. The neurologic assessment using the itemized neurology score correlated best with cortex and white matter damage if performed early,r = 0.77 (n = 18) at 8 h, r = 0.75 (n= 11) at 24 h, and 0.51 (n = 7) at 48 h.
DISCUSSION
Potential neuroprotective therapies after birth asphyxia must be evaluated in newborn animal models because the mechanisms of damage and repair changes with maturation. Younger animals have a relatively greater resistance to hypoxemic and ischemic injury than older animals(13, 14). On the other hand, groups of neurons are more vulnerable to injury (mainly by activation of excitatory amino acid receptors) in the immature animal than similar neurons in the adult(15). The lower cerebral rate of energy utilization in the young animal(16) is considered to be secondary to the anatomical and electrophysiologic immaturity of the immature brain. The immature brain differs from the mature brain in many respects; neuronal proliferation, ongoing myelination(17), on-going apoptosis(18), lower proportion of glial cells, lower metabolic needs(19, 20), different substrate dependency, and enzyme activity(21). The response to ischemia is different in the immature brain and shows species differences: the injury is not enhanced by an increased blood glucose in newborn rats(22) in contrast to the newborn pig(23) and adolescent monkey(24). The exitotoxicity via N-methyl-D-aspartate receptors is much higher in the immature brain compared with that of the adult(25), the relative vulnerability of white matter is higher, and the inflammatory response/gliotic reactions appear to be different(26). Transferring results from adults to newborns may therefore be misleading.
Shortcomings of other animal models of neonatal hypoxic ischemic encephalopathy. No model is perfect in reflecting the complexity of the human brain. A major limitation with most models is the use of vessel occlusion with regional ischemia, which is not part of clinical asphyxia in the newborn. In the 7-d-old rat with unilateral common carotid ligation exposed to 2-3 h of hypoxia (8% oxygen)(27), gross motor performance is not affected(28). The model is well characterized, and animals are available in large numbers and particularly useful for screening new treatment strategies, but it cannot be used in studies where physiologic monitoring is important.
Myers(29) work on fetal and neonatal monkeys has contributed extensively to the understanding of the pathophysiology of perinatal brain damage. However, the newborn monkey brain is more mature than that of the human, and the cost and availability of this animal limits its use.
Of survival piglet models, two models of transient bilateral carotid occlusion combined with either mild hypoxia (Fio2 = 0.12-0.16) until 70% ATP depletion(30) or 30 min of bilateral carotid occlusion with moderate hypotension and 15 min of severe hypoxia (Fio2= 0.06)(31) have proven useful in studies of post hypoxic-ischemic neuroprotection(32, 33). In a third model, a cuff around the neck inflated to a pressure above the systolic blood pressure renders the brain partially ischemic(34). None of these models exposes the rest of the body to significant hypoxia as seen during birth asphyxia. A survival model of global hypoxia was recently published(5). In piglets aged 2-5 d, the insult was produced by ventilating the piglets with 8% oxygen until the blood pressure reached 2.7 kPa before resuscitation. The time until target hypotension was reached varied from 10 to 180 min, and no relationship to subsequent outcome nor to any development of seizures was presented. The damage to the brain was generally small and affected mainly cortex, white matter, and cerebellum. The variable vulnerability of the heart(6) to hypoxia and hypotension limits the ability of this model to produce severe insults with encephalopathy.
Of acute hypoxia models without survival, the group from Philadelphia has for some years used a global hypoxia model where they ventilate the pigs with 5-7% oxygen for 60 min(35, 36). The degree of hypoxia during the insult is similar to that in our model, the differences being that we regulate the Fio2, according the individual cerebral response to hypoxia, and we have a significant reduction of MABP during the insult and substantial acidosis at the end of the insult. Their model, with invasive monitoring and sampling, has mapped out changes of the NA+K+ATPase activity and lipid peroxydation during the insult. However, these insults have not been shown to be severe enough to produce encephalopathy or permanent damage.
The distribution of brain damage. Our model is characterized by1) clinical and electrophysiologic encephalopathy (with seizures in severe cases, 39%); 2) cardiovascular and biochemical changes; and3) distribution of brain pathology similar to that seen in the full-term infant. In humans, the distribution of brain pathology in response to hypoxia-ischemia is age-dependent with significant differences between the preterm and term newborn and the adult(12). Typical in the term infant suffering prolonged asphyxia are cortical lesions in the watershed areas of the hemispheres (parasagittal region, depths of the sulci), the hippocampus, and the cerebellum, with less damage to thalamus and the brain stem(37). In the present study, cortical lesions demonstrated the typical pattern of ulegyria with subjacent white matter lesions. In animals with overall minor injury, there were several small but separate lesions in both gray and white matter. In newborns and children less than 2-3 mo, white matter lesions are frequent, particularly in the periventricular region in the premature infant or subcortically in the term newborn(38, 39). Incomplete myelinization is a prerequisite for white matter damage(40, 41), which is rarely found in primate models whose brains are much more mature at birth than are those of humans. We assume the subcortical white matter lesions found in this model to correspond to the subcortical leukomalacia typically found in the term infant.
Of the cerebral cortical regions, the hippocampus is the area particularly vulnerable to hypoxic-ischemic injury(2), and in most animals all regions of the hippocampus (except the dentate gyrus) were uniformly damaged. Sommer's sector in humans, which corresponds to the superior region in piglet hippocampus(42) and the CA1 region in rats, is typically the most sensitive to hypoxic damage in term infants as well as adults(2), and in agreement with this, the superior region was the first to be damaged in piglets with minor damage.
The cerebellum is especially vulnerable to hypoxic ischemic injury and in particular the Purkinje cells(2). We observed selective Purkinje cell necrosis in all animals with cerebellar damage. Unlike the adult, cerebellar infarcts in the watershed area are common in severely damaged newborns(2) and were a consistent finding in the most damaged animals in the present study.
EEG characteristics of the hypoxic insult. By monitoring the response of the EEG signal and the cardiovascular response to hypoxia and adjusting the Fio2 accordingly, we were able to expose each piglet to an individually required level of hypoxia that produced a LA EEG. We found 23 min to be the minimum duration of LA EEG to produce damage, and this is in agreement with a monkey model of systemic hypotension where 25 min of EEG flattening were necessary to produce marked brain injury(43). In human infants who have suffered severe birth asphyxia, one of the most reliable predictors of later mortality and morbidity is changes in background EEG activity within hours after birth(44–46). In the present study reduced postinsult EEG amplitude for the first 24 h correlated well with later neuropathologic damage.
Posthypoxic seizures and clinical encephalopathy. In infants, hypoxic ischemic encephalopathy is usually secondary to perinatal asphyxia(47) and is the single most common cause of neonatal seizures(48). The fact that 39% of the piglets developed posthypoxic seizures is thus another point of similarity to the birth-asphyxiated newborn. In several species, the clinical appearance of seizures varies with neuronal maturation. For instance, the newborn monkey, which has proper orientation alignment and layering of cortical neurons, demonstrates the adult pattern of seizures with well organized rhythmic movements(49). In human infants, seizure movements are less organized the shorter the gestational age. Synchronous discharges recorded from surface EEG may not correlate with behavioral seizure phenomena because of deficient myelinization of cerebral efferent systems. We saw both disorganized and rhythmic movement patterns in the piglets, and there was not a consistent coupling of clinical and electrical seizures. There could be pedaling movements for hours or sucking, chewing, drooling, and apnea without an EEG correlate. We also saw generalized seizures with tonic extension, particularly of the upper part of the body-mimicking decerebrate posturing-usually not accompanied by EEG seizures. Such clinical phenomena may be epileptic and arise from subcortical structures not recorded on surface EEG, or they could be primitive brain stem and spinal motor pattern release phenomena.
We treated seizure activity with conventional therapy using several different drugs and repeated dosages and found the seizure activity to be quite resistant to anticonvulsive drug therapy, just like neonatal seizures after birth asphyxia(50). If the anticonvulsant therapy also was cerebroprotective, that effect was not apparent, as the animals with seizures were the ones most severely damaged, and many died early.
To document the clinical condition of the animals, we developed a scoring system of 9 items for neurologic assessment. At 8, 16 and 24 h, this correlated well with later pathologic outcome. Before 24 h, a score of less than 6 was highly predictive of the development of seizures and/or early death. At 3 d the score was near normal also in piglets with moderate damage, including one piglet with posthypoxic seizures (Fig. 2). This suggests that the score is more suitable as an acute encephalopathy score than as a neurologic outcome measure.
Choice of inhaled gases in our model. An increased Paco2 during the insult would render the animal more acidotic, which is known to further impair cardiac function(51). Although hypercapnia occurs in human perinatal asphyxia, we therefore chose not to increase Paco2, as lowering pH would limit the hypoxic tolerance of the animal. Recently, Vannucci et al.(52) published convincing evidence from the 7-d-old rat model that 6% carbon dioxide significantly reduced the development of brain damage as examined 3 wk after the insult. If adding CO2 during the insult had the same protective effect in our model, we might not have succeeded in producing severe brain damage with low Fio2 alone.
We chose halothane as the anesthetic for several reasons. With halothane, only one substance is necessary for induction, anesthesia, and analgesia and, unlike i.v. drugs, it can be continuously monitored and kept at constant and standard levels in all animals. Although there is a significant drop in MABP due to halothane anesthesia, halothane is shown to exert a protective cardiac effect in the failing heart(53). This might add to the hypoxic tolerance of the heart, allowing longer and more severe hypoxic exposure to the brain. In our study all animals were exposed to the same concentrations of halothane. To our knowledge, a neuroprotective effect of halothane has not been shown in any newborn animal model.
Conclusion. We have developed a model of newborn hypoxic-ischemic brain damage where the whole body is exposed to the same hypoxic stress and without involving ligation of precerebral vessels. The model is reproducible in a dose-dependent way with a distribution of histologic brain damage similar to that in the asphyxiated term infant. There is clinical and EEG evidence of encephalopathy that correlates with subsequent neuropathology. As in severe birth asphyxia we have complications in other organs than the brain. We have developed a neurology score which in the early posthypoxic phase predicts pathologic damage in cortex and white matter at 3 d. This model should be suitable for examining mechanisms of damage and repair and testing out new treatment strategies.
Abbreviations
- LA:
-
low amplitude
- Fio2:
-
fraction of inspiratory oxygen
- PAco2:
-
end-tidal CO2
- PAo2:
-
end-tidal O2
- PacoO2:
-
arterial CO2
- Pao2:
-
arterial O2
- Sao2:
-
transcutaneous arterial oxygen saturation
- MABP:
-
mean arterial blood pressure
References
Nelson KB, Ellenberg JH 1986 Antecedents of cerebral palsy. Multivariate analysis of risk. N Engl J Med 315: 81–86.
Volpe JJ 1995 Hypoxic-ischemic encephalopathy: neuropathology and pathogenesis. In: Volpe JJ (ed) Neurology of the Newborn. WB Saunders, Philadelphia, pp 279–313.
Hagberg H, Gilland E, Diemer NH, Andiné P 1994 Hypoxia-ischemia in the neonatal rat brain: histopathology after post-treatment with NMDA and non-NMDA receptor antagonists. Biol Neonate 66: 205–213.
Bigge CF, Boxer PA 1994 Neuronal cell death and strategies for neuroprotection. In: Bristol JA (eds) Annual Reports in Medicinal Chemistry. Academic Press, San Diego, CA, pp 13–22.
Rootwelt T, Løberg EM, Moen A,Øyasæther S, Saugstad OD 1992 Hypoxemia and reoxygenation with 21% or 100% oxygen in newborn pigs: changes in blood pressure, base deficit, and hypoxanthine and brain morphology. Pediatr Res 32: 107–113.
Jacobson HE, Windle WF 1960 Responses of foetal and newborn monkeys to asphyxia. J Physiol 153: 447–456.
Myers RE, Kopf GS, Mirvis DM 1980 Hemodynamic response to profound hypoxia in intact rhesus monkeys. Stroke 11: 389–393.
Haaland K, Steen PA, Thoresen M 1996 Cerebral, tympanic and colon thermometry in the piglet. Reprod Fertil Dev 8: 125–128.
Wass CT, Lanier WL, Hofer RE, Scheithauer BW, Andrews AG 1995 Temperature changes of ≥1 °C alter functional neurologic outcome and histopathology in a canine model of complete cerebral ischemia. Anesthesiology 83: 325–335.
Brierley JB, Brown AW, Meldrum BS 1971 The nature and time course of the neuronal alterations resulting from oligaemia and hypoglycemia in the brain of Macaca mulatta. Brain Res 25: 483–499.
Løberg EM, Hassel B, Fonnum F, Torvik A 1994 Early entry of plasma proteins into damaged neurons in brain infarcts. APMIS 102: 771–776.
Friede RL 1991 Some features of basics reactions characteristic for immature nervous tissue. In: Friede RL (ed) Developmental Neuropathology. Springer-Verlag, Berlin, pp 21–25.
Kabat H 1940 The greater resistance of very young animals to arrest of the brain circulation. Am J Physiol 122: 588–599.
Duffy TE 1975 Carbohydrate and energy metabolism in perinatal rat brain: relation to survival in anoxia. J Neurochem 24: 271–276.
Ikonomidou C, Mosinger JL, Shahis SK, Labruyere J, Olney JW 1989 Sensitivity of the developing rat brain to hypobaric/ischemic damage parallels sensitivity to N-methyl-D-aspartate neurotoxicity. J Neurosci 9: 2809–2818.
Volpe JJ 1995 Specialized studies in the neurological evaluation. In: Volpe JJ (ed) Neurology of the Newborn. WB Saunders, Philadelphia, pp 160–171.
Sarnat HB 1992 Embryology and Clinical Expression. Oxford University Press, Oxford, pp 1–473.
Oppenheim RW 1985 Naturally occurring cell death during neural development. Trends Neurosci 8: 487–493.
Duffy TE, Nelson TR, Lowry OH 1972 Cerebral carbohydrate metabolism during acute hypoxia and recovery. J Neurochem 19: 959–977.
Chugani HT, Phelps M 1986 Maturational changes in cerebral function in infants determined by 18FDG positron emission tomography. Science 231: 840–843.
Ostwald K, Hagberg H, Andiné P, Karlsson J-O 1993 Upregulation of calpain activity in neonatal rat brain after hypoxia-ischemia. Brain Res 630: 2849–294.
Vanucci RC, Yager JY 1992 Glucose, lactic acid and perinatal hypoxic-ischemic brain damage. Pediatr Neurol 8: 3–12.
LeBlanc MH, Huang M, Vig V, Patel D, Smith EE 1993 Glucose affects the severity of hypoxic-ischemic brain injury in newborn pigs. Stroke 24: 1055–1062.
Myers RE 1976 Anoxic brain pathology and blood glucose. Neurology 26: 345–354.
McDonald JW, Johnston MV 1990 Physiological and pathological roles of excitatory amino acids during central nervous system development. Brain Res Rev 15: 41–70.
McRae A, Gilland E, Bona E, Hagberg H 1995 Microglia activation after neonatal hypoxic-ischemia. Dev Brain Res 84: 245–252.
Reynolds EOR, Wyatt JS, Azzopardi D, Delpy DT, Cady EB, Cope M, Wray S 1988 New non-invasive methods for assessing brain oxygenation and haemodynamics. Br Med Bull 44: 1052–1075.
Rice JEI, Vannucci RC, Brierley JB 1981 The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 9: 131–141.
Myers RE 1977 Experimental models of perinatal brain damage: relevance to human pathology. In: Gluck L (ed) Intrauterine Asphyxia and the Developing Fetal Brain. Year Book, Chicago, pp 37–97.
Lorek A, Takei Y, Cady E, Wyatt J, Penrice J, Edwards A, Peebles D, Wylezinska M, Owen-Reece H, Kirkbridge V, Cooper C, Aldrige R, Roth S, Brown G, Delphy D, Reynolds EOR 1994 Delayed (“secondary”) cerebral energy failure following acute hypoxia-ischemia in the newborn piglet: continuous 48-hour studies by phosphorus magnetic resonance spectroscopy. Pediatr Res 36: 699–706.
LeBlanc MH, Farias LA, Evans OB, Vig V, Smith EE, Markov AK 1991 Fructose-1,6-bisphosphate, when given immediately before reoxygenation, or before injury, does not ameliorate hypoxic ischemic injury to the central nervous system in the newborn pig. Crit Care Med 19: 75–83.
LeBlanc MH, Vig V, Smith B, Parker CC, Evans OB, Smith EE 1991 MK-801 does not protect against hypoxic-ischemic brain injury in piglets. Stroke 22: 1270–1275.
Thoresen M, Penrice J, Lorek A, Wyatt J, Cady EB, Wylezinska M, Kirkbridge V, Cooper C, Edwards D, Brown G, Reynolds EOR 1995 Mild hypothermia following severe transient hypoxia-ischemia ameliorates delayed (“secondary”) cerebral energy failure in the newborn piglet. Pediatr Res 5: 667–670.
Laptook AR, Corbett JT, Nguyen NT, Peterson J, Nunnally RL 1988 Alterations in cerebral blood flow and phosphorylated metabolites in piglets during and after partial ischaemia. Pediatr Res 23: 206–211.
DiGiacomo JE, Pane CR, Gwiazdowski S, Mishra OP, Delivoria-Papadopoulos M 1992 Effect of graded hypoxia on brain cell membrane injury in newborn piglets. Biol Neonate 61: 25–32.
Hoffman DJ, McGowan JE, Marro PJ, Mihsra OP, Delivoria-Papadopoulos M 1994 Hypoxia-induced modification of theN- methyl-D-aspartate receptor in the brain of the newborn piglet. Neurosci Lett 167: 156–160.
Leech R, Alvord EC 1977 Anoxic-ischemic encephalopathy in the human neonatal period: The significance of brain stem involvement. Arch Neurol 34: 109–113.
Banker BQ, Larroche J-C 1962 Periventricular leukomalacia of infancy. A form of neonatal anoxic encephalopathy. Arch Neurol 7: 386–410.
Takashima S, Armstrong DL, Becker LE 1978 Subcortical leucomalacia. Relationship to development of the cerebral sulcus and its vascular supply. Arch Neurol 35: 470–472.
Azzarelli B, Meade P, Mulelr J 1980 Hypoxic lesions in areas of primary myelination Childs B. rain 7: 132–145.
Friede RL 1991 Perinatal lesions of white matter. In: Friede RL (eds) Developmental Neuropathology. Springer-Verlag, Berlin, pp 69–81.
Holm IE, West M 1994 Hippocampus of the domestic pig: a stereological study of subdivisional volumes and neuron numbers. Hippocampus 4: 115–126.
Selkoe DJ, Myers RE 1979 Neurologic and cardiovascular effects of hypotension in the monkey. Stroke 10: 147–157.
Holmes G, Rowe J, Hafford J, Schmidt R, Testa M, Zimmerman A 1982 Prognostic value of the electroencephalogram in neonatal asphyxia. Electroencephalogr Clin Neurophysiol 53: 60–72.
Hellström Westaas L, Rosén I, Svenningsen NW 1995 Predictive value of early continuous amplitude integrated EEG recordings on outcome after severe birth asphyxia in full term infants. Arch Dis Child 72:F34–F38.
Van Lieshout HBM, Jacobs JWFM, Rotteveel JJ, Geven W, Hof MV 1995 The prognostic value of the EEG in asphyxiated newborns. Acta Neurol Scand 91: 203–207.
Levene MI 1995 Management and outcome of birth ashyxia. In: Levene MI, Lilford RJ (eds) Fetal and Neonatal Neurology and Neurosurgery. Churchill Livingstone, Edinburgh, pp 427–443.
Calciolari G, Perlman JM, Volpe J 1988 Seizures in the Neonatal Unit of the 1989. Types, etiologies, timing. Clin Pediatr 27: 119–123.
Caveness W, Nielsen KC, Yakovlev PI 1962 Electroencephalographic and clinical studies of epilepsy during the maturation of the monkey. Epilepsia 3: 137 15150
Connell J, Oozeer R, de Vries L, Dubowitz LM, Dubowitz V 1989 Continuous EEG monitoring of neonatal seizures: diagnostic and prognostic considerations. Arch Dis Child 64: 452–458.
Kette F, Weil MIL, Gazmuri RJ, Bisera J, Rackow EC 1993 Intramyocardial hyperbaric acidosis during cardiac arrest and resuscitation. Crit Care Med 21: 901–906.
Vannucci RC, Towfighi J, Heitjan DF, Brucklacher RM 1995 Carbon dioxide protects the perinatal brain from hypoxic-ischemic damage: an experimental study in the immature rat. Pediatrics 95: 868–874.
Schmidt U, Schwinger RHG, Bohm M 1995 Halothane restores the altered force-frequency relationship in failing human myocardium. Anesthesiology 82: 1456–1462.
Acknowledgements
Invaluable technical advice was received from SINTEF. At the National Hospital, the technical department and the medical library have provided continuous support. We particularly thank the chief veterinarian Dag Sørensen for excellent assistance and advice.
Author information
Authors and Affiliations
Additional information
Supported by the Norwegian Research Council (M.T, K.H, F.A), the Lærdal Foundation for Acute Medicine, the Norwegian SIDS Society, and the Norwegian Council for Cardiovascular Diseases.
Rights and permissions
About this article
Cite this article
Thoresen, M., Haaland, K., Løberg, E. et al. A Piglet Survival Model of Posthypoxic Encephalopathy. Pediatr Res 40, 738–748 (1996). https://doi.org/10.1203/00006450-199611000-00014
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199611000-00014
This article is cited by
-
Hydrogen gas can ameliorate seizure burden during therapeutic hypothermia in asphyxiated newborn piglets
Pediatric Research (2024)
-
Conflicting findings on the effectiveness of hydrogen therapy for ameliorating vascular leakage in a 5-day post hypoxic-ischemic survival piglet model
Scientific Reports (2023)
-
Hypothermia and heart rate variability in a healthy newborn piglet model
Scientific Reports (2022)
-
Cerebral hemodynamic response during the resuscitation period after hypoxic-ischemic insult predicts brain injury on day 5 after insult in newborn piglets
Scientific Reports (2022)
-
Short-term outcomes of remote ischemic postconditioning 1 h after perinatal hypoxia–ischemia in term piglets
Pediatric Research (2021)
