
Abstract
Heme oxygenase (HO) is the rate-limiting enzyme in the catabolism of heme to bilirubin. Cobalt chloride (CoCl2) and many other agents that generate oxidant stresses induce the HO-1 isoform. Furthermore, HO-1 has been shown to protect against oxidant stress in vitro and in vivo by mechanisms involving increased ferritin synthesis. However, little is known about the inducibility of hepatic HO-1 during the very early postnatal period, and whether HO-1 induction is associated with increased ferritin synthesis in neonates. Therefore, we studied hepatic HO-1 mRNA, HO-1 protein concentration, total HO activity, and ferritin protein levels in neonatal rats. Neonatal rats 0-5 d of age were injected with 250 μmol/kg body weight of CoCl2 · 6H2O in saline or with an equal volume of saline in age-matched controls. Liver samples were collected 4 h after injection for HO-1 mRNA analysis and 20 h after injection for analysis of HO-1 protein concentration, total HO activity, and ferritin protein levels. In CoCl2-treated rats, hepatic HO-1 mRNA was 3-10 times the levels in control rats (p < 0.05), HO-1 protein concentration was 2-5 times the levels in control rats (p < 0.05), and total HO activity was higher by 20-80% than in control rats (p < 0.05). There were no differences in hepatic ferritin protein levels between CoCl2-treated neonatal rats and controls; however, in CoCl2-treated adult rats, hepatic ferritin protein levels were 1.6 times the levels in controls(p < 0.05). Thus, neonatal rats can up-regulate hepatic HO-1 mRNA. HO-1 protein concentration, and total HO activity in response to CoCl2; however, no up-regulation of hepatic ferritin protein levels was observed in neonatal rats after CoCl2 administration or subsequent HO-1 induction. We speculate that neonatal rats induce hepatic HO-1 and up-regulate ferritin by different mechanisms than do adult rats.
Similar content being viewed by others

Main
Microsomal HO is the rate-limiting enzyme in the catabolism of heme, in a process that results in the production of biliverdin, carbon monoxide, non-heme iron, and NADP+(1–3). Biliverdin is subsequently reduced to bilirubin by the cytosolic enzyme, biliverdin reductase(4). The inducible isoform, HO-1, is an early stress response protein that is stimulated by a variety of agents and manipulations including heme(5), heavy metals such as CoCl2(6–8) ultraviolet A (UVA) radiation(9), hydrogen peroxide(9), and GSH depletion(10). Many of the agents that up-regulate HO-1 are oxidants or generate reactive oxygen species, which has led some investigators to propose that the induction of HO-1 may play a role in cellular antioxidant defenses(11, 12). Support for the theory that HO-1 induction serves an important role in cellular antioxidant defense is provided by experiments in vivo(13) and in vitro(14–16), indicating antioxidant protection by production of biliverdin and bilirubin. Our group has reported that jaundiced neonatal Gunn rats exposed to hyperoxia have less serum lipid peroxidation and protein oxidation than their nonjaundiced littermates(13). Increased HO-1 activity also has been shown to protect against oxidant stress in vivo and in vitro by mechanisms involving increased ferritin synthesis(11, 12). Thus, several avenues exist through which HO-1 induction could enhance antioxidant protection.
Although HO-1 induction may be a general and adaptive response to oxidant stress(17), the inducibility of this enzyme in the early postnatal period remains to be established. Maines and Kappas(6) found that administration of CoCl2 to pregnant dams did not increase HO activity in fetal rats, suggesting that fetal rats may be resistant to HO induction. Basal hepatic HO activity is higher in neonatal than in adult rats(6), possibly secondary to the higher heme load that neonates have as a result of ineffective erythropoiesis and a shorter red blood cell lifespan than in adults(18). Despite greater basal hepatic HO activity in neonatal rats than in adults, increases in hepatic HO activity and HO-1 mRNA after CoCl2 administration were observed in 4- and 7-d-old rats, respectively, indicating that hepatic HO-1 was inducible in these older neonatal rats(6, 7). However, the studies published to date have not addressed the basal expression and inducibility of hepatic HO-1 in the immediate postnatal period. In addition, questions remain whether the increase in hepatic HO-1 in older neonatal rats is associated with an increase in ferritin protein synthesis as was shown in adult rats andin vitro(11, 12). Therefore, we investigated the effects of CoCl2 on hepatic HO-1 expression and hepatic ferritin protein levels during the early postnatal period in neonatal rats.
METHODS
Chemicals and materials. SDS was purchased from BDH Chemicals(Poole, England). Agarose was purchased from Kodak International Biotechnologies (New Haven, CT). All other chemicals including hemin were obtained from Sigma Chemical Co. (St. Louis, MO). All enzymes were purchased from Life Technologies, Inc. (Gaithersburg, MD). CoCl2 · 6H2O was purchased from J. T. Baker Chemical (Phillipsburg, NJ). CoCl2 solution was prepared as 24 mg of CoCl2 · 6H2O dissolved in 1 ml of 0.9% saline.
Animals. Wistar rats were obtained from Simonsen (Gilroy, CA). Neonatal rats were housed with their mothers in a temperature controlled environment with a 12-h light/dark cycle. The pups were allowed free access to their mothers. The dams were given food and water ad libitium. The care and treatment of animals was in accordance with NIH guidelines and under institutional animal care and use committee approval.
Experimental design. Adult and neonatal rats aged 0, 1, 2, 3, 4, and 5 d of life were injected subcutaneously with CoCl2 (250μmol/kg, 60 mg/kg body weight) in 0.9% saline(6, 7). Between 3 and 12 rats were used for each group. On d 0, the rats were no more than 12-16 h old at dosing. This dose of CoCl2 used has been reported to cause induction of HO-1 mRNA and total HO activity in adult and suckling rats(6, 7). Control animals received equal volumes of 0.9% saline. Four hours after CoCl2 injection, some of the animals were sacrificed, and the livers were removed for HO-1 mRNA analysis. Twenty hours after CoCl2 injection the remaining animals were sacrificed, and the livers were removed for analysis of HO-1 and ferritin protein concentrations and total HO activity. The experimental time points were chosen based on previously published literature on the time course of HO-1 induction by CoCl2(6, 7).
Tissue preparation. Livers collected for HO-1 mRNA analysis were homogenized immediately in a 4 M guanidinium isothiocyanate, 25 mM sodium citrate, and 0.5% sarcosyl solution, quick-frozen in liquid nitrogen, and stored at -80°C(19).
Livers collected for quantitation of HO-1 and ferritin protein concentrations and total HO activity were homogenized in 4 volumes of 0.1 M potassium phosphate buffer (pH 7.4) and centrifuged, and the 13 000 ×g supernatant fractions were used for analyses.
Probes. The cDNA for human HO-1 was a 1.1-kb EcoRI fragment derived from the pHHO1 plasmid (pHHO1; gift of S. Shibahara, Tohoku University School of Medicine, Sendai, Japan) by standard methods(20, 21). The preparation and sequence of the human HO-1 cDNA has been described previously by Shibahara et al.(21). The cDNA for GAPDH(22)(American Type Culture Collection; Rockville, MD) was prepared as anEco RI digest by standard methods(20). The probes were labeled with 32[P]dATP (Amersham, Indianapolis, IN) by the random primer method(23) using the GIBCO BRL Random Primers DNA labeling system (Life Technologies, Inc.) according to the manufacturer's recommendations. The specific activity of the HO-1 and GAPDH probes ranged from 2 × 109 to 7 × 1010 cpm/μg, and the probes were used at concentrations of 2 × 106 cpm/mL for HO-1 and 1 × 106 cpm/mL for GAPDH.
Antibodies. Antiserum was prepared by Berkeley Antibodies, Inc.(Berkeley, CA). Polyclonal rabbit anti-rat HO-1 antibodies were raised against a 30-kD soluble HO-1 protein expressed in Escherichia coli using rat liver cDNA (gift of A. Wilks, University of California, San Francisco, CA)(24). Antiserum was precleared by incubating at 4°C with normal rat serum for 2 h. After centrifugation at 6000 ×g for 5 min, the supernatant was recovered and used as a source of HO-1 antibody for Western analysis. Rabbit anti-human ferritin antibodies were purchased from Boehringer Mannheim Corp. (Indianapolis, IN).
Northern Hybridization. Total RNA was extracted from liver tissue with a guanidinium thiocyanate:phenol extraction according to the method of Chomczynski and Sacchi(19) and quantitated spectrophotometrically at 260 nm. Formaldehyde-denatured RNA (20 μg) was electrophoresed on 1.2% agarose gels. To ensure equal loading of RNA, half of the gel was loaded, stained with ethidium bromide, and viewed under UV. The other half of the gel, which had been loaded identically, was used for capillary transfer onto Genescreen membrane (NEN Research Products, Boston, MA). The membranes were cross-linked by UV irradiation (UV Stratalinker; Stratagene, La Jolla, CA). The membranes were prehybridized for 4 h at 42°C in a solution of 5× SSPE, 5× Denhardt's solution, 0.1% SDS, 100 μg/mL denatured salmon sperm DNA, and 50% deionized formamide. The membranes were hybridized in the same solution for 24-36 h at 42°C with a32 P-labeled cDNA probe, and then washed three times at 25°C (15 min each) with a 1× SSC and 1% SDS solution, followed by three washes at 65°C (15 min each) with 0.1% SSC and 0.1% SDS. The washed membranes were exposed to Kodak film (Eastman Kodak Co., Rochester, NY) at -80°C with intensifying screens. Exposure times were 3-5 d for assessments of HO-1 and 1-2 days for GAPDH.
Autoradiograms were quantitated by densitometry using the Quantity One Densitometer (Protein and DNA Imageware Systems, Huntington Station, NY). The densitometer HO-1 values were normalized to the GAPDH values before comparisons between the treated and control groups. All comparisons were made between samples on the same gel, and each gel had samples from only 1 d of life.
Western analysis. Protein samples for HO-1 analysis (100 μg) were fractionated on a 12% polyacrylamide gel electrophoresis according to the method of Laemmli(25). The proteins in the gel were transferred to a Bio-Rad polyvinylidene difluoride membrane using a Bio-Rad Mini Trans-Blot transfer cell (Bio-Rad, Hercules, CA) according to the method of Towbin et al.(26). The membranes were blocked overnight at 25°C in solutions containing 10% powdered skim milk. Tris-buffered saline (TBS, 20 mM Tris, 500 mM NaCl, pH 7.5) and 0.1% Tween-20. The membranes were rinsed in TBS/0.1% Tween-20 and incubated for 2 h with a 1:300 dilution of rabbit anti-rat HO-1-purified antibody. After three washes at 25°C in TBS/0.1% Tween 20, the membranes were incubated for 1 h with goat anti-rabbit IgG antibody conjugated with alkaline phosphatase. The immunoreactive enzyme proteins were visualized by chemiluminescence (Bio-Rad Immun-Lite Enhancer, Bio-Rad, Hercules, CA). Microsomal protein from a CoCl2-induced adult rat liver was used as a positive control for identification of the HO-1 protein. The autoradiograms were quantitated by densitometry, and data were expressed as densitometry units. Comparisons were made between samples on the same gel, and each gel had protein samples from only 1 d of life.
Protein samples for ferritin analysis (50 μg) were fractionated on a 4-20% gradient gel (Bio-Rad) by gel electrophoresis. Thereafter, the protocol described above for HO-1 protein analysis was used for transfer and blocking. The primary antibody was rabbit anti-human ferritin, and the secondary antibody was a goat anti-rabbit IgG alkaline phosphatase conjugate. Human liver ferritin purified antigen (Medix Biotech Inc., Foster City, CA) was used as a positive control. The autoradiograms were quantitated by densitometry, and data expressed as densitometry units. Comparisons were made between samples on the same gel, and each gel had protein samples from only 1 d of life.
Total HO activity assay. HO activity in rat liver homogenate fractions were determined by quantifying the carbon monoxide (CO) produced by the degradation of heme(27). Postmitochondrial supernatants (13 000 × g) were prepared from liver homogenates. Twenty microliters each of methemalbumin (150 μM of hemin) and tissue supernatant were pipetted into reaction vials. Either 20 μL of NADPH(4.5 mM) or 20 μL of potassium phosphate buffer (0.1 M, pH 7.4) were added to the experimental and blank reaction vials, respectively. The vials were equilibrated for 5 min at 37°C and purged with carbon monoxide-free gas, and the reaction was allowed to proceed for 15 min. After terminating the reaction by transferring the vials to a powdered dry ice bath (-78°C), the headspace gas was injected into a reduction gas analyzer (Trace Analytical, Menlo Park, CA) to quantify the carbon monoxide generated by reaction. HO activity was determined from the difference between the experimental and respective blank vials and is expressed as nanomoles of carbon monoxide/h/mg of protein. Protein concentrations of the tissue preparations were determined by the method of Lowry et al.(28) using bovine serum albumin as the standard.
Statistical analysis. For comparison between treatment groups, the null hypothesis that there were no differences between treatment means was tested using the unpaired t test. Statistical comparisons of basal HO activity in control neonatal rats and days were performed by ANOVA using the Fischer's exact test of multiple comparisons (Statview 4.02, Abacus Concepts, Inc., Berkeley, CA). Statistical significance was determined atp < 0.05. All data in the text, tables, and figures are expressed as the means ± SEM.
RESULTS
Hepatic HO-1 mRNA. Neonatal rats aged 0-5 d that were treated with CoCl2 had greater HO-1 mRNA levels than were observed in age-matched saline-treated controls (p < 0.05)(Fig. 1). The increases in HO-1 mRNA levels in CoCl2-treated rats ranged from 3 to 10 times the levels observed in controls (Table 1). To allow for comparison of HO-1 mRNA within each day, the HO-1 mRNA values were normalized to GAPDH values. Although different intensities for GAPDH were also observed between the days, the intensity of GAPDH from a treated and control rat from the same day were not affected by CoCl2.

Northern analysis of steady state hepatic HO-1 and GAPDH mRNA levels in neonatal rats on d 0-5 of age. Rats were injected with 250 μmol/kg body weight of cobalt chloride in saline (T) or equal volumes of saline in the controls (C). Liver homogenates were prepared for analysis 4 h after injection. The membranes were probed with a32 P-labeled HO-1 cDNA fragment (top panel). The same membranes were probed with GAPDH to normalize the data (bottom panel). Equal amounts of total RNA (20 μg) were loaded in each lane. A representative sample for each day of life is shown(n = 5-6 per treatment group). All comparisons were made between samples on the same gel, and each gel had samples from only 1 d of life.
Hepatic HO-1 protein. Neonatal rats aged 0-5 d that were treated with CoCl2 also had greater HO-1 protein concentration than were observed in controls (p < 0.05) (Fig. 2A). The increases in HO-1 protein concentration in CoCl2-treated rats ranged from 2 to 5 times the levels observed in controls(Table 2).

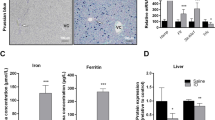
(A) Western analysis of immunoreactive hepatic HO-1 protein in 13 000 × g supernatants prepared from the livers of neonatal rats on d 0-5 of age. Rats were injected with 250μmol/kg body weight of cobalt chloride in saline (T) or equal volumes of saline in the controls (C). Liver homogenates were prepared 20 h after injection for analysis with rabbit anti-rat HO-1 antibody. Equal amounts of protein (100 μg) were loaded in each lane. A representative sample for each day of life is shown (n = 3-6 per treatment group). All comparisons were made between samples on the same gel, and each gel had samples from only 1 d of life. (B) Western analysis of immunoreactive hepatic ferritin protein in 13 000 × g supernatants prepared from the livers of neonatal rats on d 0-5 of age. Rats were injected with 250 μmol/kg body weight of cobalt chloride in saline(T) or equal volumes of saline in the controls (C). Liver homogenates were prepared 20 h after injection for analysis with rabbit anti-human ferritin antibody. Equal amounts of protein (50 μg) were loaded in each lane. A representative sample is for each day of life is shown(n = 4 per treatment group). All comparisons were made between samples on the same gel, and each gel had samples from only 1 d of life.
Hepatic ferritin protein. Neonatal rats aged 0-5 d treated with CoCl2 had no differences in ferritin protein levels(Fig. 2B) compared with controls (Table 2). In contrast, ferritin protein levels in CoCl2-treated adult rats were 1.6 times greater than in adult controls (densitometric quantitation: 2.35 ± 0.12 versus 1.48 ± 0.25, p < 0.05,n = 3 per group).
Total hepatic HO activity. Neonatal rats aged 0-5 d that were treated with CoCl2 had 20-80% increases in enzyme activity compared with control rats (p < 0.05) (Fig. 3). Higher basal levels of HO activity were seen in control rats from birth to 2 d of age; thereafter, HO activity declined gradually. HO activity from 4- and 5-d-old control rats was lower by 26-33% than HO activity from 0-, 1-, and 2-d-old control rats (p < 0.05). HO activity in 5-d-old control rats was also lower by 27% than HO activity in 3-d-old control rats(p < 0.05).

Total HO activity in 13 000 × g supernatants prepared from the livers of neonatal rats on d 0-5 of age. Rats were injected with 250 μmol/kg body weight of cobalt chloride in saline or equal volumes of saline in the controls. Liver homogenates were prepared for analysis 20 h after injection. Data are graphed as mean ± SEM(n = 6-12 per treatment group). * p < 0.05 vs same day control. The data sets for basal HO activity (control rats,light bars) sharing the same letter (a, b, or c) indicate no differences between the days, and data sets without the same letter indicate differences between the days (p < 0.05).
DISCUSSION
The present study demonstrates hepatic HO-1 induction by a heavy metal in neonatal rats less than 4 d of age. In neonatal rats during the early postnatal period, we have shown that administration of CoCl2 increases hepatic HO-1 expression without a concomitant increase in ferritin protein. In contrast to fetal rat studies where no increases in hepatic HO activity were seen in rat embryos (-8 d) and fetuses (0 d or 21 d gestation) 24 h after CoCl2 administration to pregnant rat dams(6), the present study clearly demonstrates increases in hepatic HO-1 mRNA, HO-1 protein concentration, and total HO activity in CoCl2-treated neonatal rats within 24 h of birth. Our results in the 4- and 5-d-old rats are in agreement with work done by previous investigators demonstrating induction of hepatic HO activity and HO-1 mRNA in neonatal rats as young as 4 and 7 d of age, respectively(6, 7). We found 20-80% increases in hepatic HO activity similar to the 50-70% increases seen in slightly older neonatal rats (4-7 d of age) injected with similar doses of CoCl2(6). Additionally, we found HO-1 mRNA induction in 4-d-old rats similar to that previously reported in 7-d-old rats treated with the same dose of CoCl2(7), and our data showed HO-1 mRNA in the CoCl2-treated neonatal rats was 3-10 times the levels in controls.
In the rat, basal hepatic HO activity is higher in the neonatal period than in adulthood(6). In our experiments, basal hepatic HO activity in control neonatal rats was higher from birth through the 2nd d of life than on subsequent days, with a gradual decline observed on d 4 and 5 of life. This is in contrast to other reports of peak hepatic HO activity on d 4 or 7 of life(6, 29, 30), but in agreement with the work of Sun and Maines(7) who measured basal hepatic HO activity in fetal rats 1 d before parturition (-1 d), and neonatal rats on 1, 4, and 7 d of life. They observed that hepatic HO activity was low in -1-d-old fetuses (similar to adult HO activity), high in 1-d-old rats, and was declining by 4 and 7 d of life toward low adult levels(7). Although we did not measure HO activity from -1-or 7-d-old rats, our data were consistent with higher hepatic HO activity from birth through the first 3 d of life, then decreasing HO activity by d 4 of life.
We used a gas chromatographic method to measure CO production as an assay of HO activity(27). Gas chromatographic assays of HO activity in rat tissues have been reported to correlate well with spectrophotometric values of HO activity based on bilirubin production and by the more specific HPLC determination of HO activity based on bilirubin production(31, 32). In our studies, the HO activity values in control neonatal rats (1.1-1.8 nmol CO/h/mg of protein) are lower than the HO activity values obtained by spectrophotometry (≅4-8 nmol of bilirubin/h/mg of protein) reported previously by Maines and Kappas(6). The HO activity values they report may be higher because they used liver microsomes, whereas we used total liver homogenates for the assay. To support this idea, the HO activity values we report were similar to the spectrophotometric values (≅1-2 nmol of bilirubin/h/mg of protein) obtained by Thaler et al.(30), who also used total liver homogenates. Furthermore, we observe peak hepatic HO activity in neonatal rats to be 4-9 times higher than adult HO activity values(≅0.32 ± 0.1 nmol of CO/h/mg of protein) obtained by gas chromatography that have been previously reported(33). This difference is similar to previous reports that peak hepatic HO activity in neonates is 3-9 times higher than in adults, when HO activity is measured spectrophotometrically(6, 7, 29, 30).
CoCl2 is a potent inducer of HO-1 in vivo(6, 7, 8, 34). Some authors have suggested that cobalt (II) ion reacts with hydrogen peroxide under physiologic conditions to form reactive species(35, 36). In adult rats, CoCl2 has been shown to increase HO-1 gene transcription and activity(7, 8). Tomaro et al.(34) have suggested the mechanism of CoCl2 induction of hepatic HO-1 in adult rats involves the formation of active oxidant species from cobalt derivatives, which simultaneously decrease hepatic GSH. In neonatal rats on d 0-5 of life at 2.5 h after CoCl2 injection, we found that total GSH is decreased by 12% when compared with age-matched saline-injected controls (D. J. Tom et al., unpublished results). The effects of CoCl2 on hepatic GSH are statistically significant; whether the 12% decrease in GSH in CoCl2-treated neonatal rats is of sufficient magnitude to account for up-regulation of HO-1 and whether the decrease in GSH suggests formation of reactive oxygen species is unclear. Maines and Kappas(37) have suggested that Co2+ complexes with thiol groups, such as with GSH, and this complex is directly or indirectly involved in the mechanism of HO-1 induction in vivo. Alternatively, CoCl2 may increase HO activity through direct interactions with HO-1 regulatory elements(8). Furthermore, Sinclair et al.(38) have reported that cobalt protoporphyrin is formed in the livers of rats treated with CoCl2 in vivo, and cobalt protoporphyrin administration to adult rats induces HO-1 mRNA by increases in gene transcription(8). Thus, cobalt protoporphyrin formed in vivo after CoCl2 administration may act directly with HO-1 regulatory elements(8). The contribution of each of the proposed mechanisms for the up-regulation of HO-1 remains to be determined.
We investigated whether changes in hepatic ferritin protein levels occur with CoCl2-mediated hepatic HO-1 induction in neonatal rats. Under conditions of oxidant stress, HO-1 induction has been associated with increased ferritin synthesis both in vitro and in vivo(11, 12). Vile et al.(11) demonstrated that heme-treated fibroblasts preirradiated with UVA, had increased HO-1 expression and ferritin protein concentrations. Using a glycerol model of rhabdomyolysis, Nath et al.(12) demonstrated in vivo that ferritin content was increased in the kidney as HO activity was induced, and ferritin content did not increase as HO activity was inhibited. In CoCl2-treated adult rats, we found increases in hepatic ferritin protein; however, in the CoCl2-treated neonatal rats we found no changes in ferritin protein. The fact that we observe no increases in ferritin protein levels in the CoCl2-treated neonatal rat may be due to the differences in our experimental model compared with previous models(11, 12). For example, in previous studies(11, 12), supplemental heme was available and may have contributed to the increase in ferritin protein secondary to an increase in intracellular iron from heme breakdown. Although we did not provide supplemental heme to the adult rats, the increase in hepatic ferritin protein that we observed in CoCl2-treated adult rats may be related to the different degree of HO activity induced by CoCl2 between adult and neonatal rats. CoCl2 administration has been shown to induce HO activity much more in adult (500%) than in neonatal rats (50-70%)(6). Because the increase in HO activity is so much higher in CoCl2-treated adults than in neonates, the adults may have greater heme degradation and iron release with subsequent increases in ferritin. The smaller increases in HO activity observed with CoCl2 administration to neonatal rats may not result in an increase in ferritin if the amount of iron released from the HO reaction did not exceed the capacity of preformed ferritin from intracellular sources. In addition, neonatal rats may be less responsive to up-regulation of ferritin by release of iron. Little is known about the inducibility of ferritin synthesis in neonatal rat liver; however, data suggest that ferritin synthesis can be induced during development(39). In summary, during the very early postnatal period, neonatal rats respond to CoCl2 administration with induction of hepatic HO-1; however, in contrast to adult models, no increases in hepatic ferritin protein levels were observed in neonatal rats after CoCl2 administration and subsequent HO-1 induction. We speculate that neonatal rats induce hepatic HO-1 and up-regulate ferritin by different mechanisms than do adult rats.
Abbreviations
- HO:
-
heme oxygenase
- GSH:
-
glutathione
- GAPDH:
-
glyceraldehyde-3-phosphate dehydrogenase
References
Yoshida T, Kikuchi G 1978 Purification of heme oxygenase from pig spleen microsomes. J Biol Chem 253: 4224–4229
Yoshida T, Kikuchi G 1978 Features of the reaction of heme degradation catalyzed by the reconstituted microsomal heme oxygenase system. J Biol Chem 253: 4230–4236
Tenhunen R, Marver HS, Schmid R 1969 Microsomal heme oxygenase. J Biol Chem 244: 6388–6394
Yoshinaga T, Shigeru S, Kappas A 1982 The oxidative degradation of heme c by the microsomal heme oxygenase system. J Biol Chem 257: 7803–7807
Maines MD, Mayer RD, Ewing JF, McCoubrey WK 1993 Induction of kidney heme oxygenase-1 (HSP 32) mRNA and protein by ischemia reperfusion: possible role of heme as both promoter of tissue damage and regulator of HSP32. J Pharmacol Exp Ther 264: 457–462
Maines MD, Kappas A 1975 Study of the developmental pattern of heme catabolism in liver and the effects of cobalt on cytochrome P450 and the rate of heme oxidation during the neonatal period. J Exp Med 141: 1400–1410
Sun Y, Maines MD 1990 Heme oxygenase-2 mRNA: developmental expression in the rat liver and response to cobalt chloride. Arch Biochem Biophys 282: 340–345
Lin JHC, Villalon P, Martasek P, Abraham NG 1990 Regulation of heme oxygenase gene expression by cobalt in rat liver and kidney. Eur J Biochem 192: 577–582
Keyse SM, Tyrrell RM 1989 Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc Natl Acad Sci USA 86: 99–103
Saunder EL, Maines MD, Meredith MJ, Freeman ML 1991 Enhancement of heme oxygenase-1 synthesis by glutathione depletion in Chinese hamster ovary cells. Arch Biochem Biophys 288: 368–373
Vile GF, Basu-Modak S, Waltner C, Tyrrell RM 1994 Heme oxygenase 1 mediates an adaptive response to oxidative stress in human skin fibroblasts. Proc Natl Acad Sci USA 91: 2607–2610
Nath KA, Balla G, Vercellotti GM, Balla J, Jacob HS, Levitt MD, Rosenberg ME 1992 Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J Clin Invest 90: 267–270
Dennery PA, McDonagh AF, Spitz DR, Rodgers PA, Stevenson DK 1995 Hyperbilirubinemia results in reduced oxidative injury in neonatal Gunn rats exposed to hyperoxia. Free Radic Biol Med 19: 395–404
Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN 1987 Bilirubin is an antioxidant of possible physiological importance. Science 235: 1043–1046
Stocker R, Glazer AN, Ames BN 1987 Antioxidant activity of albumin-bound bilirubin. Proc Natl Acad Sci USA 84: 5918–5922
McDonagh AF 1990 Is bilirubin good for you?. Clin Perinatol 17: 359–369
Applegate LA, Luscher P, Tyrrell RM 1991 Induction of heme oxygenase: a general response to oxidant stress in cultured mammalian cells. Cancer Res 51: 974–978
Fanaroff AA, Martin RJ 1992 Neonatal-Perinatal Medicine. Diseases of the Fetus and Infant, Mosby Year Book, St. Louis, pp 941–946
Chomczynski P, Sacchi N 1987 Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162: 256–259
Sambrook J, Fritsch E, Maniatis T 1989 Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, pp 1.21–1.105
Yoshida T, Biro P, Cohen T, Muller RM, Shibahara S 1988 Human heme oxygenase cDNA and induction of its mRNA by hemin. Eur Biochem 171: 457–461
Adams M, Dubnick M, Kerlavage A, Moreno R, Kelley J, Utterback T, Nagle J 1992 Sequence identification of 2.375 human brain genes. Nature 355: 632–634
Feinberg AP, Vogelstein B 1984 A technique for radiolabelling DNA restriction endonuclease fragments to high specific activity. Anal Biochem 137: 266–272
Wilks A, Ortiz de Montellano PR 1993 Rat liver heme oxygenase. High level expression of a truncated soluble form and nature of the meso-hydroxylating species. J Biol Chem 268: 22357–22362
Laemmli UK 1970 Cleavage of structural proteins during their assembly of the head of bacteriophage T4. Nature 227: 680–685
Towbin H, Stachelin T, Gordon J 1979 Electrophoretic transfer of protein from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76: 4350–4354
Vreman HJ, Stevenson DK 1988 Heme oxygenase activity as measured by carbon monoxide production. Anal Biochem 168: 31–38
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ 1951 Protein measurement with the folin phenol reagent. J Biol Chem 193: 265–275
Lin JH, Villalon P, Nelson JC, Abraham NG 1989 Expression of rat liver heme oxygenase gene during development. Arch Biochem Biophys 270: 623–629
Thaler MM, Gemes DL, Bakken AF 1972 Enzymatic conversion of heme to bilirubin in normal and starved fetuses and newborn rats. Pediatr Res 6: 197–201
Sunderman FW, Downs JR, Reid MC, Bibeau LM 1982 Gas-chromatographic assay for heme oxygenase activity. Clin Chem 28: 2026–2032
Vreman HJ, Stevenson DK 1988 Correlation of carbon monoxide and bilirubin production by tissue homogenates. J Chromatogr 427: 315–319
Vreman HJ, Ekstrand BC, Stevenson DK 1993 Selection of metalloporphyrin heme oxygenase inhibitors based on potency and photoreactivity. Pediatr Res 33: 195–200
Tomaro ML, Frydman J, Frydman RB 1991 Heme oxygenase induction by CoCl2, coprotoporphyrin IX, phenylhydrazine, and diamide: evidence for oxidative stress involvement. Arch Biochem Biophys 286: 610–617
Moorhouse CP, Halliwell B, Grootveld M, Gutteridge JMC 1985 Cobalt (II) ion as a promoter of hydroxyl radical and possiblecrypto-hydroxyl radical formation under physiological conditions. Differential effects of hydroxyl radical scavengers. Biochim Biophys Acta 843: 261–268
Kadiiska MB, Maples KR, Mason RP 1989 A comparison of cobalt (II) and iron (II) hydroxyl and superoxide free radical formation. Arch Biochem Biophys 275: 98–111
Maines MD, Kappas A 1976 Studies on the mechanism of induction of haem oxygenase by cobalt and other metal ions. Biochem J 154: 125–131
Sinclair P, Gibbs AH, Sinclair JF, De Matteis F 1979 Formation of cobalt protoporphyrin in the liver of rats. Biochem J 178: 529–538
Schaefer FV, Theil EC 1981 The effect of iron on the synthesis and amount of ferritin in red blood cells during ontogeny. J Biol Chem 256: 1711–1715
Author information
Authors and Affiliations
Additional information
This work was done while Dr. Tom was a postdoctoral fellow in the Department of Pediatrics, Division of Neonatal and Developmental Medicine, Stanford University School of Medicine.
Supported by National Institutes of Health (NIH) Grant HD 01124, NIH Training Grant HD 07249, NIH Grant HD 14426, and the Mary L. Johnson Research Fund.
Rights and permissions
About this article
Cite this article
Tom, D., Rodgers, P., Shokoohi, V. et al. Hepatic Heme Oxygenase Is Inducible in Neonatal Rats during the Early Postnatal Period. Pediatr Res 40, 288–293 (1996). https://doi.org/10.1203/00006450-199608000-00016
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199608000-00016
