
Abstract
Non-woven fiber mats were fabricated with the electrospinning method using poly(methyl methacrylate)-co-poly(2-(2-bromoisobutyryloxy)ethyl methacrylate) in various solvents. The surface morphology of the electrospun fibers depended on the solvent vapor pressure and polymer concentration. Surface-initiated atom transfer radical polymerization (ATRP) with 3-(N-2-methacryloyloxyethyl-N,N-dimethyl) ammonatopropanesulfonate), 2-hydroxyethyl methacrylate or 2-(perfluorooctyl)ethyl acrylate generated surface-grafted polymers, as determined by X-ray photoelectron spectroscopy. Scanning electron microscopic observation revealed that the apparent fiber morphology did not change upon modification. The atomic force microscopic images of the grafted fiber cross-sections indicated that monomers and solvents penetrated slightly into the fibers and polymerization occurred at both internal and external initiation sites. Physicochemical properties, such as contact angle, hydrophobicity and hydrophilicity, and wettability, could be altered by the proper selection of substrates (spin-coated flat films or the variously prepared non-woven fiber mats). We have successfully prepared hydrophilic and hydrophobic fiber surfaces by a combination of the electrospinning protocol and surface-initiated ATRP.
Similar content being viewed by others

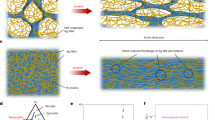

Introduction
Nano- and micro-fibers have potential applications in various fields including the environmental, medical and nanoscience fields,1, 2 because of certain physical characteristics, including their high surface area to volume ratio. For this reason, fiber-processing methods have been actively investigated. Electrospinning is a versatile method for fiber fabrication. The method allows easy control of fiber size, from the nano to the sub-micron scale, under mild conditions (atmospheric pressure and temperature). Moreover, various types of solvents can be used. Therefore, electrospinning is an attractive fiber preparation method.3, 4 Typically, the polymer solution or melt polymer that is used as the starting material for electrospinning is loaded into a syringe, and a high voltage is applied. The charged polymer solution is jetted out to the collector surface. Through the procedure of spraying into an electric field, the solvent evaporates and dry fibers are formed. Electrospinning studies have become prevalent in polymer science since the 2000s,5, 6 and complex fibers, including aligned fibers,7 core/shell fibers8 and composite fibers,9 can currently be prepared after appropriate modifications to the electrospinning apparatus.
In the last decade, surface-grafted polymers (polymer brushes) have attracted attention as a useful chemical surface-modification process. A polymer brush, tethered at one end to a solid surface, exhibits several physical advantages such as mechanical, thermal and chemical stability.10, 11 Polymer brushes are fabricated via either the grafting-to or the grafting-from approaches. In the former case, end-functionalized polymers are reacted with a solid surface to prepare the polymer brushes.12, 13 Generally, only a small number of polymer chains are immobilized on the solid surface using this approach because macromolecular polymer chains must diffuse onto the polymer brush and attach to the solid-surface functional groups despite a high degree of steric hindrance. Thus, the grafting-to method produces polymer brushes that exhibit a low grafting density and a low film thickness.
Fu et al.14 reported the surface modification of electrospun fibers by click chemistry. They synthesized poly(4-vinylbenzyl chloride) (PVBC)-b-poly(glycidyl methacrylate) (PGMA) by reversible addition-fragmentation chain-transfer polymerization, and prepared nanofibers of the block copolymer by the electrospinning method. Exposure of the PVBC-b-PGMA nanofibers to a solution of sodium azide in N,N-dimethylformamide (DMF)/water solvent (1/1, v/v) induced the reaction at the epoxy groups of PGMA on the nanofiber surface and led to surface crosslinking between the epoxy and benzyl chloride groups. Finally, alkyne-terminated poly(N-isopropylacrylamide), which was prepared by atom transfer radical polymerization (ATRP), was immobilized on electrospun fiber surfaces by the reaction of the alkyne and azide groups. A thermally sensitive non-woven fiber mat was successfully fabricated by the grafting-to method. The morphology of the nanofibers before and after the crosslinking was observed by scanning electron microscopy (SEM), and the thermoresponsive behavior of poly(N-isopropylacrylamide) on the nanofiber surface was evaluated by contact angle measurement against water.
The grafting-from method, by which polymer brushes with high grafting densities and film thicknesses can be prepared, has been actively used in recent years to modify surfaces. In this approach, initiators are immobilized on a solid surface and the controlled polymerization proceeds from those sites to form well-defined polymers that are immobilized on the surface with a high grafting density.15 This method can be applied to various substrates; for example, flat substrates,16 nanoparticles17 and several functional monomers have been used for the surface-initiated polymerization to generate products with varying surface physicochemical properties.18, 19 Takahara and colleagues reported the preparation of high-density poly(methyl methacrylate) (PMMA),20 hydrophobic poly(2-(perfluorooctyl)ethyl acrylate) (PFA-C8)21 and hydrophilic polyelectrolyte brushes22 on silicon wafers by surface-initiated ATRP. Core–sheath nanofibers were prepared by a combination of the electrospinning method and surface-initiated ATRP.23 Polystyrene (PS) containing a terminal ATRP initiator was synthesized by ATRP, and the nanofibers of polystyrene were prepared by the electrospinnnig method. The radical polymerization of acrylamide and N-(hydroxymethyl)acrylamide was induced at the nanofiber surface initiators, in water medium as a nonsolvent for polystyrene. X-ray photoelectron spectroscopy (XPS) and dynamic contact angle measurements against water were performed to confirm the successful preparation of the core-sheath nanofibers.
In this study, we prepared functional non-woven fiber mats by combining electrospinning with surface-initiated polymerization. Previous studies have reported the precise control of surface physicochemical properties by direct surface-initiated ATRP from a polymer surface bearing halogen groups.10, 11 We synthesized 2-(2-bromoisobutyryloxy)ethyl methacrylate (BIEM) as an ATRP initiator. BIEM and methyl methacrylate (MMA) monomers were polymerized by free-radical polymerization. The PMMA-co-poly(BIEM) (PMMA-co-PBIEM) obtained contains many ATRP initiation points in its side chains as shown in Scheme 1. Therefore, our method can afford polymer brushes with a high grafting density on the fiber surface. The electrospun non-woven fiber mats of the synthesized copolymer were prepared by dissolution of the copolymer in different solvents at varying concentrations. The formation of a porous or smooth structure on the fiber surface is discussed. In our previous study,24 we reported the ATRP grafting of the ionic, hydrophilic polymer poly(3-(N-2-methacryloyloxyethyl-N,N-dimethyl) ammonatopropanesulfonate) (PMAPS) onto a non-woven fiber mat, and confirmed the changes in the physicochemical properties of polymer after the modification. However, we could not discuss the fiber diameters, the formation of a porous or smooth structure and the precise effect of surface area of the PMAPS-grafted fiber mat on wettability against water because the contact angles of the spin-coated film and the mat were less than 10°. 2-Hydroxyethyl methacrylate (HEMA) was chosen as a neutral hydrophilic monomer to demonstrate the effect of the large surface area of a non-woven fiber mat by evaluating the static contact angle. We have characterized the detailed physical properties and morphologies of the electrospun fibers before and after surface modification, and the effects of the surface morphology of the non-woven electrospun fiber mat on the static contact angle are discussed.
Experimental procedure
Materials
Azobisisobutyronitrile (Wako Pure Chemicals, Tokyo, Japan, 98.0%), tetrahydrofuran (THF, Wako Pure Chemicals, 97.0%), chloroform (Wako Pure Chemicals, 99.0%), hexafluoroisopropanol (HFIP, Wako Pure Chemicals, 99.0%), toluene (Wako Pure Chemicals, 99.5%), 2-(dimethylamino)ethyl methacrylate (Wako Pure Chemicals, 99.0%), 2,2′-bipyridyl (Wako Pure Chemicals, 99.5%), DMF (Kishida Chemicals, Osaka, Japan, 99.5%), HEMA (Tokyo Chemical Industry, Tokyo, Japan, 95.0%), 2-bromoisobutyrylbromide (Aldrich, St Louis, MO, USA, 98.0%), 1,3-propanesultone (Aldrich, 99.0%) and 4,4′-dinonyl-2,2′-dipyridyl (Aldrich, 99.5%) were used without further purification. Triethylamine (Wako Pure Chemicals, 99.0%) and acetone (Tokyo Chemical Industry, 99.0%) were distilled under normal pressure over calcium hydride (CaH2). Methanol (Tokyo Chemical Industry, 99.0%) was refluxed over magnesium and distilled under atmospheric pressure. Water was purified by a Nano Pure Water System (Millipore, Billerica, MA, USA). Dichloromethane (DCM, Wako Pure Chemicals, 99.5%) was purified by a solvent purification apparatus (Solvent Dispensing System, Nikko Hansen Co., Osaka, Japan). MMA (Wako Pure Chemicals, 98.0%) was washed with sodium hydroxide solution and water. The organic phase was dried with anhydrous magnesium sulfate and distilled under reduced pressure over CaH2. Anisole (Wako Pure Chemicals, 99.0%) was distilled under reduced pressure over sodium. Copper (I) bromide (CuBr, Wako Pure Chemicals, 99.9%) was repeatedly washed with acetic acid and ethanol, and dried under vacuum. 2-(Perfluorooctyl)ethyl acrylate (FA-C8) containing 2-(perfluorodecyl)ethyl acrylate was provided by Daikin Industries (Osaka, Japan) and was distilled under reduced pressure over CaH2. BIEM25 was prepared by the reaction of HEMA and 2-bromoisobutyrylbromide. 3-(N-2-Methacryloyloxyethyl-N,N-dimethyl) ammonatopropanesulfonate) (MAPS)26 was synthesized by the reaction of 2-(dimethylamino)ethyl methacrylate and 1,3-propanesultone in acetone.
Characterization
1H nuclear magnetic resonance spectra were recorded in chloroform-d at 25 °C using a JNM-AL300 (JEOL, Tokyo, Japan, 1H 300 MHz). Size exclusion chromatography was performed at 40 °C on a Tosoh HLC-8220 GPC system (Tokyo, Japan) equipped with a guard column (Tosoh TSK guard column Super H-L, Tosoh, Yamaguchi, Japan), three columns (Tosoh TSK gel SuperH 6000, 4000 and 2500), and an ultraviolet–visible detector. THF was used as the eluent at a flow rate of 0.6 ml min−1. Polystyrene standards (Mn=1060–1,090,000; Mw/Mn=1.02–1.08) were used for calibration to estimate the number-average molecular weight (Mn) and molecular weight distributions (Mw/Mn) of the polymers. Differential scanning calorimetry measurements were performed with an EXSTAR6000 (Seiko Instruments Industries, Co., Chiba, Japan) in a temperature range of 303–523 K at a heating rate of 10 K min−1. The morphology and diameter of the electrospun fibers were analyzed by SEM. The electrospun fibers were coated with an osmium layer (∼3 nm) using a HPC-1SW Hollow Cathode Plasma CVD (Shinkuu Device Co., Mito, Japan). A voltage of 1–10 kV was applied to the target and adjusted using by Real Surface View VE7800 (Keyence Co., Osaka, Japan) to obtain precise SEM images. XPS measurements were performed by XPS-APEX (Physical Electronics Co., Chanhanssen, MN, USA) with an Al-Kα X-ray source. The take-off angles of the photoelectrons were maintained at 45°. Survey spectra were obtained in the range of 0–1000 eV at 1.0 eV energy steps; narrow scans were operated at 0.1 eV energy steps. The static contact angles were measured with a drop-shape analysis system DSA10 Mk2 (KRÜSS, Hamburg, Germany) equipped with a video camera. A 2-μl water droplet was placed on various surfaces and the static contact angles were recorded. The wettability behaviors of the non-woven fiber mat against a water droplet (6–8 μl) were monitored with a high-speed camera (VW-6000, Keyence Co.) at a frame rate of 2.0 or 4.0 ms. The surface-modified and non-modified fibers were embedded in epoxy resin (LCR D-800, Toagosei Co., Tokyo, Japan) and sliced with an ultramicrotome to obtain cross-sections for atomic force microscopy (AFM) observation. The cross-sections of the fibers were observed by AFM (Nano-R, Pacific Nanotechnology, Santa Clara, CA, USA) with a PPP-FMR-20 (NanoSensors, Neuchatel, Switzerland, resonance frequency 73 kHz, spring constant 2.3 N m−1) as a cantilever. Close contact mode operation under normal pressure was used for this study. Spin-coated film thickness measurements and observations of the surface morphology were performed with an SPI4000 (SII, NanoTechnology) using a Si tip on a cantilever (SI-DF20 (Al coated)) with a spring constant of 16 N m−1 at ambient pressure. The thicknesses of the spin-coated films were determined by AFM scanning on the scratched surfaces. We defined the difference of the spin-coated thickness pre-modification and post-modification as the grafted thickness.
Synthesis of PMMA-co-PBIEM
Azobisisobutyronitrile (7.0 mg, 0.043 mmol), purified MMA (25 ml, 234 mmol), BIEM (2.5 ml, 21 mmol) and anisole (10 ml) were combined in a glass tube and degassed by the freeze-pump-thaw cycle. The solution was stirred at 353 K for 3 h. The obtained polymer was purified by precipitation in methanol. The polymer was dried under vacuum (yield: 6.97 g, 27%, PMMA/PBIEM=0.94/0.06 (mol/mol, 1H nuclear magnetic resonance), Mn=179,000, Mw/Mn=1.99, Tg=391 K).
Preparation of electrospun fibers
Silicon wafers were cleaned with piranha solution (H2O2/H2SO4=30/70, v/v). The copolymer was dissolved in THF, chloroform, DCM, DMF or HFIP at 5.0, 7.5 or 10.0 wt% by stirring at a temperature of 313–333 K. High viscosity copolymer solutions were obtained. Electrospun non-woven fiber mats were fabricated on the cleaned silicon wafer with a NANON-01A (MECC Co., Fukuoka, Japan) nanofiber electrospinning device under ambient conditions (Figure 1). The electrospinning apparatus consisted of a syringe, needle, Teflon tube and collector. A copolymer solution was loaded into a 5 ml syringe connected to the Teflon tube with a needle tip (0.41 mm) attached to the spinneret. A voltage of 15–30 kV was applied to the solution in the syringe. The feed rate was varied from 0.25 to 3.0 ml h−1, whereas the collection distance between the syringe and the plate collector was fixed at 15.0 cm.

Photographic image of poly(methyl methacrylate)-co-poly(2-(2-bromoisobutyryloxy)ethyl methacrylate) non-woven fiber mat fabricated on cleaned silicon wafers (10 × 10 mm2) from hexafluoroisopropanol solution. A full color version of this figure is available at Polymer Journal online.
Surface-initiated ATRP
Figure 2 shows the schematic representation of surface-initiated ATRP from the fiber surface. The non-woven fiber mat fabricated from the HFIP or chloroform solution was used for surface modification. An electrospun non-woven fiber mat consisting of PMMA-co-PBIEM and a spin-coated film (prepared from 3.0 wt% toluene solution (3000 r.p.m., 30 s) on a silicon wafer (general-use size=10 × 10 mm2)) were simultaneously placed in a well-dried glass vessel equipped with a stopcock and degassed by five cycles of vacuum pumping and flushing with argon. CuBr (1.9 mg, 0.011 mmol) and 2,2′-bipyridyl (3.4 mg, 0.023 mmol) were introduced to another glass tube that was degassed by the vacuum and argon purge process. The MAPS monomer (1.40 g, 5.03 mmol) diluted with methanol (1.7 ml) and water (8.8 ml) was immediately added to the copper catalyst to give a homogeneous solution with a characteristic red color. The resulting solution was degassed by repeated freeze-pump-thaw cycles and then injected into the glass vessel containing the electrospun non-woven fiber mat and the spin-coated film. The reaction mixture was stirred at 303 K for 3 h under argon atmosphere. The reaction was stopped by opening the glass vessel to air. The resultant grafted products were rinsed in the following manner. Aqueous NaCl solution (100 mM) or water was poured slowly into a Petri dish placed on the non-woven fiber mat obtained and spin-coated film. After the fiber mat and spin-coated film were immersed in NaCl solution and water for a sufficient period, they were transferred to another Petri dish. Then, NaCl solution or water was again poured slowly into the Petri dish. These processes were repeated numerous times to eliminate the monomers. After this process, the washed electrospun non-woven fiber mat and spin-coated film were dried under reduced pressure. The white fiber mat maintained its original shape even after it was washed.

Surface-initiated atom transfer radical polymerization of various monomers from electrospun fibers. A full color version of this figure is available at Polymer Journal online.
The surface-initiated ATRP of HEMA proceeded in the same manner. The reaction mixture containing the non-woven fiber mat, the spin-coated film, CuBr (0.020 mmol), 2,2′-bipyridyl (0.040 mmol), HEMA (1.0 mmol) and water (5.0 ml) was stirred at 303 K for 3 h under argon atmosphere. The electrospun non-woven fiber mat and spin-coated film were washed with water. The surface-initiated ATRP of FA-C8 (32.0 mmol) was performed with CuBr (0.076 mmol) and 4,4′-dinonyl-2,2′-dipyridyl (0.15 mmol), in the presence of the fiber mat and spin-coated film at 333 K for 3 h under argon atmosphere. The resulting PFA-C8-grafted non-woven fiber mat was washed with methanol and dried under reduced pressure.
Results and Discussion
Preparation of the electrospun non-woven fiber mats
The quality of fibers fabricated by the electrospinning method is strongly influenced by the physical properties of the solvent, such as boiling point, volatility, viscosity and surface tension. The solvent should dissolve the polymer to give a homogeneous solution, which discharges from the spray nozzle. In general, the polymer solution of a volatile solvent does not form fibers by the electrospinning method because the solvent quickly evaporates just after it sprays from the nozzle, which results in the dried polymer blocking the needle hole. A solvent with a much higher boiling point, however, evaporates slowly from the polymer solution during the electrospinning process and will not give a dried polymer fiber deposited on the surface of a target substrate. The viscosity of the polymer solution is strongly related to the fiber diameter.27 A polymer solution dissolved in a high-surface tension solvent like water forms a pendant droplet at the edge of the syringe nozzle, even if a much higher voltage is applied. The fiber structure is broken by the drop of the pendant-like liquid from the edge of the needle.
Consequently, predicting which solvent is most suitable for the successful fabrication of fibers by the electrospinning method is a complex process. Therefore, we used various organic solvents in this work to understand the effects of the physical properties of the solvents, as shown in Table 1, on the fiber fabrication of our bromide-containing copolymer.28, 29
The SEM images in Figure 3 show the morphology of the PMMA-co-PBIEM electrospun fibers. The beaded PMMA-co-PBIEM fibers resulted only from THF solution with a polymer concentration of 5.0 wt%. The tendency for bead formation was not observed at higher polymer concentrations. The fiber diameters prepared from 7.5 and 10.0 wt% THF solutions were 1.7 and 1.8 μm, respectively. In the cases of fibers prepared from chloroform and DCM solutions, ribbon-like fibers were obtained at all polymer concentrations. Widths of 1.3, 1.7 and 2.3 μm were obtained from 5.0, 7.5 and 10.0 wt% chloroform solutions, and widths of 2.2, 3.3 and 3.9 μm were obtained from 5.0, 7.5 and 10.0 wt% DCM solutions. The diameters or widths for the ribbon-like fibers increased with increasing polymer concentration. When THF, chloroform and DCM, which exhibits a low boiling point and high vapor pressure, were used as solvents, porous fibers were obtained. In addition, the polymer solidified at the edge of the needle within a few minutes because of the quick evaporation of the solvent. Fibers with average diameters of 0.74, 0.97 and 0.92 μm were prepared from 5.0, 7.5 and 10.0 wt% DMF solutions. DMF exhibits a high boiling point and low vapor pressure; however, the resulting fiber diameters were not uniform. The non-woven fiber mat contains fibers with both thin and thick diameters. In a single fiber, the distribution of the fiber diameter was wide. The fiber diameters from the HFIP solutions increased (from 0.80 to 0.85 μm) with increasing polymer concentration. HFIP is known as a sterilizing agent and is often used as a solvent for non-woven textiles with biomaterial applications that are fabricated by the electrospinning method.30 In this work, fibers with a smooth surface and uniform diameter were obtained from the polymer/HFIP solution, even though HFIP exhibits a low boiling point. The unexpected fiber properties are probably a consequence of HFIP being a unique solvent with a low boiling point and high viscosity. The fiber diameters increased with an increase in the polymer concentration in all solvent cases. These results suggested that the diameter or width of the electrospun fiber was strongly dependent on the viscosity of the polymer solution. Shivkumar et al.27 have plotted the fiber diameters of various polymers against the Berry number (a product of the concentration of a solution and its inherent viscosity) and observed a similar relationship for electrospun poly(vinyl alcohol) fibers. Electrospinning was conducted in an aqueous solvent for various molecular weights of poly(vinyl alcohol). In their case, the fiber diameter increased in proportion to the 1.1th power of the Berry number. In our study, we expected the viscosity originating from the entanglement of the polymer chains to influence the fiber diameters.

Scanning electron microscopy images of electrospun poly(methyl methacrylate)-co-poly(2-(2-bromoisobutyryloxy)ethyl methacrylate) fibers prepared from (a) 5.0 wt%, (b) 7.5 wt% and (c) 10.0 wt% tetrahydrofuran (THF) solution; (d) 5.0 wt%, (e) 7.5 wt% and (f) 10.0 wt% chloroform solution; (g) 5.0 wt%, (h) 7.5 wt% and (i) 10.0 wt% dichloromethane (DCM) solution; (j) 5.0 wt%, (k) 7.5 wt% and (l) 10.0 wt% N,N-dimethylformamide (DMF) solution; (m) 5.0 wt% and (n) 7.5 wt% hexafluoroisopropanol (HFIP) solution.
Porous structures were observed on the fiber surfaces prepared from the chloroform, DCM and THF polymer solutions, and smooth structures on the fiber surfaces fabricated from the HFIP and DMF polymer solutions were confirmed by SEM. Two possibilities can explain the formation of the porous structure on the fiber surface. First, the formation of the porous structure may have been induced by phase separation during the electrospinning process. It has been reported that the surface temperature of a sprayed polymer solution quickly cools as a result of evaporation heat. The decrease in the surface temperature for the polymer solution would lead to thermally induced phase separation.31, 32, 33
Second, the condensation of atmospheric moisture may have induced phase separation. This moisture would condense at the outermost surface of the sprayed polymer solution as its surface temperature decreases. The polymer solution would absorb the water during the electrospinning process, thereby inducing phase separation due to the insolubility of PMMA in water. Rabolt and colleagues,32 by controlling humidity around the electrospinning instrument, confirmed vapor-induced phase separation. In our study, the formation of the porous structure would be affected both by thermally induced phase separation and by vapor-induced phase separation.
The fibers fabricated from the HFIP and DMF solutions had smooth surfaces, indicating that these solvents tend to suppress phase separation between the polymer and the solvent. In the case of DMF, the low vapor pressure would contribute to phase separation suppression of the polymer. However, the high viscosity of HFIP might suppress the phase separation during solvent evaporation. These facts suggested that we could prepare electrospun fibers with different surface structures by using different solvents.
The spin-coated films of PMMA-co-PBIEM were prepared from three different solutions (THF, chloroform or DCM) to allow a comparison of the surface morphologies with the electrospun fiber results. PMMA-co-PBIEM was dissolved in THF, chloroform or DCM (1.0 wt%) by heating at a temperature of 313 K. Silicon wafers (10 × 10 mm2) were cleaned with piranha solution (H2O2/H2SO4=30/70, v/v) and then exposed to vacuum ultraviolet rays (λ=172 nm) for 10 min under reduced pressure (30 Pa). Silicon wafers were coated with the solution by the spin-coating method (3000 r.p.m, 50 s). Figure 4 shows AFM images of the spin-coated films that were not annealed. The roughness factors (root-mean-square) of the smooth films obtained from THF, chloroform or DCM solutions were <0.3 nm in 1.0 × 1.0 μm2. Although nanopores of ca 50 nm were sporadically observed, the number of nanopores per unit area was clearly lower than that on the surfaces of the corresponding electrospun fibers. Therefore, the formation of nanopores must be unique to the electrospinning process, because small fiber diameters with relatively wide surface areas enhanced the rapid solvent evaporation immediately after discharge from the narrow nozzle.

Atomic force microscopic images of spin-coated films prepared from (a) tetrahydrofuran (THF), (b) chloroform and (c) dichloromethane (DCM) solution, and the cross-section profiles of spin-coated films prepared from (d) THF, (e) chloroform and (f) DCM solution. RMS, root-mean-square. A full color version of this figure is available at Polymer Journal online.
Surface modification of the electrospun non-woven fiber mats
The atomic compositions at the surfaces of the electrospun fiber mats were confirmed by XPS measurements before and after surface-initiated ATRP (Figure 5). The XPS spectrum of the electrospun non-woven fiber mat of PMMA-co-PBIEM showed characteristic Br3d, C1s and O1s peaks that corresponded to the theoretical atomic composition of the copolymer (Table 2). After the surface-initiated ATRP of MAPS, the S2p and N1s peaks of sulfonic and ammonium groups were observed at 168.2 and 403.1 eV, respectively. The surface atomic ratios of carbon, oxygen, nitrogen and sulfur observed by narrow-scan XPS corresponded to the theoretical values calculated from the atomic composition of PMAPS. The same analysis method was applied to the grafting product of PFA-C8; a new F1s peak at 690.4 eV was detected in the XPS spectrum of the non-woven fiber mat that had been modified by surface-initiated ATRP. Peaks corresponding to the C–C, C–O, C=O, CF2 and CF3 bonds appeared at 287.4, 288.6, 290.7, 293.1 and 295.3 eV, respectively. In the case of grafted poly(HEMA) (PHEMA), no new atom peaks were observed when compared with the spectrum of PMMA-co-PBIEM. Therefore, a typical curve fitting of the C1s peak region was performed for the PMMA-co-PBIEM non-woven fiber mat and the PHEMA-grafted non-woven fiber mat. The C1s peaks could be fitted to three line shapes at 285.5, 287 and 289.3 eV by Gaussian–Lorenz functions (Table 3). These peaks corresponded to the binding energy peaks of C=O, C–O and C–C, respectively. The intensity of the peak attributed to the C–O bond derived from hydroxyl groups after the surface modification of PHEMA increased from 0.288 to 0.368 compared with that of the PMMA-co-PBIEM non-woven fiber mat. Additionally, successful polymerization could be evaluated from the intensity of the Br3d peak that corresponded to the end point of PHEMA. The results showed that the intensity of the Br3d peak in the XPS spectrum decreased compared with that of the non-modified fiber mat, which indicated that polymerization of PHEMA had occurred.

X-ray photoelectron spectra of (a) poly(methyl methacrylate)-co-poly(2-(2-bromoisobutyryloxy)ethyl methacrylate) fiber surface survey scan spectrum; high-resolution spectra of the (b) O1s, (c) C1s and (d) Br3d peak region; (e) poly(3-(N-2-methacryloyloxyethyl-N,N-dimethyl) ammonatopropanesulfonate)-grafted fiber surface survey scan spectrum; high-resolution spectra of the (f) O1s, (g) N1s, (h) C1s and (i) S2p peak region; (j) poly(2-(perfluorooctyl)ethyl acrylate)-grafted fiber surface survey scan spectrum; high-resolution spectra of the (k) F1s, (l) O1s and (m) C1s peak region; (n) poly(2-hydroxyethyl methacrylate)-grafted fiber surface survey scan spectrum; high-resolution spectra of the (o) O1s and (p) C1s peak region. Take-off angles were kept constant at 45°.
The XPS-determined atomic ratios of C/O/N/S in the PMAPS-grafted non-woven fiber mat were estimated to be 0.669/0.235/0.046/0.048, which are in relatively close agreement with the theoretical values calculated from the atomic composition of PMAPS (Table 2). Similarly, the observed atomic ratios of C/O/F (0.439/0.062/0.498) corresponded to the theoretical values for the PFA-C8-grafted non-woven fiber mat. The atomic ratios of C/O (0.698/0.302) for the PHEMA-modified fiber mat were also in accord with theoretical values. These surface atomic ratios of the different fibers suggested that the surfaces of PMMA-co-PBIEM fibers were completely covered with grafted layers of PMAPS, PHEMA and PFA-C8, which were sufficiently thick to prevent detection of the core PMMA-co-PBIEM fiber components.
To confirm the fiber shapes and surface morphologies, we used SEM to observe the electrospun fibers obtained after surface-initiated ATRP (Figure 6). Because the methanol/water, water and FA-C8 solvents in the polymerization were poor solvents or nonsolvents for PMMA-co-PBIEM, fiber shapes and surface morphologies were preserved after the surface-initiated polymerization. After the ATRP reaction of MAPS, a fiber diameter of 1.2 μm was observed, in comparison to the SEM-determined diameter of 0.85 μm of the starting material (prepared from 7.5 wt% HFIP solution). The expected grafted thickness of the fiber from the surface-initiated ATRP was approximately 100–300 nm. Similarly, diameters of 1.1 and 1.2 μm were observed in the cases of PHEMA and PFA-C8. Tanaka et al.34 have investigated the interfaces between deuterated PMMA (dPMMA) film and poor or nonsolvents by neutron reflectivity and AFM. Although water is a typical nonsolvent for dPMMA, the liquid/solid interfaces were diffuse in comparison with the interface between air and dPMMA. For water, the interfacial swollen (diffuse) layer was observed in contrast to dPMMA/methanol, in which an interfacial swollen layer could not be confirmed because of the solvent’s deep penetration into the dPMMA film. In our study, solvents or monomers might penetrate into the fibers, and allow polymerization to proceed from both internal and surface initiation sites. Surface-initiated ATRP of MAPS was performed for the porous fiber fabricated from 7.5 wt% chloroform solution. The nanopores on the fiber surface remained intact after the surface modification, as shown in Figure 6, and pore sizes were unchanged. The fiber shape and morphology were clearly maintained after the surface-initiated ATRP of MAPS, HEMA and FA-C8.

Scanning electron microscopy (SEM) images of (a) poly(3-(N-2-methacryloyloxyethyl-N,N-dimethyl) ammonatopropanesulfonate) (PMAPS)-grafted electrospun fibers, (b) poly(2-hydroxyethyl methacrylate)-grafted electrospun fibers, and (c) poly(2-(perfluorooctyl)ethyl acrylate)-grafted electrospun fibers. These fibers were prepared from hexafluoroisopropanol solution. (d) SEM image of PMAPS-grafted porous fibers. The porous fibers were prepared from poly(methyl methacrylate)-co-poly(2-(2-bromoisobutyryloxy)ethyl methacrylate) chloroform solution.
To examine the grafted thickness of the fiber and its distribution of graft initiation points, we prepared cross-section samples via ultramicrotome to analyze the inside and outside of the fibers by AFM. The surface-modified and non-modified fibers were embedded in epoxy resin (LCR D-800, Toagosei Co.) and the resin was crosslinked by ultraviolet irradiation for 10 min. The resultant resin was sliced by ultramicrotome (slice thickness=50–100 nm; slice velocity=0.2–1.0 mm s−1) to produce a cross-section for AFM observation. Figure 7 shows the cross-section image of non-modified fiber observed by AFM (tapping mode). Diagonally cut fiber was observed because of random embedding in the epoxy resin. For the core fiber of PMMA-co-PBIEM, no grafted layer was observed between the non-modified fiber surface and the epoxy resin. Figure 8 shows the cross-section images of fibers modified with PMAPS, PHEMA and PFA-C8. Elliptic fiber cross-sections were observed. The grafted layer of PMAPS was identifiable in the magnified AFM image at the interface between the fiber and epoxy resin, and was estimated to be ∼82 nm thick. The grafted thickness of the spin-coated film was 37 nm, as evaluated from scratched AFM images. The surface-initiated ATRP was performed on the spin-coated PMMA-co-PBIEM film by the same surface modification procedure used on the fibers to measure the grafted thickness. The spin-coated PMMA-co-PBIEM and PMAPS-grafted films were scratched with a needle. The heights between the flat spin-coated layer and the scratched bottom area of the silicon wafer were estimated from the AFM images. The thickness of the PMAPS-grafted spin-coated film was 162 nm, whereas that of the PMMA-co-PBIEM spin-coated film was 125 nm. The grafted thickness of 37 nm was calculated by the difference between these values. Similarly, the grafted layers of the PHEMA- and PFA-C8-grafted fibers were estimated from the cross-section images of the grafted fibers obtained by AFM. The grafted thicknesses of PHEMA- and PFA-C8-grafted spin-coated films were measured from the images of the scratched spin-coated films. In the case of PHEMA and PFA-C8, similar tendencies were confirmed. The grafted layers measured from the AFM cross-section images were ∼40 nm for grafted PHEMA and 70 nm for PFA-C8 fibers. These thicknesses are greater than those of the layers of the grafted spin-coated film. These differences in the grafted thicknesses were derived from the affinity between PMMA-co-PBIEM and the solvent or monomer. The root-mean-square value of the PMMA-co-PBIEM spin-coated film prepared from toluene solution was 0.27 nm (1.0 × 1.0 μm2), as measured by AFM, which suggested that a smooth surface was obtained. However, the root-mean-square value of the fiber surface was estimated to be 9.9 nm (0.5 × 0.5 μm2). We expected the surface area per unit volume of the fibers to be larger than that of spin-coated film. The solvent or monomer molecules might penetrate further into the polymer chains at a surface with a large roughness factor than at a smooth surface; the monomers may then polymerize from the inside.34 The fibers inside the PMMA-co-PBIEM may have been swelled by the solvent or monomer. The fiber was swelled in a concentric circle, whereas the swelling area of the spin-coated film expanded in a perpendicular direction. Consequently, the large grafted layers of the fibers were observed in comparison with the grafted thicknesses of the spin-coated films.

The cross-sectional image of poly(methyl methacrylate)-co-poly(2-(2-bromoisobutyryloxy)ethyl methacrylate) fiber observed by atomic force microscopy. A full color version of this figure is available at Polymer Journal online.

Atomic force microscopic images of the grafted areas after the surface modification of poly(methyl methacrylate)-co-poly(2-(2-bromoisobutyryloxy)ethyl methacrylate) with (a) poly(3-(N-2-methacryloyloxyethyl-N,N-dimethyl) ammonatopropanesulfonate), (b) poly(2-hydroxyethyl methacrylate) and (c) poly(2-(perfluorooctyl)ethyl acrylate). A full color version of this figure is available at Polymer Journal online.
Wettabilities of the non-woven fiber mats
The wettabilities of electrospun non-woven fiber mats were characterized by static contact angle measurements against water before and after the surface modification (Figure 9). The PMMA-co-PBIEM spin-coated film was modified by surface-initiated ATRP of three monomers using the same procedure as for the control samples. The contact angle of the PMMA-co-PBIEM electrospun fiber mat was 127°, whereas that for the spin-coated film was 76°. The contact angle of the non-woven fiber mat should increase in comparison to that of the spin-coated film because the non-woven fiber mat, which exhibits a large roughness and surface area, can trap large amounts of air in the fiber mat. This effect can be explained by Cassie–Baxter35 theory. The apparent contact angle depends on the roughness of the surface and surface-free energy of the materials that form the surfaces.36, 37 The Cassie–Baxter state (Equation (1)) indicates the composite surface of air and the solid surface. In the Cassie–Baxter state, a liquid droplet is resting partly on the features of a solid material and bridging the air in between these features. The apparent contact angle increases on the rough surface compared with the flat surface because of the presence of air. The Cassie–Baxter model is described by the following equation:


Water static contact angle images of (a) poly(methyl methacrylate)-co-poly(2-(2-bromoisobutyryloxy)ethyl methacrylate) film, (b) poly(3-(N-2-methacryloyloxyethyl-N,N-dimethyl) ammonatopropanesulfonate)-grafted film, (c) poly(2-hydroxyethyl methacrylate)-grafted film and (d) poly(2-(perfluorooctyl)ethyl acrylate)-grafted film; 1 and 2 are spin-coated films and electrospun non-woven fiber mats, respectively.
Here, θCB is the apparent contact angle on the rough surface; θF is the contact angle on the flat surface; and f is the fraction of solid remaining in contact with the droplet. The factor f (0.36) was calculated from the contact angle values. After hydrophobic PFA-C8 was grafted onto the surfaces, the contact angles of the spin-coated film and the non-woven fiber mat dramatically increased to 129° and ca 150°, respectively. These results suggested that the wettability of the PFA-C8-grafted non-woven fiber mat showed Cassie–Baxter wetting in the same manner as PMMA-co-PBIEM. The f value, 0.32, was obtained from Cassie–Baxter equation. These results suggested that the decrease in f value from 0.36 (PMMA-co-PBIEM non-woven fiber mat) to 0.32 (PFA-C8-modified non-woven fiber mat) was caused by the hydrophobicity of the perfluoroalkyl chains, despite the equivalent surface areas of the fiber mats. The wettability behavior against a water droplet was confirmed by high-speed camera. The water droplet landed on the PMMA-co-PBIEM non-woven fiber mat (Supplementary Figure S1); however, it was quickly repelled several times on the PFA-C8-grafted non-woven fiber mat (Supplementary Figure S2). A water-repellent non-woven fiber mat was therefore successfully prepared by surface modification of PFA-C8.
In contrast, the contact angles decreased on PMAPS-grafted and PHEMA-grafted spin-coated surfaces and fiber mats. In the case of grafted PHEMA, the contact angle value for the non-woven fiber mat decreased to less than 10°, whereas a contact angle of 57° was obtained for the spin-coated film. Contact angle values of both the spin-coated film and the non-woven fiber mat decreased because of the increase in the surface-free energy that was induced by the PHEMA hydroxyl groups. The difference in the contact angles between the spin-coated film and the non-woven fiber mat was attributed to an increase in the surface area: water droplets penetrated without repulsion into the large space of the hydrophilic-polymer-modified, non-woven fiber mat. This behavior can be explained by Wenzel theory.38 This model proposes that surface penetration occurs when a liquid droplet falls onto a rough surface. The contact angle in Wenzel wetting is described by the following (Equation (2)):

Here, θF is the contact angle on the flat surface; θW is the apparent contact angle on the rough surface of the same material; and r is the roughness factor, which is defined as the ratio of the actual area to its projection on the flat surface. In Wenzel theory, the apparent contact angle decreases if the flat surface contact angle is less than 90° and increases if the flat surface contact angle is greater than 90°. For our PHEMA-grafted substrate, the roughness factor, r, in the Wenzel equation was calculated from the contact angle values on the PHEMA-grafted spin-coated film and on the non-woven fiber mat to be greater than 1.8. This result suggested that the contact area against water of the non-woven fiber mat was dramatically increased compared with that of the spin-coated film. Lower contact angles (less than 10°) were obtained on PMAPS-grafted surfaces for both the spin-coated film and the non-woven fiber mat. It has been reported that a betaine-type polyelectrolyte used as polymer brush exhibits very high wettability against water because of the presence of hydrated ionic groups.39 It was difficult to determine the roughness effect on the PMAPS-grafted fiber mat because of the low contact angle on the PMAPS-grafted spin-coated film. However, neutral and hydrophilic PHEMA showed relatively larger contact angle than that of PMAPS. Therefore, the effect of the large surface area was confirmed by a comparison of the contact angles between the PHEMA-grafted non-woven fiber mat and spin-coated film. Supplementary Figures S3 and S4 show the water-droplet movies on the PMAPS- and PHEMA-grafted non-woven fiber mats. The liquid droplet penetrated into the fiber mat, and expanded in the horizontal direction due to the large surface area and hydrophilicity. Water repellency and wettability were enhanced by the roughness of the electrospun non-woven fiber mat.
Conclusion
We have reported the combination of electrospinning and surface-initiated ATRP methods. PMMA-co-PBIEM electrospun fibers with smooth surfaces were fabricated from DMF and HFIP solutions, whereas porous structures on the fiber surface were obtained from THF, chloroform and DCM solutions because of the phase separation and the low vapor pressures of the solvents. The fibers with porous surface structures exhibit larger surface areas than the fibers with smooth surfaces. Therefore, the non-woven fiber mat prepared by the porous fibers is expected to improve the efficiency of filtration and the sensitivity of sensors. However, the physical properties of the porous fiber mat would be inferior to that of the smooth fiber because of the presence of mechanical defects. Surface-initiated ATRP of three types of monomers (MAPS, HEMA and FA-C8) were performed in poor solvents or monomer (water/methanol, water, FA-C8, respectively) at the electrospun fiber surface to assess the surface physicochemical properties. XPS results indicated that the surfaces of PMMA-co-PBIEM fibers were completely covered with grafted layers of PMAPS, PHEMA and PFA-C8. The SEM observation revealed that the fiber morphology was maintained even after the polymer grafting process in the solution. The images of grafted fiber cross-sections were observed by AFM. The grafted thicknesses of PMAPS, PHEMA, PFA-C8 were estimated to be approximately 82, 40 and 70 nm, respectively. These values were larger than the grafted thicknesses of the spin-coated films, which indicated that the monomers or solvents slightly penetrated into the fiber and polymerized not only at the outermost surface, but also from near the surface of the fibers. The fibers inside the PMMA-co-PBIEM were possibly swelled by the solvent or monomer. The fibers were swelled in a concentric circle to the radial direction, whereas the swelling area of the spin-coated film expanded only in the perpendicular direction. The wettabilities before and after the surface modification were investigated by static contact angle measurements against water. The repellency against water was enhanced on the PFA-C8-grafted non-woven fiber mat in comparison to the PFA-C8-grafted spin-coated film. However, the hydrophilicities of the PMAPS- and PHEMA-grafted non-woven fiber mats increased dramatically. In conclusion, we have successfully prepared fibers with hydrophilic and hydrophobic surfaces by combining electrospinning with surface-initiated ATRP. This versatile technique may allow the preparation of many types of fiber mats with different surface properties and expand the applications of electrospun fibers.

Synthesis of poly(methyl methacrylate)-co-poly(2-(2-bromoisobutyryloxy)ethyl methacrylate) . AIBN, azobisisobutyronitrile.
References
Zusssman, E., Yarin, A. L., Bazilevsky, A. V., Avrahami, R. & Feldman, M. Electrospun polyacrylonitrile/poly(methyl methacrylate)-derived turbostratic carbon micro-/nanotubes. Adv. Mater. 18, 348–353 (2006).
Ding, Z., Salim, A. & Ziaie, B. Selective nanofiber deposition through field-enhanced electrospinning. Langmuir 25, 9648–9652 (2009).
Baumgarten, P. K. Electrostatic spinning of acrylic microfibers. J. Colloid Interface Sci. 36, 71–79 (1971).
Doshi, J. & Reneker, D. H. Electrospinning process and applications of electrospun fibers. J. Electrostatics 35, 151–160 (1995).
Son, W. K., Youk, J. H., Lee, T. S. & Park, W. H. The effects of solution properties and polyelectrolyte on electrospinning of ultrafine poly(ethylene oxide) fibers. Polymer 45, 2959–2966 (2004).
Jarusuwannapoom, T., Hongrojjanawiwat, W., Jitjaicham, S., Wannatong, L., Nithitanakul, M., Pattamaprom, C., Koombhongse, P., Rangkupan, R. & Supaphol, P. Effect of solvents on electro-spinnability of polystyrene solutions and morphological appearance of resulting electrospun polystyrene fibers. Eur. Polym. J. 41, 409–421 (2005).
Lee, K. H., Kim, K. W., Pesapane, A., Kim, H. Y. & Rabolt, J. F. Polarized FT-IR study of macroscopically oriented electrospun nylon-6 nanofibers. Macromolecules 41, 1494–1498 (2008).
Chen, H., Wang, N., Di, J., Zhao, Y., Song, Y. & Jiang, L. Nanowire-in-microtube structured core/shell fibers via multifluidic coaxial electrospinning. Langmuir 26, 11291–11296 (2010).
Zhang, L., Chandrasekar, R., Howe, J. Y., West, M. K., Hedin, N. E., Arbegast, W. J. & Fong, H. A metal matrix composite prepared from electrospun TiO2 nanofibers and an Al 1100 alloy via friction stir processing. ACS Appl. Mater. Interfaces 1, 987–991 (2009).
Kimura, T., Kobayashi, M., Morita, M. & Takahara, A. Preparation of poly(vinylidene fluoride-co-trifluoroethylene) film with a hydrophilic surface by direct surface-initiated atom transfer radical polymerization without pretreatment. Chem. Lett. 38, 446–447 (2009).
Matsugi, T., Saito, J., Kawahara, N., Matsuo, S., Kaneko, H., Kashiwa, N., Kobayashi, M. & Takahara, A. Surface modification of polypropylene molded sheets by means of surface-initiated ATRP of methacrylates. Polym. J. 41, 547–554 (2009).
Klein, J., Kumacheva, E., Mahalu, D., Perahia, D. & Fetters, L. J. Reduction of frictional forces between solid surfaces bearing polymer brushes. Nature 370, 634–636 (1994).
Ishida, N. & Biggs, S. Direct observation of the phase transition for a poly(N-isopropylacryamide) layer grafted onto a solid surface by AFM and QCM-D. Langmuir 23, 11083–11088 (2007).
Fu, G. D., Xu, L. Q., Yao, F., Zhang, K., Wang, X. F., Zhu, M. F. & Nie, S. Z. Smart nanofibers from combined living radical polymerization, ‘Click Chemistry’, and electrospinning. ACS Appl. Mater. Interfaces 1, 239–243 (2009).
Tsujii, Y., Ohno, K., Yamamoto, S., Goto, A. & Fukuda, T. Structure and properties of high-density polymer brushes prepared by surface-initiated living radical polymerization. Adv. Polm. Sci. 197, 1–45 (2006).
Advincula, R., Zhou, Q., Park, M., Wang, S., Mays, J., Sakellariou, G., Pispas, S. & Hadjichristidis, N. Polymer brushes by living anionic surface initiated polymerization on flat silicon (SiOx) and gold surfaces: homopolymers and block copolymers. Langmuir 18, 8672–8684 (2002).
Li, C., Han, J., Ryu, C.Y. & Benicewicz, B. C. A versatile method to prepare RAFT agent anchored substrates and the preparation of PMMA grafted nanoparticles. Macromolecules 39, 3175–3183 (2006).
Ishikawa, T., Kobayashi, M. & Takahara, A. Macroscopic frictional properties of poly(1-(2-methacryloyloxy)ethyl-3-butyl imidazolium bis(trifluoromethanesulfonyl)-imide) brush surfaces in an ionic liquid. ACS Appl. Mater. Interfaces 2, 1120–1128 (2010).
Malham, I.B. & Bureau, L. Density effects on collapse, compression, and adhesion of thermoresponsive polymer brushes. Langmuir 26, 4762–4768 (2010).
Sakata, H., Kobayashi, M., Otsuka, H. & Takahara, A. Tribological properties of poly(methyl methacrylate) brushes prepared by surface-initiated atom transfer radical polymerization. Polym. J. 37, 767–775 (2005).
Yamaguchi, H ., Honda, K ., Kobayashi, M ., Morita, M ., Masunaga, H ., Sakata, O ., Sasaki, S . & Takahara, A . Molecular aggregation state of surface-grafted poly{2-(perfluorooctyl)ethyl acrylate} thin film analyzed by grazing incidence X-ray diffraction. Polym. J. 40, 854–860 (2008).
Kobayashi, M., Yamaguchi, H., Terayama, Y., Wang, Z., Ishihara, K., Hino, M. & Takahara, A . Structure and surface properties of high-density polyelectrolyte brushes at the interface of aqueous solution. Macromol. Symp. 279, 79–87 (2009).
Fu, G. D., Lei, J. Y., Yao, C., Li, X. S., Yao, F., Nie, S. Z., Kang, E. T. & Neoh, K. G. Core-sheath nanofibers from combined atom transfer radical polymerization and electrospinning. Macromolecules 41, 6854–6858 (2008).
Yano, T., Yah, W. O., Yamaguchi, H ., Terayama, Y., Nishihara, M., Kobayashi, M. & Takahara, A. Preparation and surface characterization of surface-modified electrospun poly(methyl methacrylate) copolymer nanofibers. Chem. Lett. 39, 1110–1111 (2010).
Matyjaszewski, K., Gaynor, S. G., Kulfan, A. & Podwika, M. Preparation of hyperbranched polyacrylates by atom transfer radical polymerization. 1. Acrylic AB* monomers in ‘Living’ radical polymerizations. Macromolecules 30, 5192–5194 (1997).
Duann, Y. F., Chen, Y. C., Shen, J. T. & Lin, Y. H. Thermal induced graft polymerization using peroxide onto polypropylene fiber. Polymer 45, 6839–6843 (2004).
Tao, J. & Shivkumar, S. Molecular weight dependent structural regimes during the electrospinning of PVA. Mater. Lett. 61, 2325–2328 (2007).
Qian, Y.F., Su, Y., Li, X.Q., Wang, H.S. & He, C.L. Electrospinning of polymethyl methacrylate nanofibres in different solvents. Iran. Polym. J. 19, 123–129 (2010).
Sung, J.H., Kim, H.S., Jin, H.J., Choi, H.J. & Chin, I.J. Nanofibrous membranes prepared by multiwalled carbon nanotube/poly(methyl methacrylate) composites. Macromolecules 37, 9899–9902 (2004).
Nam, J., Huang, Y., Agarwal, S. & Lannutti, J. Materials selection and residual solvent retention in biodegradable electrospun fibers. J. Appl. Polym. Sci. 107, 1547–1554 (2008).
Han, S. O., Son, W. K., Youk, J. H., Lee, T. S. & Park, W. H. Ultrafine porous fibers electrospun from cellulose triacetate. Mater. Lett. 59, 2998–3001 (2005).
Megelski, S., Stephens, J. S., Chase, D. B. & Rabolt, J. F. Micro- and nanostructured surface morphology on electrospun polymer fibers. Macromolecules 35, 8456–8466 (2002).
Kongkhlang, T., Kotaki, M., Kousaka, Y., Umemura, T., Nakaya, D. & Chirachanchai, S. Electrospun polyoxymethylene: spinning conditions and its consequent nanoporous nanofiber. Macromolecules 41, 4746–4752 (2008).
Tanaka, K., Fujii, Y., Atarashi, H., Akabori, K., Hino, M. & Nagamura, T. Nonsolvents cause swelling at the interface with poly(methyl methacrylate) films. Langmuir 24, 296–301 (2008).
Cassie, A. B. D. & Baxter, S. Wettability of porous surfaces. Trans. Faraday Soc. 40, 546–551 (1944).
Feng, X. & Jiang, L. Design and creation of superwetting/antiwetting surfaces. Adv. Mater. 18, 3063–3078 (2006).
Roach, P., Shirtcliffe, N. J. & Newton, M. I. Progess in superhydrophobic surface development. Soft Matter 4, 224–240 (2008).
Wenzel, R. N. Resistance of solid surfaces to wetting by water. Ind. Eng. Chem. 28, 988–994 (1936).
Kobayashi, M. & Takahara, A. Tribological properties of hydrophilic polymer brushes under wet conditions. Chem. Rec. 10, 208–216 (2010).
Acknowledgements
The present work is supported by a Grant-in-Aid for the Global COE Program, ‘Science for Future Molecular Systems’ and a Grant-in-Aid for Scientific Research on Innovative Area (20106001) from the MEXT.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Polymer Journal website
Rights and permissions
About this article
Cite this article
Yano, T., Yah, W., Yamaguchi, H. et al. Precise control of surface physicochemical properties for electrospun fiber mats by surface-initiated radical polymerization. Polym J 43, 838–848 (2011). https://doi.org/10.1038/pj.2011.80
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pj.2011.80
Keywords
This article is cited by
-
Development of silane/acrylate-based hybrid polymer coating through sol–gel technique for anti-corrosive application
Iranian Polymer Journal (2023)
-
Direct polymer brush grafting to polymer fibers and films by surface-initiated polymerization
Polymer Journal (2018)
-
Anti-fouling behavior of polymer brush immobilized surfaces
Polymer Journal (2016)
-
Immobilization of Carbonic Anhydrase on Modified Electrospun Poly(Lactic Acid) Membranes: Quest for Optimum Biocatalytic Performance
Catalysis Letters (2015)
-
Surface grafting of electrospun fibers using ATRP and RAFT for the control of biointerfacial interactions
Biointerphases (2013)
