
Abstract
Using 7-d-old rat pups, the neuroprotective efficacy of the lipid peroxidation inhibitor tirilazad mesylate (U-74006F) was tested in a model of perinatal hypoxic-ischemic (HI) brain damage. The experimental protocol was divided into five parts: 1) pre- plus post-HI treatment or 2) only post-HI treatment with tirilazad (7.5 mg/kg intraperitoneally) or vehicle with evaluation of hemispheric weight deficit 14 d after the insult; 3) post-HI treatment with tirilazad or vehicle with histopathologic evaluation 14 d after the insult; 4) pre- plus post-HI treatment; or 5) posthypoxic treatment with tirilazad or vehicle with evaluation of brain edema 20 h after the insult. In the pre- plus post-HI treatment group, the mean left hemispheric weight deficit was 20.7%± 17.8 (mean ± SD) in tirilazad-treated rats and 27.5% ± 20.4 in vehicle-treated rats (p = 0.032). Corresponding values for the post-HI treated animals were 19.6% ± 16.0 and 28.6% ± 15.4(p = 0.043). Histopathologic injury assessed as pathology score on a scale of 0-5 was less extensive in tirilazad-treated animals compared with controls (p = 0.038). There was a significant increase in water content in the HI hemisphere compared with the contralateral (hypoxic) hemispheres in tirilazad- and vehicle-treated animals. This increase of water content in the HI hemispheres did not differ between tirilazad- and vehicle-treated animals. The lipid peroxidation inhibitor tirilazad administered after perinatal HI reduced brain damage by 30%, but no effect was found on early postinsult edema.
Similar content being viewed by others
Main
Perinatal HI is a well recognized cause of brain damage that can contribute to subsequent neurologic impairment or to death(1). The generation of oxygen-derived free radicals during HI has been implicated in the pathogenesis of brain injury in both term and preterm infants(2). Oxygen-derived free radicals can increase capillary permeability(3), impair microcirculation(4), damage biologic membranes with lipid peroxidation(5) and destroy cellular DNA(6, 7). Because it is self-perpetuating and leads to a cascade of self-generating processes, lipid peroxidation may be particularly damaging(8–10). Thus, inhibition of lipid peroxidation may be a potential mechanism for reducing the pathophysiologic effects of HI.
Tirilazad mesylate (U-74006F) is the first compound of a new class of agents, the lazaroids, which inhibit lipid peroxidation via a combination of chemical antioxidant and membrane stabilizing mechanisms(11–13). Tirilazad is a 21-amino steroid closely related to the glucocorticoid steroids but without their hormonal activity. The compound has been shown to be protective in a variety of experimental models of head and spinal injury, subarachnoid hemorrhage, and focal and global cerebral ischemia related to its antioxidant mechanisms(14). Therefore, we studied the neuroprotective effects of tirilazad in a rat model of perinatal HI.
METHODS
Experimental Procedures
A model of perinatal HI that combines reduced concentrations of ambient oxygen and unilateral common carotid artery ligation was used to produce brain damage(15, 16). In this study we used 293 7-d-old Sprague-Dawley rat pups from 26 litters and 42 7-d-old inbred Wistar F rat pups from five litters. The rats suckled until the start of the experiment. Under enflurane anesthesia (3-3.5% for induction, 1-1.5% for maintenance) the left common carotid artery was ligated and cut between ligatures, and after 1.5 h recovery the rats were exposed to 7.8 ± 0.01% oxygen in nitrogen for 2 h in a humidified chamber at 36 °C. In all experiments, half of the animals in each litter were treated with 7.5 mg of tirilazad/kg intraperitoneally and the other half with vehicle for tirilazad intraperitoneally (control animals). Tirilazad was kindly supplied by Dr. Edward D. Hall, Upjohn Co., Kalamazoo, MI.
Evaluation of the Brain Damage
Weighing. The rats were divided into two groups. Group 1 included animals from seven litters. They were treated with 7.5 mg of tirilazad/kg (n = 40) or with vehicle (n = 42) 15 min before and again immediately after exposure to HI. Group 2 included animals from seven litters. They were treated with 7.5 mg of tirilazad/kg (n= 31) or with vehicle (n = 31) immediately after exposure to HI.
Brain damage in groups 1 and 2 is expressed as the percent-age of weight deficit of the hemisphere ipsilateral to the carotid artery ligation compared with the weight of the contralateral hemisphere. Two weeks after the insult, the animals were killed by cervical dislocation and decapitated, and the brain was dissected out. The olfactorial tubercles, cerebellum, and the brainstem were discarded. The hemispheres were then separated in the midline and weighed with an accuracy of 10-4 g. A close correlation between hemispheric weight deficit and estimated brain damage has been reported previously(16–18).
Morphology. Due to a temporary disease in the Sprague-Dawley strain, we used inbred Wistar F in this protocol. In group 3, animals from five litters were treated with 7.5 mg of tirilazad/kg (n = 19) or vehicle (n = 19) immediately after exposure to HI, and 14 d later the surviving rat pups were anesthetized with 0.1 mL methohexital (Brietal) and perfusion-fixed with 4% paraformaldehyde in a 0.1 M phosphate buffer through the ascending aorta before the brain was removed. The brains were thereafter immersion-fixed in paraformaldehyde. Specimens were embedded in paraffin, sectioned in three coronal planes (+1, -2.5, and -4 mm from bregma), and stained with hematoxylin-eosin. Brain sections were evaluated by an investigator blinded to the treatment. A 5-point, graded scoring system was used as follows: 0 = indicates no visible pathology; 1 = single infarcts or regions with selective neuronal necrosis restricted to the lateral cortex in posterior sections; 2 = multiple or larger infarcts in the lateral cortex in posterior sections and in the medial habenular nucleus of the thalamus; 3 = confluent cortical and medial thalamic infarction, selective neuronal necrosis in striatum and hippocampus; 4 = extensive infarction in medial and lateral cortex in anterior and posterior sections, infarction in hippocampus and in several thalamic nuclei, selective neuronal necrosis, or infarction in the striatum; 5 = infarction exceeding 60% of the hemispheric area in two of three coronal planes (see Fig. 3).

Histopathology scoring. A is an example of a score of 1, B a score of 3, and C a score of 5.
Evaluation of the Brain Edema
The rats were divided into two groups. Group 4 included animals from four litters. They were treated with tirilazad (n = 23) or vehicle(n = 20) 15 min before and again immediately after exposure to HI. Group 5 included animals from eight litters. They were treated with 7.5 mg of tirilazad/kg (n = 39) or vehicle (n = 37) immediately after exposure to HI. Twenty hours after the insult, the animals were decapitated, and the brain was dissected out as described above. The hemispheres were weighed and then desiccated at 92 °C for 24 h. Percentage brain water content was determined according to the formula: wet weight - dry weight/wet weight × 100 = water content (%).
Statistical Analysis
Of 335 animals used, five were excluded before the start of the experiment due to low body weight, 29 died during operation or exposure, 39 died before evaluation, and five were excluded due to illegible marking. Thus, 257 animals were analyzed statistically. A multivariate analysis of vriance test was performed to test the difference between the treatments on the reduction of left versus right hemispheric weight and development of cerebral edema. The brain pathology score was evaluated with the Mann-Whitney U test. Mortality frequency was evaluated with χ2 analysis. Commercial software (Statistical/w, StaSoft, Tulsa, OK) was used for all calculations. A p value less than 0.05 was regarded as statistically significant.
RESULTS
Body Weight
There was no difference in mean body weight at 7 d between the tirilazad-treated animals and the vehicle-treated animals in groups 1 and 2(Table 1). Neither were any differences found in mean body weights of the rat pups at 7 d in group 3 (tirilazad, 10.3 g ± 1.0, mean ± SD; vehicle, 9.9 g ± 1.0); group 4 (tirilazad, 13.0 g± 1.2, vehicle, 12.9 g ± 1.1); and group 5 (tirilazad, 13.5 g± 1.9; vehicle; 13.6 g ± 1.7).
Brain Damage
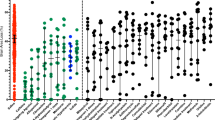
Weighing (groups 1 and 2). In group 1, in which the animals received treatment before and after HI, the mean left hemisphere weight deficit at 14 d after HI was 20.3% ± 18.3 (mean ± SD) in tirilazad-treated rats and 27.4% ± 20.5 in vehicle-treated rats(p = 0.032) (Fig. 1 and Table 1). In group 2, in which the animals received treatment only after HI, the mean left hemispheric weight deficit was 19.6% ± 16.0 in tirilazad-treated rats and 28.6% ± 15.4 in vehicle-treated rats(p = 0.043) (Fig. 1 and Table 1). The mortality frequency was not significantly different in rat pups treated with tirilazad (27.5%) compared with those receiving vehicle (21.4%) in group 1. Corresponding values in group 2 were 19.4% (tirilazad) and 19.4% (vehicle).

Neuroprotective effect of tirilazad after HI. Treatment with tirilazad or vehicle before and after or only after HI. Brain damage was evaluated 14 d after HI and expressed as percent weight deficit of left hemisphere (HI) compared with right hemisphere (hypoxia only). Values are mean± SEM. *p < 0.05.
Morphology (group 3). The brain pathology score was lower in the group treated with tirilazad compared with controls (p = 0.038) (Fig. 2). Extensive confluent infarction in the cerebral cortex, thalamus, hippocampus, and striatum was common in the control group, whereas in the tirilazad-treated animals localized single or multiple areas of infarction or selective neuronal necrosis predominated (Figs. 2 and 3). The hemisphere contralateral to the carotid ligation was not damaged by the hypoxic exposure. The mortality was not significantly different in rat pups treated with tirilazad (8.3%) compared with those receiving vehicle (8.3%).

Distribution of histopathology scores for tirilazad- and vehicle-treated animals. Evaluation of brain damage was performed 14 d after HI.
Brain Edema
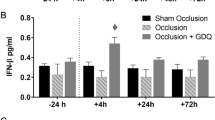
In this model there was considerable development of edema in the ipsilateral hemisphere, which was already significant 30 min after HI, reaching a maximum at 6 h to 3 d and decreasing thereafter(15, 19–21). The extent of edema amounted to an increase in water content of about 1-2% which equated to about a 10-20% increase in cerebral tissue(19, 22). The water content did not change in the contralateral hemisphere or in the sham-operated animal. There was a significant increase in water content in the left (HI) hemisphere compared with the right hemisphere (hypoxia) at 20 h after the insult in both groups of rats. There was no significant difference in left hemispheric water content between tirilazad- and vehicle-treated animals in groups 4 and 5 (Fig. 4). No animals died before evaluation.

The effect of tirilazad on left cerebral hemisphere edema. Treatment with tirilazad or vehicle before and after HI or only after HI. Cerebral edema was evaluated 20 h after HI. Values are mean ± SD.
DISCUSSION
Either pre- plus posthypoxic or posthypoxic treatment with tirilazad provided statistically significant neuroprotection against HI in this neonatal rat model. In both cases, the left hemispheric weight deficit was significantly less 14 d after HI in rats treated with tirilazad compared with those treated with vehicle. No added neuroprotective effect of pre-HI treatment with tirilazad was seen, compared with only post-HI administration. These results support the concept of a post-HI burst of radical formation and are in agreement with previous studies with allopurinol(23), the spin-trapping agentα-phenyl-N-tert-butylnitrone(24), and iron chelator(25). In adult rats receiving 10 mg of tirilazad/kg of body weight by intraperitoneal injection, tirilazad mesylate had a half-life of 2.4 h in the plasma and 1.9 h in the brain(13). At the start of the posthypoxic recovery, when the free radical cascade is about to start(10), a considerable amount of tirilazad mesylate might already have been metabolized during the 2 h of hypoxic exposure, which may explain the lack of increased neuroprotection.
The major histopathologic changes in the model consisted of infarction and selective neuronal necrosis in the cortex, hippocampus, thalamus, and striatum with the predominate lesion situated in the cerebral cortex. Evaluation of the histopathologic changes was made 14 d after the HI, and our findings are in agreement with the distribution of the injury found in previous studies where histologic examinations were performed 2, 3, and 23 d after HI in this model(15, 18, 26). The neuroprotective effect of tirilazad in group 3, expressed as reduction of histopathologic score in tirilazad-treated animals 14 d after HI, could also be demonstrated by significantly reduced left cerebral hemisphere weight deficit 14 d after HI in tirilazad-treated animals. This is in accordance with previous studies suggesting a close correlation between weight deficit and histopathology of the injured hemisphere(16, 18, 27).
In this study the increase in water content at 20 h was 0.4%, which was significant but less pronounced than in previous studies(15, 19–21). It is possible that the insult was less severe in those specific litters and that the increase in water content reflected vascular rather then cytotoxic edema formation. In contrast to the neuroprotective effect of tirilazad with respect to a reduction in delayed neuronal damage at 14 d, the magnitude of edema at 20 h after HI was not significantly reduced by treatment with tirilazad compared with vehicle either prehypoxia/posthypoxia or posthypoxia only. Perhaps the pathophysiology of vascular brain edema in the setting of HI involves mechanisms other than or in addition to the production of oxygen-derived free radicals. However, Palmer et al.(23, 28) demonstrated in two separate studies that prehypoxic and posthypoxic treatment with a xanthine oxidase inhibitor, allopurinol, reduced both cerebral edema, measured 42 h after HI, and the extent of perinatal hypoxic-ischemic brain damage, but on the other hand, reducing the extent of cerebral edema with mannitol after perinatal HI did not affect the severity of the brain damage(20). These data suggest that an early reduction in edema is not a prerequisite for subsequent neuroprotection.
Oxygen-derived free radicals are formed during reperfusion after ischemia via different pathways, including oxidation of accumulated hypoxanthine by xanthine oxidase and oxidation of arachidonic acid in the presence of lipoxygenase and cyclooxygenases(29, 30). Studies in fetal sheep have shown hypoxanthine production by the brain during oxygen deficiency(31), and the interstitial concentrations were quadrupled during severe asphyxia(32). Other studies that support the hypothesis of free radical production in the neonatal or perinatal circulation include detection of free radical production(33, 34), neuroprotection by blocking xanthine oxidase(23, 28), and administration of antioxidants, radical scavengers, or iron chelating agents(25, 35, 36). In summary, brain injury after HI in neonatal rats was reduced by posttreatment with the lipid peroxidation inhibitor tirilazad.
Abbreviations
- HI:
-
hypoxia-ischemia
References
Volpe JJ 1995 Neurology of the Newborn, 3rd Ed. WB Saunders, Philadelphia, pp 314–369
Saugstad OD 1992 Neonatal oxygen radical disease. In: Davies TJ (ed) Recent Advances in Pediatrics, 10th Ed. Churchill Livingstone, Edinburgh, pp 173–187
Parks DA, Granger DN 1983 Ischemia-induced vascular changes: role of xanthine oxidase and hydroxyl radicals. Am J Physiol 245:G285–G289
Demopoulos HB, Flamm ES, Pietronigro DD, Seligman ML 1980 The free radical pathology and the microcirculation in the major central nervous system disorders. Acta Physiol Scand 492( suppl): 91–119
Halliwell B, Gutteridge JMC 1989 Free Radicals in Biology and Medicine, 2nd Ed. Oxford University Press, New York, pp 188–276
Lesko SA, Lorentzen RJ, Ts'o PO 1980 Role of superoxide in deoxyribonucleic acid strand scission. Biochemistry 19: 3023–3028
Brawn K, Fridovich I 1981 DNA strand scission by enzymically generated oxygen radicals. Arch Biochem Biophys 206: 414–419
Braughler JM, Hall ED 1989 Central nervous system trauma and stroke. I. Biochemical considerations for oxygen radical formation and lipid peroxidation. Free Radic Biol Med 6: 289–301
Hall ED, Braughler J 1989 Central nervous system trauma and stroke. II. Physiological and pharmacological evidence for involvement of oxygen radicals and lipid peroxidation. Free Radic Biol Med 6: 303–313
Siesjo BK, Agardh CD, Bengtsson F 1989 Free radicals and brain damage. Cerebrovasc Brain Metab Rev 1: 165–211
Braughler JM, Pregenzer JF, Chase RL, Duncan LA, Jacobsen EJ, McCall JM 1987 Novel 21-amino steroids as potent inhibitors of iron-dependent lipid peroxidation. J Biol Chem 262: 10438–10440
Hall ED, Pazara KE, Braughler JM 1988 21-amino steroid lipid peroxidation inhibitor U74006F protects against cerebral ischemia in gerbils. Stroke 19: 997–1002
Laizure SC, Franklin LK, Kaiser DG, Williams CL, Stevens RC, Sanders PL, Miller M 1993 Disposition of tirilazad (U74006F), a 21-amino steroid, in the plasma, heart, brain, and liver of the rat. Drug Metab Dispos Biol Fate Chem 21: 951–954
Hall ED, McCall JM, Means ED 1994 Therapeutic potential of the lazaroids (21-amino steroids) in acute central nervous system trauma, ischemia and subarachnoid hemorrhage. Adv Pharmacol 28: 221–268
Rice JE, Vannucci Rc, Brierley JB 1981 The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 9: 131–141
Andiné P, Thordstein M, Kjellmer I, Nordborg C, Thiringer K, Wennberg E, Hagberg H 1990 Evaluation of brain damage in a rat model of neonatal hypoxic-ischemia. J Neurosci Methods 35: 253–260
Gilland E, Puka-Sundvall M, Andiné P, Bona E, Hagberg H 1994 Hypoxic-ischemic injury in the neonatal rat brain: effects of pre- and post-treatment with the glutamate release inhibitor BW1003C87. Brain Res Dev Brain Res 83: 79–84
Hagberg H, Gilland E, Diemer NH, Andiné P 1994 Hypoxia-ischemia in the neonatal rat brain: histopathology after post-treatment with NMDA and non-NMDA receptor antagonists. Biol Neonate 66: 205–213
Mujsce DJ, Christensen MA, Vannucci RC 1990 Cerebral blood flow and edema in perinatal hypoxic-ischemic brain damage. Pediatr Res 27: 450–453
Mujsce DJ, Towfighi J, Stern D, Vannucci RC 1990 Mannitol therapy in perinatal hypoxic-ischemic brain damage in rats. Stroke 21: 1210–1214
Kalayci O, Cataltepe S, Cataltepe O 1992 The effect of bolus methylprednisolone in prevention of brain edema in hypoxic ischemic brain injury: an experimental study in 7-day-old rat pups. Brain Res 569: 112–116
Elliot KAC, Jasper H 1949 Measurement of experimental induced brain swelling and shrinkage. Am J Physiol 157: 122–129
Palmer C, Towfighi J, Roberts RL, Heitjan DF 1993 Allopurinol administered after inducing hypoxia-ischemia reduces brain injury in 7-day-old rats. Pediatr Res 33: 405–411
Cao X, Phillis JW 1994 α-Phenyl-tert-butyl-nitrone reduces cortical infarct and edema in rats subjected to focal ischemia. Brain Res 644: 267–272
Palmer C, Roberts RL, Bero C 1994 Deferoxamine posttreatment reduces ischemic brain injury in neonatal rats. Stroke 25: 1039–1045
Towfighi J, Yager JY, Housman C, Vannucci RC 1991 Neuropathology of remote hypoxic-ischemic damage in the immature rat. Acta Neuropathol 81: 578–587
Towfighi J, Housman C, Vannucci RC, Heitjan DF 1994 Effect of unilateral perinatal hypoxic-ischemic brain damage on the gross development of opposite cerebral hemisphere. Biol Neonate 65: 108–118
Palmer C, Vannucci RC, Towfighi J 1990 Reduction of perinatal hypoxic-ischemic brain damage with allopurinol. Pediatr Res 27: 332–336
McCord JM 1985 Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med 312: 159–163
Rehncrona S, Westerberg E, Akesson B, Siesjo BK 1982 Brain cortical fatty acids and phospholipids during and following complete and severe incomplete ischemia. J Neurochem 38: 84–93
Thiringer K, Blomstrand S, Hrbek A, Karlsson K, Kjellmer I 1982 Cerebral arteriovenous difference for hypoxanthine and lactate during graded asphyxia in the fetal lamb. Brain Res 239: 107–117
Kjellmer I, Andiné P, Hagberg H, Thiringer K 1989 Extracellular increase of hypoxanthine and xanthine in the cortex and basal ganglia of fetal lambs during hypoxia-ischemia. Brain Res 478: 241–247
Armstead WM, Mirro R, Busija DW, Leffler CW 1988 Postischemic generation of superoxide anion by newborn pig brain. Am J Physiol 255:H401–H403
Hasegawa K, Yoshioka H, Sawada T, Nishikawa H 1993 Direct measurement of free radicals in the neonatal mouse brain subjected to hypoxia: an electron spin resonance spectroscopic study. Brain Res 607: 161–166
Rosenberg AA, Murdaugh E, White CW 1989 The role of oxygen free radicals in postasphyxia cerebral hypoperfusion in newborn lambs. Pediatr Res 26: 215–219
Thordstein M, Bågenholm R, Thiringer K, Kjellmer I 1993 Scavengers of free oxygen radicals in combination with magnesium ameliorate perinatal hypoxic-ischemic brain damage in the rat. Pediatr Res 34: 23–26
Author information
Authors and Affiliations
Additional information
Supported by grants from the Swedish Medical Research Council (9455), Wilhelm and Martina Lundgren's Foundation, the Freemasonry Orphanage Foundation, Magnus Bergvall Foundation, Linnea and Josef Carlsson Foundation, General Maternity Hospital Foundation, the Swedish Society for Medical Research, Sigurd and Elsa Golje Foundation, Åhlén Foundation, Göteborg Medical Society, the Samariten Foundation, Sven Jerring Foundation, the Faculty of Medicine, Göteborg, the 1987 Foundation for Stokeresearch, Åke Wiberg Foundation, and Konung Gustaf V's 80 års Foundation.
Rights and permissions
About this article
Cite this article
Bågenholm, R., Andiné, P. & Hagberg, H. Effects of the 21-Amino Steroid Tirilazad Mesylate (U-74006F) on Brain Damage and Edema after Perinatal Hypoxia-Ischemia in the Rat. Pediatr Res 40, 399–403 (1996). https://doi.org/10.1203/00006450-199609000-00006
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199609000-00006
