
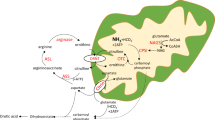
Abstract
Summary: A female and a male sibling aged 15 and 8 yr developed almost normally for 3 yr, then gradually deteriorated intellectually and physically. Both patients showed marked hyperargininuria (up to 370 times normal), prominent lysinuria, cystinuria, and ornithinuria, and a milder generalized amino aciduria. In plasma, arginine was 600–1000 μM (4–7 times normal), but other amino acids were not increased. The levels did not change in response to increased protein intake from 1–3 g/kg/day. In cerebrospinal fluid (CSF), arginine was 70–100 μM (3–5 times normal) and numerous other amino acids were also high. Plasma ammonia was generally normal, but rose to 250 μg/dl (5 times normal) after increased protein intake and, occasionally, other stresses. The erythrocyte arginase activity level was <1% of normal and half normal in the mother and three healthy siblings. White blood cell arginase activity was less than 5% of the lowest normal control. Liver obtained at open biopsy in the older sibling was deficient in arginase activity (1.5% of normal), but had levels of the other four urea cycle enzymes that were within the normal range. Electron microscopic and histochemical studies revealed patchy severe hydropic changes, increased cellular glycogen, normal mitochondria, and dilated endoplasmic reticulum.
Speculation: The pathogenesis of the neurologic abnormalities in patients with arginase deficiency may in part be the perturbation of intra-cellular amino acid levels secondary to the elevated levels of arginine.
Similar content being viewed by others


Article PDF
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Cederbaum, S., Shaw, K., Spector, E. et al. Hyperargininemia with Arginase Deficiency. Pediatr Res 13, 827–833 (1979). https://doi.org/10.1203/00006450-197907000-00007
Issue Date:
DOI: https://doi.org/10.1203/00006450-197907000-00007
Keywords
This article is cited by
-
Liver involvement in urea cycle disorders: a review of the literature
Journal of Inherited Metabolic Disease (2017)
-
Hyperargininemia due to arginase I deficiency: the original patients and their natural history, and a review of the literature
Amino Acids (2015)
-
Arginase-1 deficiency
Journal of Molecular Medicine (2015)
-
Amino acids in CSF and plasma in hyperammonaemic coma due to arginase1 deficiency
Journal of Inherited Metabolic Disease (2008)
-
NTPDase and 5′-nucleotidase Activities of Synaptosomes from Hippocampus of Rats Subjected to Hyperargininemia
Neurochemical Research (2007)
