
Abstract
The thermoreversible mechanical properties of a non-ionic polymer, lipid-based complex fluid were investigated by oscillatory and steady shear rheology, and analyzed in the context of mesophase architecture and polymer–water interactions. The sol phase (G″>G′) is observed at lower temperatures (5–17 oC) than the gel phase (G′>G″; 20–50 oC) and is driven by the temperature-induced changes in water solubility of polyethylene glycol (PEG). In the sol state, water solvated, extended PEG chains cannot interact within the confines of a two-dimensional hexagonally ordered array of prolate micelles that possess sufficient lattice dimensions to accommodate the polymer chains. With increasing temperature, the less water-soluble PEG adopts a compacted conformation that shields the polymer from bulk water. The densely coiled PEG chains localized within the interstitial water layers of a multi-lamellar structure promote chain entanglement and formation of a hydrogen-bonded network with the surrounding water layer, resulting in a soft gel. The strength of the hydrogen bonds within the network and therefore gel strength can be adjusted over an order of magnitude by temperature modulation between 20 and 50 oC. These studies identify principles useful in the preparation of stimuli-responsive mechanical materials.
Similar content being viewed by others


Introduction
Hydrogels, water-swollen polymer networks, are becoming increasingly important in a variety of technologies, ranging from biomaterials to soft electronics.1, 2, 3, 4, 5 Significant opportunities exist for developing hydrogels as protein-based materials. For example, protein-doped materials have been employed as high-throughput screens for drug discovery, heterogeneous biocatalysis or fabrication of hybrid biotic–abiotic devices.6, 7, 8, 9, 10 Another area of emerging need includes matrices suitable for the long-term storage of purified membrane proteins. Hydrogels, unlike inorganic matrices, such as sol–gel-derived silicas, have shown higher protein loading, successful entrapment of large proteins, and co-integration of both aqueous- and membrane-soluble proteins.11 Moreover, inorganic matrices often suffer from difficulty in reconstitution of proteins with retention of native structure and function.12 Numerous types of compartmentalized organic particles (for example, micelles, liposomes, polymersomes and so on) have successfully been used for the stable integration of multiple proteins.13, 14 Although the insertion of functional proteins into a variety of materials has been demonstrated, removal without detriment to the protein or matrix has not been achieved. A hydrogel possessing dynamically tunable mechanical properties may offer a pathway for achieving facile biomolecule insertion as well as extraction, allowing it to function as a re-usable protein container. The development of hydrogels with externally regulated mechanical properties offers a pathway towards production of a versatile material, useful as either a protein storage medium or as a renewable protein-based device.
Factors governing the mechanical behavior of hydrogels include the type of bonding (covalent versus non-covalent), the extent of intermolecular junction points between the polymer chains and polymer–water interactions. Although incorporation of covalent crosslinks produces durable elastic solids (chemical gels), non-covalent crosslinks readily allow for the development of dynamic systems (physical gels). Examples of physical gels with dynamically tunable mechanical properties are readily found in nature, including mucus secretions,15 the vitreous humor of the eye16 and synovial fluid of diarthrodial joints.17 Of the synthetic hydrogels capable of stimuli-responsive changes in mechanical properties, one system in particular is of interest, because of its demonstrated ability to stabilize large integral membrane proteins.9 The system, non-ionic polymer, lipid-based complex fluids, is composed of three amphiphilic molecules dispersed in ∼76 wt% water.18, 19, 20 The three amphiphiles are a saturated phospholipid (dimyristoylphosphatidylcholine, DMPC, 1), a zwitterionic co-surfactant (for example, N,N-dimethyldodecylamine-N-oxide, LDAO, 2) and a non-ionic polymer. The lipid component imparts characteristics (that is, chemical composition and structure) of a biomembrane. The non-ionic polymer controls the structure and physical properties of the complex fluid and can be introduced as either a polyethylene glycol (PEG) grafted-lipid conjugate (DMPE-EOn, 3) or as a block copolymer of PEOn-PPOm-PEOn (4). The co-surfactant softens the bilayer and guarantees membrane protein compatibility.9 The amphiphiles self-organize into macroscopically uniform, optically transparent, non-covalent aggregates when dispersed in water and undergo an inverted thermo-reversible phase transition, converting from a free-flowing liquid to a gel at higher temperatures. Despite the considerable details on the temperature-dependent lyotropic mesophase structures produced by adjustment of polymer architecture, the molecular interactions that govern the inverted, reversible gelation and mechanical properties have yet to be conclusively established.18, 19, 20 The objective of this work is to examine the viscoelastic properties of a PEGylated lipid-based complex fluid (DMPE-EO99) in the sol and gel regions by carrying out dynamic and static rheology measurements. The mechanical response is analyzed in the context of mesophase structure, determined by neutron and X-ray diffraction, and temperature-dependent water–polymer interactions as revealed by cloud-point determination and molecular-level interactions monitored by Raman spectroscopy. The composition selected for this study is the complex fluid that has been previously demonstrated to stabilize a large integral membrane protein, the photosynthetic reaction center, with retention of structure and function.9

Experimental procedure
Materials and methods
Lyophilized dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and 1,2 dimyristoyl-sn-glycerol-3-phosphoethanolamine-N-poly(ethylene glycol), (DMPE-EO99 or DMPE-PEG5000) were purchased from Avanti Polar Lipids (Alabaster, AL, USA) and used as received. Lauryldimethylamine-N-oxide (LDAO) was purchased from Calbiochem-Novabiochem Corp. (LaJolla, CA, USA). Milli-Q (18 MΩ) water was used for sample preparation.
Sample preparation
Samples were prepared as quaternary compositions as previously described.20 Samples consisted of 0.759±0.01 weight fraction (Φw) of water, Φs=0.0313±0.0005 LDAO co-surfactant and ΦL=0.148± 0.006 lipid (for example, 0.0716 g of DMPC; 0.0313 DMPE-PEG5000 in a total mass of 0.5 g), yielding a total lipid to PEG content of 7 mol%. Hydration of the solid components in de-ionized water was accomplished by repeated cycles of heating (50 oC), vortex mixing and cooling on an ice bath until sample uniformity was achieved.
Physical methods
Differential scanning calorimetry was performed on a Q100 instrument (TA Instruments, New Castle, DE, USA) interfaced with a refrigerated cooling system. Weighed amounts (1–5 mg) of the sample were hermetically sealed in aluminum pans and data were collected between −65 and 70 °C at a heating rate of 2 °C min−1. Instrument calibration was performed using an indium standard. Raman spectra were obtained using a Renishaw inVia spectrometer (Renishaw plc, Wotton-under-Edge, UK) equipped with Leica microscope (DM2500M; Leica Microsystems GmbH, Wetzlar, Germany). A standard Si sample was used for the calibration at 520.7 cm−1. A 633-nm He-Ne laser was employed as an excitation source with a maximum power of 17 mW. A microscope equipped with a × 20 objective (numerical aperture 0.4) was used to focus the laser light on the sample. Spectra were recorded by averaging 20 accumulations collected at a resolution of 3 cm−1. Samples were held in sealed thin-wall quartz capillaries (0.1 mm optical density, Charles-Supper Co., Natick, MA, USA). Temperature control was achieved using a Peltier plate equipped with temperature controller (CL-100, Warner Instruments, Hamden, CT, USA). Cloud-point (lower critical solution temperature (LCST)) measurements were performed using a custom-built fiber optic spectrometer equipped with a DT-1000-CE UV-Vis light source, and a SD2000 CCD (Ocean Optics, Dunedin, FL, USA). The change in optical density was monitored as a function of temperature from 3 to 40 °C at a heating rate of 0.5 °C min−1. The sample was sealed in a 5 mm nuclear magnetic resonance tube and temperature was regulated using a custom-built Peltier.
Rheology experiments were performed using a TA Instruments (Newark, DE, USA) AR-2000 stress controlled rheometer equipped with a 20 mm parallel plate geometry and a Peltier plate for temperature control (±0.1 °C). A solvent trap was used to prevent solvent evaporation. A 450–500 μm gap was sufficient to achieve sample distribution over the entire geometric surface. All standard calibrations were made before data collection. Samples were examined using a series of oscillatory tests. The linear viscoelastic region was first determined by carrying out stress sweeps under an applied torque between 0.1–103μN·m at constant frequency (1 Hz). The temperature sweeps were carried out from 5 to 50 °C with a rate of 3 °C min−1 at a controlled oscillatory stress within the linear viscoelastic region. Frequency sweeps were also performed from 0.1 to 100 Hz at a constant oscillatory stress within the linear viscoelastic region at select temperatures identified by the temperature ramps.
Small-angle X-ray scattering (SAXS) measurements were made using the pin-hole set-up at the undulator beamline 12ID-C (12 keV) at the Advanced Photon Source at Argonne National Laboratory (Lemont, IL, USA). The two-dimensional (2D) scattering profiles were recorded using a home built charge-coupled device detector. The detector is composed of four charge-coupled device chips and features a 175-mm square active area with 1000 × 1000 pixel resolution. The sample-to-detector distance was such as to provide a detecting range for momentum transfer of 0.01 Å−1<q<0.6 Å−1. The scattering vector, q, was calibrated using a silver behenate standard at q=1.076 Å−1. The 2D scattering images were azimuthally averaged to produce plots of scattered intensity, I(q), versus scattering vector, q, where is q=4π/λ(sinθ). The value of q is proportional to the inverse of the length scale, Å−1. Temperature control of samples was achieved using a custom-built Peltier cooler.
Small angle neutron scattering measurements were performed at the Intense Pulsed Neutron Source at the Argonne National Laboratory, using time-of-flight small-angle X-ray scattering instruments and the small-angle neutron diffractometer. The instrument used pulsed neutrons derived from spallation with a wavelength of radiation in the range of 0.5–14 Å and a fixed sample to detector distance of 2 m. The scattered neutrons were measured using a 64 × 64 array of position sensitive, gas-filled, 40 × 40 cm2, proportional counters with the wavelengths measured by time-of-flight through binning the pulse to 68 constant Δt/t=0.005–0.35 Å−1. Samples were sealed in 1-mm path length suprasil cells. Samples were equilibrated at the experimental run temperature for at least 30 min before data collection. The sample temperature was regulated by a water recirculating bath and measured using a calibrated thermocouple. Low-temperature measurements were carried out with the sample chamber under an N2, thereby preventing condensation of water onto the sample cell. Data were collected for 5–8 h and reduced using standard methods. The results were placed on the absolute scale using the known scattering cross section of a silica gel sample.
Results and discussion
Thermoreversible gelation
The simplest means by which to evaluate the mechanical characteristics of a complex fluid is by the so-called ‘tube inversion’ test, which involves simple visual assessment of a sample’s ability to resist flow to gravity. As shown in Figure 1a (inset, right side panel), the DMPE-PEG5000 complex fluid is able to hold its own weight in an inverted vial at room temperature (22 oC), demonstrating its gel-like behavior. The gel is not, however, a self-supporting material upon removal from the vial, suggesting a ‘soft’ or ‘weak’ gel. Upon cooling on ice, the sample flows within the vial (Figure 1a, left side panel). Further details on the viscoelastic response of the complex fluid can be made through dynamic and steady-shear rheological experiments analyzed in the context of the structural data determined by small-angle diffraction and local molecular environment (that is, solute–solvent interactions) as assessed by thermal analysis, cloud-point determination and vibrational spectroscopy.

(a) Temperature dependence of dynamic storage, G′ (solid circles) and loss moduli, G″ (open squares) and tan δ (crosses). Inset: photographs of complex fluid collected after incubation on ice (left) and at room temperature (right). (b) Frequency dependence of storage, G′ (solid circles) and loss, G″ (open squares) moduli recorded at selected temperatures determined by the temperature-ramp studies. (c) Shear rate dependence of steady-shear viscosity at selected temperatures determined by the temperature-ramp studies. Lines are provided as a guide to the eye. A full color version of this figure is available at Polymer Journal online.
Rheological studies using oscillatory shear are commonly used to characterize the mechanical properties of soft solids (gels). The temperature-dependent rheological characteristics of a complex fluid prepared by introducing the polymer component as a lipid conjugate, DMPE-PEG5000, was evaluated by monitoring the evolution of the moduli by heating the sample at a rate of 3 oC min−1 under a low applied oscillatory stress of 1.1 Pa (Figure 1a). Two rheologically distinct regions are observed. At reduced temperatures, between 5 and 17 oC, the viscous modulus (G″) exceeds the elastic modulus (G′), indicating liquid-like behavior of the complex fluid, which is consistent with the visual observation of the cooled sample flowing within the sample vial (Figure 1a). As the temperature is further increased, a large discontinuity is observed between 17 and 19 oC. The sol-to-gel transition, Tgel, defined as the point where the loss tangent, tan δ, is unity (for example, the point where G′∼G″) occurs at 17.5 oC.21 The location of the sol-to-gel transition determined by dynamic rheology compares well with the visual observations made during the simple tube-inversion test. At higher temperatures (19–50 oC) the elastic modulus, G′ is greater than G″, defining an elastic, gel region. Within the second region, the elastic modulus remains higher in magnitude than the loss modulus, but a slow decline in the magnitude of both moduli is observed. A G′ maximum of 730 Pa is reached, making this a ‘soft’ gel.22 The temperature variation of the moduli for the PEGylated lipid-based complex fluid resembles profiles reported for many other aqueous dispersions containing poly(oxyalkylene)s.23, 24, 25 Many of these systems, however, form hard gels, having a maximum G′ value in the kPa range.23 The thermo-inverted gelation mechanism for concentrated aqueous solutions of PEOn-PPOm-PEOn has been determined to arise from the temperature-driven hydrophobic effect of the polypropylene oxide (PPO) block, which causes the micelles to associate (hard-sphere crystallization).23
It has been well-documented that dynamic frequency sweeps can provide a more comprehensive picture of the viscoelasticity of a mesophase, providing insights into the physical network structure. For the DMPE-PEG5000 complex fluid, the elastic modulus, G′, and the viscous modulus, G″, are plotted as a function of frequency recorded at selected temperatures within each of the distinct regions identified in the temperature ramp studies (Figure 1a). In the liquid-like region (10 oC), both moduli display strong frequency dependence with G″>G′ over the entire frequency range examined (Figure 1b, left panel). The behavior is consistent with non-elastic behavior. A similar mechanical response has been associated with low-viscosity liquids composed of isolated micelles.26 As reported previously for the low molecular weight composition (DMPE-PEG2000), the sol phase structure was determined to be composed of unassociated prolate micelles.18 (The sol structure of this composition is detailed below.) The mechanical response further suggests that the grafted polymer must also be non-interacting. Thus, the local mesoscale environment in which the polymer chains reside is one where the solvated chains stretched along the direction normal to the micelle surface, like a brush in a good solvent environment, can be accommodated without overlap or entanglement.
The dynamic frequency sweep collected at a temperature above the Tgel (at 22 oC) shows the elastic modulus, G′ greater than the viscous modulus, G″ with the two moduli intersecting at f∼10 Hz. (Figure 1b, center panel). The observed absence of a low-frequency crossover is consistent with an elastic lamellar phase and compares well with similar measurements made on hydrogels prepared from a quaternary mixture of DMPC/DMPE-PEG2000/pentanol in water.27, 28 The frequency independent value of G′ is the gel modulus, Go, and is∼130 Pa.29 The magnitude and near invariance of the elastic modulus at low frequencies suggests the presence of physical crosslinks.30 Evaluation of the mechanical response at a higher temperature within the gel phase (35 oC) is presented in Figure 1b, right panel. The profile qualitatively looks similar to the response recorded at 22 oC with G′>G″ at low frequencies and convergence at higher frequencies (f∼10 Hz). The gel modulus, Go, is∼53.6 Pa, a value less than that recorded at 22 oC, suggesting a weakening in the gel strength with increasing temperature. The frequency data confirm the results of the temperature ramp study, indicating that the DMPE-PEG5000 complex fluid transitions from well separated and predominately non-interacting polymer chains (sol) to a state where physical crosslinks and entangled (interacting) polymer chains are significantly increased (elastic gel).
The mechanical characteristics of the complex fluid are further probed through steady-shear rheology (Figure 1c). Steady-shear viscosity studies were performed on the complex fluid at temperatures below and above the sol-to-gel phase transition. Below Tgel (at T=10 oC), the flow curve recorded for the DMPE-PEG5000 composition is essentially linear over the entire range of applied shear stress (Figure 1c, solid circles), consistent with isolated (non-interacting) polymer chains. At temperatures above the Tgel, (at T=28 and 40 oC), the flow curves exhibit complex flow behavior with viscosity dependent on shear stress (Figure 1c). The flow curves contain an intermediate Newtonian region separated by two shear-thinning regions (reduction in viscosity) at low and high shear stress. The observation of shear thinning suggests that under these conditions, disentanglement of polymer chains occurs. Similar profiles have been reported for telechelic or associative polymers and has been taken as an indicator of a soft-to-rigid-to-soft shifting transition.31 The yield stress is found to shift towards lower applied shear with increasing temperature, an observation consistent with the both the temperature ramp studies and frequency-dependent behavior of the moduli, indicating that the gel becomes ‘softer’ with increasing temperature.
Mesophase structure
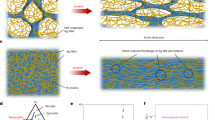
The rheological data can be interpreted in the context of the complex fluid’s mesophase structure in the sol and gel states as determined by neutron and X-ray scattering. As previously reported, the structure of the DMPE-PEG2000 complex fluid at reduced temperature was determined to be a 2D hexagonally ordered array of prolate micelles.18 The cold-phase structure of the DMPE-PEG5000 complex fluid has not been reported. Small-angle neutron scattering was employed for the determination of the sol structure at 9 oC. The advantage of neutrons over X-rays for characterization of the mesophase structure lies in the ability to contrast specific regions of a multi-component system by using isotopic substitution of a given element, which allows for varying the scattering power (scattering length density). Here, D2O was exchanged for H2O, giving an approximate six-fold enhancement in the scattering power. Thus, observed scattering arises almost exclusively from excess scattering contrast between the organic amphiphiles and D2O. The neutron scattering curve recorded on the DMPE-PEG5000 composition at 9 oC is shown in Figure 2a (black open circles). The scattering curve is dominated by a broad peak centered at Q=0.038 Å−1 and a less pronounced peak (shoulder) at higher Q. The breadth of the diffraction peak indicates poor structural coherence, consistent with that of a liquid. Structural information was derived from the experimental profile by fitting the peak at 0.038 Å−1 to a Gaussian function (Supplementary Figure S1) parameterized by position, width and amplitude using a Levenberg–Marquardt algorithm (IGOR Pro 6.0, Wavemetrics, Lake Oswego, OR, USA; Table 1).18 Subtraction of the fitted Gaussian and the incoherent background scattering, (I(Q)=0.176 Å−1), arising from the hydrogenated amphiphiles, reveals additional Bragg peaks, which can be similarly modeled with a Gaussian function. This procedure permitted deconvolution of the scattering profile into five orders of diffraction (Figure 2b, Table 1) positioned at Q=0.021, 0.038, 0.041, 0.067 and 0.092 Å−1, which can be indexed as the h,k=[1,0], [1,1], [2,0], [3,0] and [3,2] reflections of a 2D hexagonal lattice ((space group p6m), Figure 3 left). The d-spacing or hexagonal repeat distance is 299 Å. The lattice spacing (the repeat distance between cylindrical aggregates) is related to the d-spacing by s=(2/√3)d, and is 345 Å. The structure is identical to that previously determined for the DMPE-PEG2000 complex fluid.18 Unexpectedly, a doubling of the ethylene oxide (EO) repeat units did not increase the lattice dimensions. The absence of lattice expansion is contrary to prior studies examining changes in the lamellar gel-phase structure with polymers of increasing EO block length.20 Specifically, in the lamellar gel phase, increasing the EO repeat units causes a systematic increase in d-spacing. The expansion of the d-spacing was attributed to increased steric pressure between opposing lamellae, causing swelling of the interstitial water layers.20 This suggests that the micellar sol phase adopts a 2D hexagonal meosphase geometry, providing sufficient space for the PEG chains to stretch and extend without chain interaction between adjacent micelle coronas. It is further noted that the [2,1], [2,2], [3,1] and [4,0] reflections could not be fitted to the same level of accuracy as the other peaks due to their expected low relative intensity under the assigned space group and the reduction in their scattering intensity with increasing Q.18 Thus, the form factor component of the scattering profile was not evaluated.

(a) Experimental small-angle neutron scattering profile of the complex fluid at 9 oC (open circles) with the sum of the fitted five orders of diffraction and incoherent scattering (red line) and the residual scattering after removal of the five orders of diffraction (black squares). (b) The sum of the five Bragg peaks extracted from the experimental scattering profile. The dashed lines represent the nonlinear least-squares fit for each of the individual Gaussian functions extracted from the experimental small-angle neutron scattering profile. Representative SAXS azimuthally averaged data collected on the gel (c) at 22 oC, and (d) at 43 oC.

Schematic illustration of mesophase architectures as determined by neutron and X-ray scattering (left) 2D hexagonal sol, (top, right) swollen lamellar gel, (bottom, center) freeze-dried, collapsed lamellar solid.
The gel phase structure at 22 oC was determined by SAXS (Figure 2c) and revealed five orders of diffraction with integral order spacing with respect to the first peak (q=0.0393, 0.0797, 0.120, 0.161 and 0.202 Å−1) consistent with an ordered lamellar structure featuring an expanded d-spacing of 160 Å (Figure 3, right panel). Precise information, derived from the one-dimensional electron-density maps, on the lipid bilayer and intervening water channel structure, and the conformational state and projection of the PEG chains, was detailed elsewhere for a similar composition.20 Briefly, it was determined that the lamellar structure consists of amphiphilic bilayers ca. 45 Å thick (dB).20 The thickness of the interstitial water region, dW, given by dW=d–dB, would be∼115 Å. In addition, low-resolution ‘images’ of electron density changes within the interstitial water regions showed a broad region of increased electron density spanning nearly 54 Å from the bilayer surface, reflecting the region of average localization/projection of the PEG chains.20 Such mesophase architecture would promote the formation of lateral (intra-lamellar) and possible (to a lesser extent) cross-channel (inter-lamellar) physical crosslinks between grafted PEG chains (Rg=22 Å), producing a network favorable for physical gelation.
At 43 oC (Figure 2d), the structure remains lamellar with increased order as evidenced by the appearance of two additional orders of diffraction (q=0.0297, 0.0607, 0.0916, 0.124, 0.155, 0.184 and 0.213 Å−1). An increase in the lattice spacing (to d=212 Å) is also observed, suggesting that the grafted PEG chains have altered their conformational state to increase steric pressure between the opposing bilayers. The improved structural ordering and additional swelling of the interstitial water layers is attributed to the reduced water solubility of the PEG chains. The EO chains are believed to adopt a more compact mushroom conformation in this state, shielding the polymer from bulk water. Further compaction of the PEG chains would be expected to reduce the physical crosslinks and entanglement between adjacent polymer chains, thereby lowering sample viscosity and weakening the gel. This trend is clearly observed in the temperature-dependent rheological data, where the magnitude of G′ and viscosity decrease with increasing temperature in the gel state.
The effect of water removal on the hydrogel structure was also evaluated. The hydrogel was quenched from room temperature by rapid immersion in liquid nitrogen. Once completely frozen, the sample was placed under vacuum (35 mTorr) for 18 h. A white, optically birefringent (Figure 4, inset) solid was recovered and the structure determined by SAXS (Figure 4). The freeze-drying process successfully captured a well-ordered lamellar structure as evidenced by four orders of diffraction (q=0.124, 0.248, 0.370 and 0.499Å−1) in the SAXS pattern. The removal of water (from ca. 76 to 3 wt%, as determined by thermogravimetric analysis (TGA)) collapsed the d-spacing to 51 Å (Figure 3). On the basis of a bilayer thickness of 45 Å, an intervening layer of ∼6 Å exists, which may be composed of residual tightly bound water and the collapsed PEG network. The preservation of a well-ordered multilamellar structure could be facilitated by the collapsed PEG network acting to stabilize the bilayer.32 Although qualitative examination of the SAXS pattern can be used to infer the role of PEG, more precise information regarding the lipid bilayer structure, and conformational state and region of localization of the polymer is awaiting further X-ray studies and determination of the electron-density profiles.20 These studies are beyond the scope of the current work and will be reported elsewhere.

SAXS pattern recorded on the freeze-dried hydrogel. Inset: representative polarized optical micrograph (image collected using × 16 magnification). A full color version of this figure is available at Polymer Journal online.
Polymer–water interactions
The fundamental characteristics of a hydrogel can be determined by evaluation of polymer–water interactions.33 Water within a hydrogel can be broadly classified as three types: i) freezing water that is very weakly associated with the polymer and behaves as pure (bulk) water; ii) freezing-bound water, which has altered thermal properties compared with bulk water due to interactions with the polymer; iii) non-freezing bound water that does not exhibit a detectable phase transition within normal temperatures because of strong association to the polymer. One well-documented way of studying the various types of water within a hydrogel is by differential scanning calorimetry.34 The heating curve recorded on a DMPE-PEG5000 complex fluid is presented in Figure 5a. Two melting transitions from water are observed. The smaller melting point, located at −17.0 oC, is assigned to freezing-bound water. This transition has been previously assigned to arise from a PEG hydrate, which can bind to ice/water molecules via hydrogen bonds.35 It has been further determined in PEG-grafted liposomes that the freezing-bound water primarily resides with the PEG chains.34 The larger melting point, located at −1.4 oC, is assigned to bulk water and represents the majority component within the complex fluid. The existence of freezing-bound water, which is polymer associated, yet remains mobile and exhibits intermediate bonding character between polymer and bulk water, has been implicated as critical for gelation.36 Further studies conducted on a wider variety of hydogels are needed to fully understand the importance of freezing-bound water in the formation of gel states.

(a) Differential scanning calorimetry heating scan (2 oC min−1) collected on a DMPE-PEG5000-based complex fluid. (b) Intensity of transmitted visible light (λ=550 nm) versus complex fluid temperature. Line is drawn as a guide to the eye.
At higher temperatures, a complex endothermic phase transition between 17 and 24 oC is observed. The transition features two components; first, a weaker pre-transitional event at 17.7 oC, followed by a second event at 21.5 oC. Because of the small enthalpy associated with these events, that is considerably smaller than expected for the Lβ to Lα transition for a DMPC water dispersion, it is assigned to the non-bilayer to bilayer structural rearrangement occurring at the sol-to-gel phase transition.18 Last, an endothermic transition is observed at 54.5 oC and is assigned to melting of the polyethylene oxide (PEO).37
The temperature-dependent water solubility of the grafted PEG chains can be evaluated by determination of the cloud point. The water solubility of many polymers change as a function of temperature, often exhibiting phase separation as the temperature is raised above the cloud point or LCST. The observed behavior is common for polymers that form hydrogen bonds with water. Below the LCST, the polymer chains expand or swell. Above the LCST, the polymer chains collapse or shrink. The archetypical thermo-responsive polymer is poly(N-isopropylacrylamide), which exhibits an LCST near body temperature (32 oC).38 Aqueous solutions of PEO typically have a cloud point between 100 and 150 oC, but introduction of hydrophobicity can significantly lower the LCST.39 The cloud point of the complex fluid was determined by monitoring transmitted visible light as a function of increasing temperature, using a fiber optic spectrometer (Figure 5b). A sharp reduction in transmitted light was observed at 12 oC signaling the onset of the LCST. Maximum opacity is attained at 19 oC (ca. 50% T). Gradual clearing of the sample occurs as the temperature is raised to 45 oC, exceeding 73%T by 25 oC. The location of the transition region overlaps the sol-to-gel transition determined by rheology. Below the LCST, the stretched PEG chains are isolated by solvent molecules (non-interacting), a condition that serves to increase the core-corona interfacial curvature causing the structural transition into the micellar state.40 At the LCST, associated water is released, promoting polymer chain interaction (entanglement and hence gelation), leading to clouding of the complex fluid. A reduction in the size of the hydrophilic dimensions of the amphiphilic headgroup causes the structural transition to the lamellar phase.
Raman spectroscopy is the preferred technique for examining molecular structure in aqueous environments. Specifically, the O–H stretching modes can provide direct information about the local molecular-level structure of the gels and water. Five distinct Raman active modes originating from water molecules can be used to provide information on the orientation and interactions of water.33 These interactions can be evaluated by monitoring the vibrational bands in the Raman spectrum between 3000 and 3600 cm−1 (Figure 6a).41, 42 Among the five modes, two main components are observed within the spectral region at ca. 3200 and 3400 cm−1, and their relative intensity and position vary with temperature. The peak at ca. 3200 cm−1 is assigned to the collective in-phase vibration of strongly hydrogen-bonded water molecules (νip).33 The mode at ca. 3400 cm−1 is assigned to the out-of-phase vibrations between water molecules, and the first and higher shell of neighboring molecules (νoop). The mode positions, determined by fitting to Gaussian functions (Supplementary Figures S2-3, Table S1) are found to systematically shift to higher wave numbers as sample temperature is increased. The νoop mode shifts from a minimum value of 3427±1 cm−1 (at 10 oC) to a maximum of 3433±1 cm−1 (at 35 oC) from the sol-to-gel state. The bulk water νoop mode is also observed to shift to higher wave numbers with increasing temperature (from 3417±1 cm−1 at 10 oC to 3421±1 at 35 oC). The rate of change determined for the complex fluid νoop mode, however, is greater than that determined for bulk water (Δ0.0842 cm−1 K−1; Figure 7). Prior work has demonstrated that the vibrational energy of the O–H stretching mode increases with decreasing O–H bond length.33 On the basis of prior findings, the data suggests that the hydrogen bonds between water molecules within the gel phase are weaker than those found in bulk water. A similar analysis can be carried out for the νip mode, which reflects bonding interactions between PEG and water. Here, however, an opposite trend is observed. Specifically, the rate of change for the complex fluid νip mode is less than that determined for bulk water, indicating that the hydrogen bonds between PEG and water within the gels are stronger than that in bulk water. Collectively, these results indicate that an extended network structure of weakly hydrogen-bonded water that is strongly associated to the PEG is present in the gel state. (Supplementary Table S2).

Raman spectra recorded in the (a) high and (b) mid-frequency region at select temperatures in the sol (10 oC) and gel (25, 35 oC) phases of a DMPE-PEG5000-based complex fluid.

Temperature dependence of wavenumber of the (a) out-of-phase and (b) in-phase mode of O–H stretching for DMPE-PEG5000-based complex fluids compared with bulk water. Solid lines represent fitted line. A full color version of this figure is available at Polymer Journal online.
The hydrogen bond is most likely formed between water and the ether group on the PEG. Validation of the nature of the hydrogen bond can be made by examination of the vibrational modes located within the mid-frequency region of the Raman spectra.43 The diagnostic mode used to assess the nature of hydrogen bonding interactions in PEG-based systems is the asymmetric stretching mode of C–C–O (Figure 6b).43 In the sol state, the mode is positioned at 1131 cm−1 and shifts to lower wave numbers, 1129 cm−1, with increasing temperature and formation of the gel. Prior work has reported that such a shift is indicative of the formation of inter- and intra-chain hydrogen bonds.43 Thus, collectively, the Raman data indicates that the gel phase is composed of a physically crosslinked network produced by hydrogen-bonded water to the ether oxygen of the polymer chains.
Conclusion
The mechanical characteristics of a thermally driven reversible transient network were examined in the context of mesophase architecture and polymer–water interactions. Specifically, the temperature-driven water solubility and thus conformational changes of the amphiphile-tethered PEO regulates the formation and strength of a hydrogen bonded network. At temperatures below the LCST (19 oC), the hydrated EO chains stretch normal from the amphiphile surface, minimizing polymer-chain interactions. The disentangled (isolated) polymer chains lead to a marked reduction in both the elastic modulus and viscosity. The highly extended chains also drive a structural conversion to a micellar state from stacks of bilayers (lamellar). Neutron scattering on the cold phase revealed that the micelles organize in a 2D hexagonal lattice. This 2D hexagonal phase features sufficient distance between adjacent cylinders (34.5 nm) to accommodate the stretched polymer chains without entanglement at a grafting density of 7 mol%. As the temperature is increased above the LSCT, the PEO becomes less water soluble and shields itself from the bulk water by adopting a compact coiled (mushroom) state. This conformational change is accompanied by a mesophase structural transition to multilamellar. Within this geometry, the coiled polymer becomes localized at the membrane—water interface. The localization of the polymer against the bilayer—water interface fosters polymer-chain association through formation of temporary junctions. The junctions were probed by Raman spectroscopy and revealed signature modes consistent with a hydrogen-bonded network between water and the ether oxygen of PEG. Between 20 and 25 oC, the gels strengthen with increasing temperature (that is, maximum elastic modulus), driven by the decrease in PEO water solubility and chain compaction. Above ∼25 oC, further reduction in PEO water solubility results in chain collapse, but thermal weakening of the hydrogen bonds,44 acting to lessen the gel strength as evidenced by a decrease in the magnitude of the elastic modulus.
In nature, the ability to reversibly form hydrogen bonds is a key principle leading to the formation of specific tertiary structures that enable functional biomolecules. Here, the ability to reversibly form a hydrogen-bonded network, driven by the water solubility, and hence, conformation changes of PEG generates a thermo-responsive, structured lyotropic mesophase. Although earlier studies demonstrated that the existence of an inverted phase transition (sol at a lower temperature than the gel) was ideal for the facile reconstitution of membrane proteins, future studies will explore ways to exploit the temperature-induced changes in mechanical properties to extract proteins post-storage. In addition, the co-integration of both membrane- and water-soluble proteins, and synthetic modifications to the current system permitting the selective release of these components will be evaluated. It is envisioned that hybrid materials containing both covalent and non-covalent crosslinking will produce materials capable of functioning as a reusable protein storage container.
References
Peppas, N. A., Hilt, J. Z., Khademhosseini, A. & Langer, R. Hydrogels in biology and medicine: from molecular principles to bionanotechnology. Adv. Mater. 18, 1345–1360 (2006).
Grubjesic, S., Lee, B., Seifert, S. & Firestone, M. A. Preparation of a self-supporting cell architecture mimic by water channel confined photocrosslinking within a lamellar structured hydrogel. Soft Matter 7, 9695–9705 (2011).
Slaughter, B. V., Khurshid, S. S., Fisher, O. Z., Khademhosseini, A. & Peppas, N. A. Hydrogels in regenerative medicine. Adv. Mater. 21, 3307–3329 (2009).
Koo, H.-J., So, J.-H., Dickey, M. D. & Velev, O. D. Towards all-soft matter circuits: prototypes of quasi-liquid devices with memristor characteristics. Adv. Mater. 23, 3559–3564 (2011).
Koo, H.-J., Chang, S. T., Slocik, J. M., Naik, R. R. & Velev, O. D. Aqueous soft matter based photovoltaic devices. J. Mater. Chem. 21, 72–79 (2011).
Ge, X., Lebert, J. M., Monton, M. R. N., Lautens, L. L. & Brennan, J. D. Materials screening for sol-gel-derived high-density multi-kinase microarrays. Chem. Mater. 23, 3685–3691 (2011).
Brennan, J. D. Biofriendly sol-gel processing for the entrapment of soluble and membrane-bound proteins: toward novel solid-phase assays for high-throughput screening. Acc. Chem. Res. 40, 827–835 (2007).
Mertz, D., Vogt, C., Hemmerle, J., Mutterer, J., Ball, V., Voegel, J.-C., Schaaf, P. & Lavelle, P. Mechanotransductive surfaces for reversible biocatalysis activation. Nat. Mater. 8, 731–735 (2009).
Laible, P. D., Kelley, R. F., Wasielewski, M. R. & Firestone, M. A. Electron-transfer dynamics of photosynthetic reaction centers in thermoresponsive soft materials. J. Phys. Chem. B 109, 23679–23686 (2005).
Mulder, C. L., Theogarajan, L., Currie, M., Mapel, J. K., Baldo, M. A., Vaughn, M., Willard, P., Bruce, B. D., Moss, M. W., McLain, C. E. & Morseman, J. P. Luminescent solar concentrators employing phycobilisomes. Adv. Mater. 21, 3181–3185 (2009).
Wang, Y. & Caruso, F. Mesoporous silica spheres as supports for enzyme immobilization and encapsulation. Chem. Mater. 17, 953–961 (2005).
Besanger, T. & Brennan, J. Entrapment of membrane proteins in sol-gel derived silica. J. Sol-Gel Sci. Technol. 40, 209–225 (2006).
O'Neil, C. P., Suzuki, T., Demurtas, D., Finka, A., Hubbell, J. A. & Novel, A Method for the encapsulation of biomolecules into polymersomes via direct hydration. Langmuir 25, 9025–9029 (2009).
Tanner, P., Baumann, P., Enea, R., Onaca, O., Palivan, C. & Meier, W. Polymeric vesicles: from drug carriers to nanoreactors and artificial organelles. Acc. Chem. Res. 44, 1039–1049 (2011).
Taylor Nordgard, C. & Draget, K. I. Oligosaccharides as modulators of rheology in complex mucous systems. Biomacromolecules 12, 3084–3090 (2011).
Sharif-Kashani, P., Hubschman, J.-P., Sassoon, D. & Pirouz Kavehpour, H. Rheology of the vitreous gel: effects of macromolecule organization on the viscoelastic properties. J. Biomech. 44, 419–423 (2011).
Chang, D. P., Abu-Lail, N. I., Guilak, F., Jay, G. D. & Zauscher, S. Conformational mechanics, adsorption, and normal force interactions of lubricin and hyaluronic acid on model surfaces. Langmuir 24, 1183–1193 (2008).
Firestone, M. A., Thiyagarajan, P. & Tiede, D. M. Structure and optical properties of a thermoresponsive polymer-grafted, lipid-based complex fluid. Langmuir 14, 4688–4698 (1998).
Firestone, M. A., Wolf, A. C. & Seifert, S. n. Small-angle X-ray scattering study of the interaction of poly(ethylene oxide)-b-poly(propylene oxide)-b-poly(ethylene oxide) triblock copolymers with lipid bilayers. Biomacromolecules 4, 1539–1549 (2003).
Lee, B. & Firestone, M. A. Electron density mapping of triblock copolymers associated with model biomembranes: Insights into conformational states and effect on bilayer structure. Biomacromolecules 9, 1541–1550 (2008).
van de Manakker, F., van der Pot, M., Vermonden, T., van Nostrum, C. F. & Hennink, W. E. Self-Assembling hydrogels based on β-cyclodextrin/cholesterol inclusion complexes. Macromolecules 41, 1766–1773 (2008).
Kelarakis, A., Castelletto, V., Chaibundit, C., Fundin, J., Havredaki, V., Hamley, I. W. & Booth, C. Rheology and structures of aqueous gels of triblock(oxyethylene/oxybutylene/oxyethylene) copolymers with lengthy oxyethylene blocks. Langmuir 17, 4232–4239 (2001).
Harrison, W. J., Aboulgasem, G. J., Elathrem, F. A. I., Nixon, S. K., Attwood, D., Price, C. & Booth, C. Micelles and gels of mixed triblock copoly(oxyalkylene)s in aqueous solution. Langmuir 21, 6170–6178 (2005).
Grassi, G., Crevatin, A., Farra, R., Guarnieri, G., Pascotto, A., Rehimers, B., Lapasin, R. & Grassi, M. Rheological properties of aqueous Pluronic alginate systems containing liposomes. J. Colloid Interf. Sci. 301, 282–290 (2006).
Halacheva, S., Rangelov, S. & Tsvetanov, C. Rheology of aqueous solutions of polyglycidol-based analogues to pluronic block copolymers. J. Phys. Chem. B 112, 1899–1905 (2008).
Kumar, R., Ketner, A. M. & Raghavan, S. R. Nonaqueous Photorheological fluids based on light-responsive reverse wormlike micelles. Langmuir 26, 5405–5411 (2009).
Warriner, H. E., Idziak, S. H. J., Slack, N. L., Davidson, P. & Safinya, C. R. Lamellar biogels: fluid-membrane-based hydrogels containing polymer lipids. Science 271, 969–973 (1996).
Warriner, H. E., Davidson, P., Slack, N. L., Schellhorn, M., Eiselt, P., Idziak, S. H. J., Schmidt, H.-W. & Safinya, C. R. Lamellar biogels comprising fluid membranes with a newly synthesized class of polyethylene glycol-surfactants. J. Chem. Phys. 107, 3707–3722 (1997).
Sun, K. & Raghavan, S. R. Thermogelling aqueous fluids containing low concentrations of Pluronic F127 and laponite nanoparticles. Langmuir 26, 8015–8020 (2010).
Lomakin, J., Huber, P. A., Eichler, C., Arakane, Y., Kramer, K. J., Beeman, R. W., Kanost, M. R. & Gehrke, S. H. Mechanical properties of the beetle elytron, a biological composite material. Biomacromolecules 12, 321–335 (2010).
Bossard, F., Aubry, T., Gotzamanis, G. & Tsitsilianis, C. pH-Tunable rheological properties of a telechelic cationic polyelectrolyte reversible hydrogel. Soft Matter 2, 510–516 (2006).
Nykaenen, A., Nuopponen, M., Hiekkataipale, P., Hirvonen, S.-P., Soininen, A., Tenhu, H., Ikkala, O., Mezzenga, R. & Ruokolainen, J. Direct imaging of nanoscopic plastic deformation below bulk Tg and chain stretching in temperature-responsive block copolymer hydrogels by cryo-TEM. Macromolecules 41, 3243–3249 (2008).
Sekine, Y. & Ikeda-Fukazawa, T. Temperature dependence of the structure of bound water in dried glassy poly-N,N,-dimethylacrylamide. J. Phys. Chem. B 114, 3419–3425 (2010).
Tirosh, O., Barenholz, Y., Katzhendler, J. & Priev, A. Hydration of polyethylene glycol-grafted liposomes. Biophys. J. 74, 1371–1379 (1998).
Hillgren, A. & Alden, M. Differential scanning calorimetry investigation of formation of poly(ethylene glycol) hydrate with controlled freeze-thawing of aqueous protein solution. J. Appl. Polym. Sci. 91, 1626–1634 (2004).
Liu, W. G. & Yao, K. D. What causes the unfrozen water in polymers: hydrogen bonds between water and polymer chains? Polymer 42, 3943–3947 (2001).
Lutz, P. J. Structural properties of poly(ether) macromonomer based hydrogels. Polym. Bull. 58, 161–171 (2007).
Hay, D. N. T., Rickert, P. G., Seifert, S. n. & Firestone, M. A. Thermoresponsive nanostructures by self-assembly of a poly(N-isopropylacrylamide)-lipid conjugate. J. Am. Chem. Soc. 126, 2290–2291 (2004).
Rackaitis, M., Strawhecker, K. & Manias, E. Water-soluble polymers with tunable temperature sensitivity: Solution behavior. J. Polym. Sci. Polym. Phys. 40, 2339–2342 (2002).
Guillet, P., Fustin, C.-A., Mugemana, C., Ott, C., Schubert, U. S. & Gohy, J.-F. Tuning block copolymer micelles by metal-ligand interactions. Soft Matter 4, 2278–2282 (2008).
Sekine, Y. & Ikeda-Fukazawa, T. Structural changes of water in a hydrogel during dehydration. J. Chem. Phys. 130, 034501–034507 (2009).
Morikubo, S., Sekine, Y. & Ikeda-Fukazawa, T. Structure and dynamics of water in mixed solutions including laponite and PEO. J. Chem. Phys. 134, 044905–044909 (2011).
Liang, S., Wu, J., Tian, H., Zhang, L. & Xu, J. High-strength cellulose/poly(ethylene glycol) gels. ChemSusChem 1, 558–563 (2008).
Guo, C., Wang, J., Liu, H.-z. & Chen, J-y. Hydration and conformation of temperature-dependent micellization of PEO-PPO-PEO block copolymers in aqueous solutions by FT-Raman. Langmuir 15, 2703–2708 (1999).
Acknowledgements
We thank Dr Scott Brombosz for assistance in preparation of the freeze-dried hydrogels and Dr Bryan Ringstrand for editorial assistance in preparation of the manuscript. This work was performed under the auspices of the Office of Basic Energy Sciences, Division of Materials Sciences, United States Department of Energy, under contract No. DE-AC02-06CH1135.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Polymer Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Lee, S., Seifert, S. & Firestone, M. Multi-length scale evaluation of the temperature-tunable mechanical properties of a lyotropic mesophase. Polym J 45, 179–187 (2013). https://doi.org/10.1038/pj.2012.99
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pj.2012.99
