
Abstract
Metabolic reprogramming occurs in tumors to foster cancer cell proliferation, survival and metastasis, but as well at a systemic level affecting the whole organism, eventually leading to cancer cachexia. Indeed, as cancer cells rely on external sources of nitrogen and carbon skeleton to grow, systemic metabolic deregulation promoting tissue wasting and metabolites mobilization ultimately supports tumor growth. Cachectic patients experience a wide range of symptoms affecting several organ functions such as muscle, liver, brain, immune system and heart, collectively decreasing patients’ quality of life and worsening their prognosis. Moreover, cachexia is estimated to be the direct cause of at least 20% of cancer deaths. The main aspect of cachexia syndrome is the unstoppable skeletal muscle and fat storage wasting, even with an adequate caloric intake, resulting in nutrient mobilization – both directly as lipid and amino acids and indirectly as glucose derived from the exploitation of liver gluconeogenesis – that reaches the tumor through the bloodstream. From a metabolic standpoint, cachectic host develops a wide range of dysfunctions, from increased insulin and IGF-1 resistance to induction of mitochondrial uncoupling proteins and fat tissue browning resulting in an increased energy expenditure and heat generation, even at rest. For a long time, cachexia has been merely considered an epiphenomenon of end-stage tumors. However, in specific tumor types, such as pancreatic cancers, it is now clear that patients present markers of tissue wasting at a stage in which tumor is not yet clinically detectable, and that host amino acid supply is required for tumor growth. Indeed, tumor cells actively promote tissue wasting by secreting specific factors such as parathyroid hormone-related protein and micro RNAs. Understanding the molecular and metabolic mediators of cachexia will not only advance therapeutic approaches against cancer, but also improve patients’ quality of life.
Similar content being viewed by others
Introduction
Cachexia is a life-threatening condition associated with several pathologies.1 It is particularly relevant in cancer patients, where it occurs in up to 80% of cancers. Cachexia is a marker of unfavorable prognosis, it affects the majority of patients with advanced cancer2 and it represents the direct cause of at least 20% of cancer-associated deaths.1
As it affects multiple organs, cachexia is an extremely complex disease, which severity is difficult to assess objectively. Indeed, only recently a method for staging cachexia extent has been proposed and validated.3
Typical symptoms of cachexia are massive loss of total body mass, anorexia, general inflammation and pronounced muscle-wasting resulting in a drastic decrease of quality of life.4 Furthermore, as broad muscular wasting also involves chest, diaphragm and cardiac muscle, it is not surprising that the majority of cancer deaths are related to respiratory5 or cardiac failure.6
Aside from being a direct cause of cancer death, cachexia also limits the therapeutic options as cachectic patients are normally less tolerant to radio- and chemotherapy because of general weakness and discomfort.7 Moreover, cachectic patients present a reduced response to therapy.8
Despite one of the main feature of cachexia being anorexia, the mode of tissue wasting is completely different from the one induced by starvation.9 Of note, treating anorexia through parenteral nutrition does not reverse cachexia, indicating that the decreased calories intake is not the primary cause of the disease.9
Unlike starvation, which primarily affects fat tissue, skeletal muscle is the major target of wasting in cachectic patients, suggesting a different signaling pathway targeting muscle loss.10 However, even though the main tissue affected by cachexia is the skeletal muscle, cachexia cannot be reduced to a muscle-wasting syndrome. Indeed, several other organs such as liver, heart, fat tissue and brain are affected, making cachexia a true multi-organ syndrome.11
To provide a clear definition of the molecular and metabolic determinants of tissue wasting, it is vital to apply a systemic approach in defining the contribution of each single organ to the cachectic process and to understand the role of tumor in this process and the interplay between the two compartments.
Indeed, although cachexia is a metabolic disorder characterized by tissue wasting, resistance to anabolic signals and an overall catabolic state, cancers, on the other side are highly proliferating and energy-demanding tissues.12 Consequently, the metabolic alterations present in cachectic patients results in a negative energy balance and into the release of nutrients in the bloodstream, further supporting tumor growth.11
Therefore, it is important to investigate the interplay between these two compartments and to understand how cancers promote this pathologic state to foster its own progression.
Immune system
Inflammation is a double-edged sword in cancer. Aside from the natural role of immune system in controlling tumor growth, ultimate cancer cells hijack the immune system to produce specific cytokines promoting tumor growth, survival and progression.13 Chronic inflammation is also a major driver of cachexia (Table 1), as it affects the function of several tissues such as skeletal muscle, fat, brain and liver.14 Indeed, several pro-inflammatory cytokines promote cachexia: tumor necrosis factor alpha (TNFα), interleukin-6 and -1 (IL-6/IL-1) and interferon gamma.15
TNFα, initially named cachectin,16 is probably the most characterized cytokine in cachexia as it promotes anorexia17 and skeletal muscle wasting mainly through the NF-kB pathway.18 TNFα blockade (etanercept) provided promising results in improving cachexia-associated fatigue in a small cohort of cancer patients.19 However, recent trials using neutralizing antibodies against TNFα showed no benefit, suggesting that targeting TNFα alone is not sufficient to prevent cachexia.20
TNFα can also synergize with interferon gamma21 and IL-122 in promoting muscle wasting. Despite that IL-1 itself promotes anorexia23 using IL-1 receptor antagonist was not sufficient to impair cachexia progression in a rat model.24 Nevertheless, human polymorphisms in IL-1B gene, resulting in augmented levels of IL-1β, were associated with a negative prognostic value,25 indicating the involvement of IL-1 pro-inflammatory cytokines in the pathogenesis of cachexia. A study in cancer patients identified increased circulating levels of cytokines (IL-1α, IL-6 and TNFα), suggesting the presence of a robust network of cytokines collectively promoting cachexia.15 IL-6 can directly drive cachexia in specific murine models26 and acute phase protein in liver and skeletal muscle by STAT3 activation.27 Moreover, IL-6 circulating levels correlate with cachexia development and poor prognosis in prostate cancer patients.28 IL-6 can be produced not only by the immune system but also directly by the tumor,29 further highlighting the direct involvement of tumor cells in driving cachexia. Other members of the IL-6 family such as ciliary neurotrophic factor and leukemia inhibitory factor have also been associated with cachexia development.30, 31 The upregulation of pro-inflammatory cytokines co-occurs with decreased expression of the cytokines hampering inflammation, such as IL-4 -10 and -12.32 Coherently, several treatments controlling excessive inflammation provided beneficial effects on cachexia progression.32, 33, 34, 35
Skeletal muscle wasting
Skeletal muscle represents one of the major compartments of the human body, whose function is necessary for a variety of biological processes, from movement to respiration. A tight balance between protein synthesis and degradation is required to maintain muscle homeostasis36 while a decrease in synthesis or an excessive degradation results in wasting.36 The complex hormonal network of anabolic and catabolic factors normally regulating this balance36, 37 is heavily disrupted during tumor progression.
Indeed, it has been reported that during cachexia, both cancer patients and mouse models, experience a decrease in the circulating levels of the anabolic factor insulin-like growth factor-1 (IGF-1) and the development of insulin resistance.37, 38, 39, 40, 41, 42, 43
In parallel with the defective activity of anabolic factors, the production of factors promoting catabolism is augmented both in cachectic mouse models and patients, that is: angiotensin II,44, 45 IL-6,26, 46 myostatin,47, 48 activin A,49 interferon gamma and TNFα.15, 21 Blockade of activin receptor IIB (ActRIIB), the receptor for several transforming growth factor beta family ligands known to promote atrophy (as activin A and myostatin), was sufficient to reverse cachexia and prevent death in several cancer cachexia mouse models, providing the first formal proof of the direct impact of cachexia on cancer death.50 Transforming growth factor beta mechanism of action in promoting cachexia has been elucidated in a recent report. The work from Waning et al.51 showed that, in several mouse models of bone metastasis, increased transforming growth factor beta signaling (released during osteolysis caused by bone metastasis) promotes skeletal muscle oxidation of the calcium channel RyR1 (ryanodine receptor and calcium release channel), ultimately leading to leaky channels and inefficient muscle activity. Clinical relevance of this finding derives from the fact that also patients with bone metastasis present the same channel oxidation, and that drugs restoring calcium channel functionality prevented cancer-related muscle weakness. Another factor affecting skeletal muscle and upregulated during atrophy is TRAF6 (TNFα receptor adapter protein), which is also overexpressed in muscle from gastric cancer patients.52, 53 Its inhibition has been shown to prevent skeletal muscle wasting induced by cachexia in experimental models.53
Insulin resistance has been recently modeled in a tumor model of drosophila. In this animal model, insulin signaling was disrupted by the expression of ImpL2, an insulin growth factor binding protein that inhibits both insulin and IGF-1 signaling.54, 55 ImpL2 was produced directly by different tumor types, promoting peripheral organs insulin resistance and therefore systemic tissue wasting,54, 56 a phenomenon likely present also in cancer patients.57 Interestingly, in the drosophila model, insulin/IGF-1 signaling was upregulated in cancer cells, thus allowing to benefit of the systemic hyperglycemia.55 Further studies will be required understand the relevance of this mechanism in patients.
At the cellular level, three main degradation pathways have been described in skeletal muscle to account for protein degradation (Table 2), that is, ubiquitin-mediated proteasome degradation (UPR), autophagy and calcium-activated protease calpains.36, 58, 59
During tumor cachexia, skeletal muscle specifically upregulates muscle specific UPR system,60 in particular by promoting ubiquitin-ligase MurF1 and Atrogin-1 expression.4, 61 In a rat model of cachexia, induced by the Yoshida ascites hepatoma, UPR upregulation was evident following tumor growth, as shown by Atrogin-1 messenger RNA60 and increased protein ubiquitylation.60, 62 The expression of this ubiquitin ligase has been shown to be mainly regulated by the transcription factor FoxO3a (Forkhead Box (Fox) O).63 Another transcription factor involved in UPR upregulation is NF-κB, which has been shown to stimulate Murf1 expression following Lewis lung carcinoma (LLC) tumor inoculation and muscle wasting in mice.64 NF-κB inhibition with sulfasalazine (in combination with MAPK and proteasome inhibitors) prevented cachexia in a murine model of lung cancer.65
Despite the large body of evidence supporting UPR as a major driver of muscle atrophy in murine models, limited evidence are present for this mechanism in human cancer cachexia.52, 66
Besides UPR, there is a growing interest in the role of autophagy in mediating skeletal muscle wasting.67, 68, 69 Indeed, autophagy has been suggested to be upregulated during cancer cachexia in patients. In a small cohort, lung cancer patients presented increased levels of autophagy mediators BNIP3 (messenger RNA) and LC3B (protein), as well as of the transcription factor promoting autophagy FOXO1.70 Similarly, in another study performed on esophageal cancer patients versus weight-stable non-cancerous control patients, autophagy was identified as the main promoter of skeletal muscle proteolysis.71 Eventually, in a group of 92 gastrointestinal cancer patients expression of GABARAPL1 (an interactor of lysosomal vesicles and autophagy inducer72) was increased compared with healthy controls.73
Calpain proteases have been proposed to initiate the degradative process during cachexia,74 however, limited information concerning their role in muscle wasting is available.60
Adult skeletal muscle normally regenerates after injury through activation and differentiation of a resident population of stem cells called satellite cells. However, the behavior of these cells is deregulated in cachexia, both in murine cancer models and in patients.75 NF-κB induces the activation and expansion of the satellite cell pool, but these cells are unable to complete differentiation, thus further worsening the wasting process.75
Also, specific tumor-derived micro RNA promoting myoblast and skeletal muscle death have been recently identified in microvesicles,76 indicating a direct action of tumor on skeletal muscle. Further studies will be instrumental in defining the specific impact of such structures in mediating the cross-talk between tumor and skeletal muscle.
Cachectic muscle features an impaired mitochondrial metabolism associated with ineffective ATP generation,77, 78 dysfunction of the electron transport chain functionality,79 lipid alterations in the mitochondrial fraction78 and increased expression of mitochondrial uncoupling proteins (UCPs).80, 81, 82 UCPs promote proton leak across the inner mitochondrial membrane, therefore, reducing the proton gradient. UCP-1 expression has been shown to disperse proton gradient with concurrent heat generation, while UCP2 and 3 expressions has been proposed as a cellular mean to prevent excessive oxidative stress by inhibiting OXPHOS.83, 84
Accordingly, cachectic skeletal muscles and in vitro model of cachexia (C2C12-derived myotubes treated with LLC-conditioned medium) present sign of excessive oxidative stress,79, 85 responsible for the worsening wasting process, mainly by promoting protein oxidation by reactive oxygen species,86 ultimately contributing to muscle weakness.51 Mitochondrial dysfunction occurring in the skeletal muscle has been associated with alteration in the lipid content of the mitochondrial fraction, most likely affecting mitochondrial functionality by altering membrane fluidity.78
Whether these alterations in UCP levels are actively causing wasting or are an attempt to prevent it, the resulting increase in proton leak might be responsible for the energetic inefficiency typical of this condition; therefore contributing to the overall increase in the resting energy expenditure (REE) normally evident in cachectic patients.87 Intriguingly, it has been suggested by in vitro experiments that following TNFα treatment, skeletal muscle might promote a futile cycle linked to the co-activation of phosphofructokinase-1 and fructose-1,6-bisphosphatase, resulting in ATP consumption,88 further promoting REE.
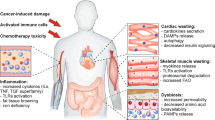
Cardiac muscle
The heart is an important target of cachexia. Cardiac alterations are typical in cancer patients11 and ultimately results in heart failure and arrhythmia, which are two of the concurring causes of death during cachexia.6 Similarly to skeletal muscle, cardiac wasting involves the activation of protein turnover mediated by the UPR system.89 Indeed, the heart weight and functionality has been reported to decrease in a murine model of colon cancer90 developing chronic heart failure. As in skeletal muscle, NF-κB inhibition has been shown to ameliorate cardiac atrophy and functionality in a mouse model of Colon-26-driven cancer cachexia,91 suggesting novel therapeutic approaches for this severe cause of cancer-cachexia death.
Chronic heart failure has been previously associated with the increase of REE,92 providing another reason for the increase of energy expenditure in cachectic patients. This increase might be at least in part directly related to an increased metabolism of cardiac tissue, as ex vivo hearts from tumor-bearing rats present an increased oxidative rate.93
Liver wasting
One of the main functions of the liver is to act as a biological factory. Indeed, it produces the majority of compound required by the organisms, including glucose, amino acids, fatty acids, cholesterol and hormones regulating several complex functions, such as hepcidin (iron homeostasis), IGF-1 (mass growth), angiotensin (blood pressure) and several coagulation cascade factors. Furthermore, the liver is an organ characterized by high-metabolic rate, substantially contributing to REE.94
Albeit neglected (compared with muscle and fat), liver mass substantially increases during cachexia progression,94, 95 strongly suggesting the involvement of this organ in cancer cachexia. There is limited evidence concerning the role of liver metabolism on cachexia development. However, liver mass increase in colorectal cancer patients has been shown to correlate with increased energy expenditure,94 increased expression of UCPs.80 Furthermore, inefficient oxidative phosphorylation, primarily related to an increased mitochondrial cardiolipin accumulation,96 has been identified ex vivo in liver hepatocytes from a cachectic rat model of peritoneal carcinoma,97 indicating a direct involvement of liver tissue in cachexia.
During tumor growth, liver tissue is actively co-opted to perform high-rate gluconeogenesis, using the lactate derived from tumor glycolysis.98, 99 This oncological version of the Cori Cycle has been reported by tracing experiments with 14C-labeled glucose in metastatic cancer patients in the 70s.98, 100
This pathway is extremely energy-demanding since, per each glucose molecule produced, 6 ATP are consumed. The resulting glucose is mostly scavenged by the highly glycolytic tumors; this results in a net negative balance further worsening the higher metabolic rate typical of cancer patients.101, 102
Another typical feature of hepatic dysfunction associated with cachexia is the onset of steatosis, present in both patient and murine model.103, 104 In fact, it has been shown that tumor-bearing patients or mice injected with the Colon-26 (C26) model are characterized by a rapid decrease in circulating very low-density lipoprotein responsible for the mobilization of lipid in the bloodstream.105 From a molecular standpoint, this downregulation has been directly associated with the increased expression of TSC22D4, a transcription factor directly induced by transforming growth factor beta. This inhibits very low-density lipoprotein secretion, lipogenesis and eventually leads to the accumulation of lipids in the liver, which ultimately might promote liver gluconeogenesis.106
Although liver contributes to cachexia by increasing energy expenditure through gluconeogenesis and reducing very low-density lipoprotein circulation, it participates as well to the worsening of inflammation by secreting acute phase proteins and reducing albumin secretion, a process mostly driven by IL-6 and TNFα.107, 108 This eventually results in muscular protein breakdown and adipocytes lipolysis.27
Cancer patients exhibit decreased albumin production, in all likelihood mediated by TNFα, as TNFα treatment per se is sufficient to inhibit albumin production in vivo (mice and rabbits), or in isolated hepatocytes in vitro.109, 110 However, a different mechanism has been proposed in pancreatic cancer, where hypoalbuminemia is not associated with decreased synthesis,111 but with increased uptake through KRAS-dependent macropinocytosis to sustain energy maintenance.112, 113
Altogether, these data indicate that liver directly contributes to cachexia by promoting hypermetabolism and increased energy expenditure. However, further studies are required to evaluate its relative contribution to the cachectic process and to define better the metabolic interplay between tumor and liver.
Lipid wasting and browning
Albeit not as penetrant as skeletal muscle wasting, adipose tissue depletion has been identified as one of the symptoms of cachexia.114, 115 Indeed, cachectic patients manifest high levels of circulating free fatty acids, glycerol and triacylglycerol.116, 117 This is linked to the increased circulation of several factors promoting lipid mobilization, such as the adipokine Zn-alpha 2-glycoprotein/lipid-mobilizing factor (ZAG/LMF), IL-1, IL-6 and TNFα.15, 16, 32
In specific mouse models of cachexia (LLC lung cancer and B16 melanoma), lipid wasting precedes skeletal muscle loss.115 Indeed, preventing triglyceride degradation and lipolysis in mice lacking key lipolytic enzymes (for example, adipose triglyceride lipase and hormone-sensitive lipase) ultimately averts skeletal muscle loss, underlining a close link between these two different tissues.115 Interestingly, also cachectic cancer patients present increased triglyceride hydrolase activity.117 In addition, the fatty acid mobilization-promoting adipokine ZAG promotes also skeletal muscle protein synthesis and fatty acid oxidation, suggesting that increased level of Zn-alpha 2-glycoprotein during tissue wasting acts as a salvage pathway.99, 118
An interesting feature of cancer cachexia is the progressive switch of fat tissue type, from white (white adipose tissue) to brown (brown adipose tissue), which derives its name from the darker color associated with the enrichment in mitochondria.119, 120 These mitochondria present high levels of the UCP-1, which directly promotes thermogenesis by uncoupling the electrochemical gradient from ATP generation.121 Browning strongly contributes to the increased energy expenditure common in cachectic patients.119 Pro-inflammatory factors either derived from the host immune system or the tumor, contribute to this switch.119, 120 In LLC tumor-bearing mice, several tumor-derived cytokines correlate with the induction of tissue browning and, therefore, with increased energy expenditure.119 In particular, cachectic lipid wasting occurs mostly in tumors actively secreting parathyroid hormone-related protein.119 This hormone is augmented as well in several cancer patients and has been previously associated with hypercalcemia, a common metabolic abnormality in many cancer types.122 Collectively, fat tissue wasting can be interpreted as a critical turning point in the cachectic process, as it further contributes to the propagation of cachexia by stimulating skeletal muscle wasting.115 As it is also emerging that tumors require fatty acid oxidation,123 it will be important to assess the impact of lipolysis on tumor progression.
Brain and food intake
Decreased in appetite and alterations in taste perception are common features in cancer patients.11, 124 In particular, cancer anorexia is a characteristic of end-stage patients contributing to the worsening of cachexia. Although this is in part related to the development of depressive disorders associated with the psychological implications of having cancer,8 it is also bound to the alterations in the complex hormonal network regulating appetite.10 A pivotal player in cachexia development is the hypothalamus, which regulates both food intake and body energy expenditure.125 Several factors regulates food intake, including the vagal stimulation induced by gastric distension bound to food intake126, 127 and hormones. Hormones produced peripherally either promote food intake (orexigenic, like ghrelin), or inhibit it (anorexigenic, such as leptin, insulin, cholecystokinin, peptide YY and glucagon-like peptide 1/GLP1).128 At central level in the hypothalamus, peripheral signals regulates the axis promoting food intake such as the Neuropeptide Y (NPY) and Agouti-related protein (AgRP) or the anorexigenic one, such as pro-opiomelanocortin (POMC) precursor related to the production of melanocyte-stimulating hormone α-MSH.129 Despite the peripheral signals triggering food intake are maintained in some cachectic conditions, it has been observed a decreased responsiveness of the hypothalamus.130 This is the case for ghrelin, whose levels are normally increased in cachectic patients, but without food intake promotion,42 a phenomenon named ‘ghrelin resistance’.42, 131
In addition, in cancer it has been reported an increased resistance to the hormones promoting food intake, such as Neuropeptide Y and Agouti-related protein.132, 133
On the contrary, an increased activation of pro-anorexigenic factors derived by hypothalamic melanocortin system has been shown.134, 135
Tumor-associated inflammation is involved in this process (or these processes) as several pro-inflammatory cytokines such as TNFα, interferon gamma, IL-1 and IL-6, directly promote these alterations136 and, coherently, anti-inflammatory drugs such as cyclooxygenase inhibitors ameliorates cancer anorexia.137, 138
In addition, TNFα has also been associated to the stimulation of bitterness perception,139 thus further inhibiting the willingness of food assumption.
Altogether these cytokines are responsible for the so-called ‘sickness behavior’, a common disease state typical of many chronic disease and proposed as an evolutionary response involved in fighting infections by depleting iron and nutrients required for bacterial growth.140
The direct involvement of tumor-derived factors in promoting anorexia has been proposed.141, 142 Coherently, the bioactive lipid sphingosine-1-phosphate, which is involved in cancer progression and is produced by several tumors,143 has been recently shown to promote energy expenditure and anorexia.135 Increased engagement of sphingosine-1-phosphate-receptor on hypothalamic neurons promotes increased temperature and oxygen consumption, while decreasing food intake.135
In fact, hypothalamus controls a wide range of biological activities, including energy expenditure regulation and glucose homeostasis.144 Coherently, during cachexia hypothalamic activity specifically mediates increased energy expenditure,135, 145 it will be important, therefore, to define the role of hypothalamus in regulating glucose homeostasis.
Appetite improvement have been achieved in cancer patients with the progesterone analog megestrol acetate (by an unclear mechanism)146 and by the ghrelin analog anamorelin,41 thus providing novel therapeutic avenues in the treatment of such complication.
Pancreas
The identification of decreased glucose tolerance in cancer patients, dating back to 1919, identified glucose metabolism as the first metabolic abnormality in cancer.147 Although insulin resistance is a strong risk factor for cancer development,148, 149 tumor progression can promote insulin resistance by itself. Indeed, it has been shown that several cancer patients present insulin resistance150, 151 that progressively worsen during cachexia development.10 Furthermore, the degree of glucose tolerance positively correlates with mortality risk.152
Decreased insulin sensitivity during tumor progression has also been reported in drosophila,54, 55 and in mice,39 where Colon-26 tumor inoculation was sufficient to induce insulin resistance before cachexia onset,39 while in Walker 256 tumor-bearing rats isolated Langerhans islet were more resistant to glucose challenge, resulting in decreased insulin secretion.153
One of the factors identified for the induction of insulin resistance is TNF-α, which directly impairs insulin signaling and IRS-1 activation.154
Insulin has several metabolic activities that can affect tumor progression.155, 156 The increase in insulin level, as it happens in the onset of insulin resistance, per se promotes directly tumor growth by acting as growth factors. Coherently, several tumors overexpress the insulin receptor and IGF-1 receptor.157, 158 However, insulin resistance can also promote tumor growth indirectly by modulating host metabolism in at least two independent manners. On one side, as insulin is an anabolic factor that normally blocks protein breakdown and promotes protein synthesis;159, 160 insulin resistance (and similarly IGF-1-resistance) might promote muscle wasting, hence, amino acid mobilization into the circulation, potentially fueling cancer. On the other, insulin signaling impairment also promotes liver gluconeogenesis,161 further increasing REE, tissue wasting and ultimately fueling cancer aerobic glycolysis.
The role of insulin signaling in preventing cachexia is further stressed by experimental data showing that mice treated with insulin sensitizers (rosiglitazone)39, 162 and patients treated with insulin ameliorate cachexia symptoms.163
Not only insulin secretion is affected by tumor progression, but also glucagon levels are increased.164 The increased production of glucagon in the alpha islet of pancreas during cancer progression further promotes liver gluconeogenesis as reported in different tumor models, both in humans and in animal models.164, 165, 166
Albeit there is no clear mechanism behind the induction of this hormone by the tumor, normalizing its levels has been suggested to impair cachexia progression.166
Gastrointestinal tract
Gut functionality contributes to cachexia. This is particularly relevant in gastrointestinal tumors, as proven in a mouse model of colon cancer (transgenic APC+/min strain), where gut barrier was disrupted along with tumor growth, resulting in increased systemic inflammation and endotoxemia.167, 168
Other than colon cancers, a broader impact of gut on cachexia is bound to gut microbiota.169 The human body is in symbiosis with the gut microbiota, which identify the portion of microorganism residing in the intestinal tract outnumbering human cells by a 10-fold factor.170 Alteration of the gut flora due to undernutrition and chemotherapy ultimately affects specific metabolite availability and absorption,169, 171 which in turn affects tumor growth and cachexia.172, 173 The gastrointestinal tract, and mainly the stomach, is also the source of the orexigenic peptide ghrelin, which is strongly increased during cachexia.42 As ghrelin exerts several other activities, ranging from increasing adiposity,174 reducing REE175 and impairing muscle atrophy,176 it is possible that its expression is induced as a compensatory mechanism to buffer cachexia. Owing to the pleiotropic effects of ghrelin (partly shared with its unacylated form through an unknown receptor176, 177, 178), the use of ghrelin analog, Anamorelin, has a strong therapeutic potential. Indeed, early clinical trials are suggesting beneficial effect of Anamorelin in improving both appetite and skeletal muscle mass.41, 179
Cachexia: a cancer target or an innocent bystander?
As cachexia associates with several pathological conditions, such as chronic inflammation, cardiac disease and AIDS,37 cancer cachexia has always been regarded as an epiphenomenon of tumor progression,6 and even its role on death promotion has only been formally proven recently.50
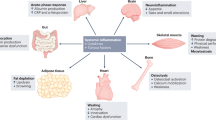
However, mounting evidence supports the notion that cachexia is not only a severe complication of tumor growth, but results from the systemic metabolic reprogramming of the host to grow and progress180 (Figure 1).

The simplified scheme represents the major organs commonly affected during cachexia progression and how they fuel tumor growth. In brief, tumor tissue and the co-opted immune system secrete specific factors, thus promoting skeletal muscle wasting and lipolysis. Pro-inflammatory cytokines contribute to develop anorexia and insulin resistance, ultimately worsening skeletal muscle wasting. Gastrointestinal tract tumors and bone metastasis can promote further cachexia by causing endotoxemia and transforming growth factor beta release, respectively. (Adapted from Servier Medical Art, www.servier.com).
In fact, tumor can promote cachexia by secreting tissue-wasting factors10, 76, 87, 119, 120 and by promoting dysfunction in specific organs, such as liver,105 gut, immune system, brain and pancreas.
Interesting data concerning the active interplay between tumor and cachexia derive from the study of pancreatic ductal adenocarcinoma, where cachexia development occurs in almost 90% of the cases.1 In this tumor type, increased amino acid levels are an early marker of disease occurrence181 in patients, years before pancreatic ductal adenocarcinoma diagnosis. Coherently, in a K-RAS-driven transgenic model of pancreatic cancer the increase in circulating amino acid was present before the actual tumor was detectable.181 This effect on the host, even at early phase of tumor growth, when no evidence of discomfort is present, indicates the importance of actively understanding the interplay between tumor and the host on a systemic level, with a particular focus toward the metabolic dependencies of growing tumors. Further studies will be required to evaluate if this phenomenon is specific to pancreatic ductal adenocarcinoma or if it is a feature of different tumor types.
As tumor is a highly energy-demanding tissue, energy and metabolic intermediates are required to sustain proliferation and cell-death resistance,182 it is not surprising, therefore, that tumor cells promote metabolic reprogramming not only cell-autonomously, but of the whole organism.
Although the study of cancer metabolism has been mostly focused toward the definition of aerobic glycolysis, several other metabolic pathways are emerging as important for tumor development. Because of the inefficient perfusion typical of cancers183 along with the avidity of cancer cells for glucose,184 other carbon sources are required for tumor growth.185
For instance, several studies indicate that specific metabolite uptake is required for tumor growth and progression in some cases, such as lipids,186, 187 branched amino acids,188 glutamine,189, 190 serine191 or even entire proteins by macropinocytosis.112, 113 By actively scavenging different nutrients from the bloodstream, cancer cells manage severe nutrient deprivation.113 Altogether, tumor cells have a metabolic advantage from the induction of systemic tissue wasting, prompting to reconsider cachexia as a part of the metabolic program in tumor development. A further indication for this mechanism derives from the work of Luo et al.192 who identified a metabolic cross-talk occurring in colorectal cancers between tumor and skeletal muscle. To aim this, colon cancer cells release high-motility group box 1 (HMGB1), which contributes to the metabolic reprogramming of skeletal muscle through RAGE (Receptor for Advanced Glycation End-products), inducing autophagy and release of free amino acids in the plasma. This results in the transfer of carbon skeleton from the muscle to the tumor as demonstrated by 13C-glutamine tracing.192 Considering that high-motility group box 1 circulating levels correlate with the severity in different cancer types,193, 194, 195 it will be important to explore this cross-talk in different tumor types.
Cancer cachexia it is not merely a complication of tumor progression, as cancer cells induce and exploit systemic functions. It would be reductive to consider cachexia as solely caused by tumor metabolism acting as ‘energy sink’ as initially proposed,1, 196 as evidences by the fact that tumor mass hardly correspond to the severity of cachexia.10, 105 Indeed, it is a syndrome induced also by several other noxious factor, from chronic infections to cardiac or respiratory failure.1
Concluding remarks
As cancer cachexia is being progressively defined, also from a molecular standpoint, it will be important to discriminate between the alterations actually promoting tumor progression and the one which are simple off-targets of the abnormal levels of factors presents during tumor growth. Understanding the effects of tumor on the entire organism and identify the signaling pathways involved will allow more effective cancer therapies and, ultimately, a better quality of life for patients.
Although several molecular mechanisms driving cachexia have been identified by using murine models, it will be vital to define the real impact of such processes in human patients. Moreover, relatively few murine models are used to generate cachexia (the vast majority being LLC in C57BL/6, and Colon-26 in Balb/c, tumor cells injection), while the use of transgenic mice is generally restricted to C57BL/6 APC+/min.26 The use of different strains and different cancer types will be essential to model the variety of the cachectic processes occurring in cancer patients, especially considering that cachexia has different penetrance according to the associated cancer type.10
Further efforts will be required to define the pathogenesis of cachexia in patients at early stages (that is, the pre-cachectic stage197), where systemic alterations are more likely to be reversible. As cancer cachexia affects different tissue at the same time, it will also be pivotal to devise therapeutic strategies with multiple targets.
Abbreviations
- IGF-1:
-
insulin-like growth factor-1
- IL-:
-
interleukin-
- LLC:
-
lewis lung carcinoma
- REE:
-
resting energy expenditure
- TNFα:
-
tumor necrosis factor alpha
- UCPs:
-
uncoupling proteins
- UPR:
-
Ubiquitin proteasome degradation.
References
Fearon KC, Glass DJ, Guttridge DC . Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 2012; 16: 153–166.
Melstrom LG, Melstrom KA Jr, Ding XZ, Adrian TE . Mechanisms of skeletal muscle degradation and its therapy in cancer cachexia. Histol Histopathol 2007; 22: 805–814.
Argiles JM, Lopez-Soriano FJ, Toledo M, Betancourt A, Serpe R, Busquets S . The cachexia score (CASCO): a new tool for staging cachectic cancer patients. J Cachexia Sarcopenia Muscle 2011; 2: 87–93.
Johns N, Stephens NA, Fearon KC . Muscle wasting in cancer. Int J Biochem Cell Biol 2013; 45: 2215–2229.
Houten L, Reilley AA . An investigation of the cause of death from cancer. J Surg Oncol 1980; 13: 111–116.
Kalantar-Zadeh K, Rhee C, Sim JJ, Stenvinkel P, Anker SD, Kovesdy CP . Why cachexia kills: examining the causality of poor outcomes in wasting conditions. J Cachexia Sarcopenia Muscle 2013; 4: 89–94.
Fearon KC, Baracos VE . Cachexia in pancreatic cancer: new treatment options and measures of success. HPB (Oxford) 2010; 12: 323–324.
Vaughan VC, Martin P, Lewandowski PA . Cancer cachexia: impact, mechanisms and emerging treatments. J Cachexia Sarcopenia Muscle 2013; 4: 95–109.
Evans WJ, Morley JE, Argiles J, Bales C, Baracos V, Guttridge D et al. Cachexia: a new definition. Clin Nutr 2008; 27: 793–799.
Tisdale MJ . Wasting in cancer. J Nutr 1999; 129 (Suppl 1): 243S–246S.
Argiles JM, Busquets S, Stemmler B, Lopez-Soriano FJ . Cancer cachexia: understanding the molecular basis. Nat Rev Cancer 2014; 14: 754–762.
Vander Heiden MG, Cantley LC, Thompson CB . Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324: 1029–1033.
Coussens LM, Werb Z . Inflammation and cancer. Nature 2002; 420: 860–867.
Argiles JM, Lopez-Soriano FJ, Busquets S . Counteracting inflammation: a promising therapy in cachexia. Crit Rev Oncog 2012; 17: 253–262.
Mantovani G, Maccio A, Mura L, Massa E, Mudu MC, Mulas C et al. Serum levels of leptin and proinflammatory cytokines in patients with advanced-stage cancer at different sites. J Mol Med (Berl) 2000; 78: 554–561.
Torti FM, Dieckmann B, Beutler B, Cerami A, Ringold GM . A macrophage factor inhibits adipocyte gene expression: an in vitro model of cachexia. Science 1985; 229: 867–869.
Jakubowski AA, Casper ES, Gabrilove JL, Templeton MA, Sherwin SA, Oettgen HF . Phase I trial of intramuscularly administered tumor necrosis factor in patients with advanced cancer. J Clin Oncol 1989; 7: 298–303.
Han Y, Weinman S, Boldogh I, Walker RK, Brasier AR . Tumor necrosis factor-alpha-inducible IkappaBalpha proteolysis mediated by cytosolic m-calpain. A mechanism parallel to the ubiquitin-proteasome pathway for nuclear factor-kappab activation. J Biol Chem 1999; 274: 787–794.
Monk JP, Phillips G, Waite R, Kuhn J, Schaaf LJ, Otterson GA et al. Assessment of tumor necrosis factor alpha blockade as an intervention to improve tolerability of dose-intensive chemotherapy in cancer patients. J Clin Oncol 2006; 24: 1852–1859.
Jatoi A, Ritter HL, Dueck A, Nguyen PL, Nikcevich DA, Luyun RF et al. A placebo-controlled, double-blind trial of infliximab for cancer-associated weight loss in elderly and/or poor performance non-small cell lung cancer patients (N01C9). Lung Cancer 2010; 68: 234–239.
Acharyya S, Ladner KJ, Nelsen LL, Damrauer J, Reiser PJ, Swoap S et al. Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J Clin Invest 2004; 114: 370–378.
Flores EA, Bistrian BR, Pomposelli JJ, Dinarello CA, Blackburn GL, Istfan NW . Infusion of tumor necrosis factor/cachectin promotes muscle catabolism in the rat. A synergistic effect with interleukin 1. J Clin Invest 1989; 83: 1614–1622.
Uehara A, Sekiya C, Takasugi Y, Namiki M, Arimura A . Anorexia induced by interleukin 1: involvement of corticotropin-releasing factor. Am J Physiol 1989; 257: R613–R617.
Costelli P, Llovera M, Carbo N, Garcia-Martinez C, Lopez-Sorianoq FJ, Argiles JM . Interleukin-1 receptor antagonist (IL-1ra) is unable to reverse cachexia in rats bearing an ascites hepatoma (Yoshida AH-130). Cancer Lett 1995; 95: 33–38.
Graziano F, Ruzzo A, Santini D, Humar B, Tonini G, Catalano V et al. Prognostic role of interleukin-1beta gene and interleukin-1 receptor antagonist gene polymorphisms in patients with advanced gastric cancer. J Clin Oncol 2005; 23: 2339–2345.
Baltgalvis KA, Berger FG, Pena MM, Davis JM, Muga SJ, Carson JA . Interleukin-6 and cachexia in ApcMin/+ mice. Am J Physiol Regul Integr Comp Physiol 2008; 294: R393–R401.
Bonetto A, Aydogdu T, Kunzevitzky N, Guttridge DC, Khuri S, Koniaris LG et al. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS ONE 2011; 6: e22538.
Kuroda K, Nakashima J, Kanao K, Kikuchi E, Miyajima A, Horiguchi Y et al. Interleukin 6 is associated with cachexia in patients with prostate cancer. Urology 2007; 69: 113–117.
Zaki MH, Nemeth JA, Trikha M . CNTO 328, a monoclonal antibody to IL-6, inhibits human tumor-induced cachexia in nude mice. Int J Cancer 2004; 111: 592–595.
Henderson JT, Seniuk NA, Richardson PM, Gauldie J, Roder JC . Systemic administration of ciliary neurotrophic factor induces cachexia in rodents. J Clin Invest 1994; 93: 2632–2638.
Seto DN, Kandarian SC, Jackman RW . A key role for leukemia inhibitory factor in C26 cancer cachexia. J Biol Chem 2015; 290: 19976–19986.
Argiles JM, Busquets S, Lopez-Soriano FJ . Anti-inflammatory therapies in cancer cachexia. Eur J Pharmacol 2011; 668 (Suppl 1): S81–S86.
Fearon K, Arends J, Baracos V . Understanding the mechanisms and treatment options in cancer cachexia. Nat Rev Clin Oncol 2013; 10: 90–99.
Lundholm K, Gelin J, Hyltander A, Lonnroth C, Sandstrom R, Svaninger G et al. Anti-inflammatory treatment may prolong survival in undernourished patients with metastatic solid tumors. Cancer Res 1994; 54: 5602–5606.
Cerchietti LC, Navigante AH, Castro MA . Effects of eicosapentaenoic and docosahexaenoic n-3 fatty acids from fish oil and preferential Cox-2 inhibition on systemic syndromes in patients with advanced lung cancer. Nutr Cancer 2007; 59: 14–20.
Bonaldo P, Sandri M . Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech 2013; 6: 25–39.
Cohen S, Nathan JA, Goldberg AL . Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov 2015; 14: 58–74.
Penna F, Bonetto A, Muscaritoli M, Costamagna D, Minero VG, Bonelli G et al. Muscle atrophy in experimental cancer cachexia: is the IGF-1 signaling pathway involved? Int J Cancer 2010; 127: 1706–1717.
Asp ML, Tian M, Wendel AA, Belury MA . Evidence for the contribution of insulin resistance to the development of cachexia in tumor-bearing mice. Int J Cancer 2010; 126: 756–763.
Costelli P, Muscaritoli M, Bossola M, Penna F, Reffo P, Bonetto A et al. IGF-1 is downregulated in experimental cancer cachexia. Am J Physiol Regul Integr Comp Physiol 2006; 291: R674–R683.
Garcia JM, Friend J, Allen S . Therapeutic potential of anamorelin, a novel, oral ghrelin mimetic, in patients with cancer-related cachexia: a multicenter, randomized, double-blind, crossover, pilot study. Support Care Cancer 2013; 21: 129–137.
Garcia JM, Garcia-Touza M, Hijazi RA, Taffet G, Epner D, Mann D et al. Active ghrelin levels and active to total ghrelin ratio in cancer-induced cachexia. J Clin Endocrinol Metab 2005; 90: 2920–2926.
Rofe AM, Bourgeois CS, Coyle P, Taylor A, Abdi EA . Altered insulin response to glucose in weight-losing cancer patients. Anticancer Res 1994; 14: 647–650.
Yoshida T, Delafontaine P . Mechanisms of cachexia in chronic disease states. Am J Med Sci 2015; 350: 250–256.
Nagaya N, Uematsu M, Kojima M, Date Y, Nakazato M, Okumura H et al. Elevated circulating level of ghrelin in cachexia associated with chronic heart failure: relationships between ghrelin and anabolic/catabolic factors. Circulation 2001; 104: 2034–2038.
Mantovani G, Maccio A, Madeddu C, Serpe R, Massa E, Dessi M et al. Randomized phase III clinical trial of five different arms of treatment in 332 patients with cancer cachexia. Oncologist 2010; 15: 200–211.
Lokireddy S, Wijesoma IW, Bonala S, Wei M, Sze SK, McFarlane C et al. Myostatin is a novel tumoral factor that induces cancer cachexia. Biochem J 2012; 446: 23–36.
Gallot YS, Durieux AC, Castells J, Desgeorges MM, Vernus B, Plantureux L et al. Myostatin gene inactivation prevents skeletal muscle wasting in cancer. Cancer Res 2014; 74: 7344–7356.
Loumaye A, de Barsy M, Nachit M, Lause P, Frateur L, van Maanen A et al. Role of activin a and myostatin in human cancer cachexia. J Clin Endocrinol Metab 2015; 100: 2030–2038.
Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010; 142: 531–543.
Waning DL, Mohammad KS, Reiken S, Xie W, Andersson DC, John S et al. Excess TGF-beta mediates muscle weakness associated with bone metastases in mice. Nat Med 2015; 21: 1262–1271.
Sun YS, Ye ZY, Qian ZY, Xu XD, Hu JF . Expression of TRAF6 and ubiquitin mRNA in skeletal muscle of gastric cancer patients. J Exp Clin Cancer Res 2012; 31: 81.
Paul PK, Gupta SK, Bhatnagar S, Panguluri SK, Darnay BG, Choi Y et al. Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J Cell Biol 2010; 191: 1395–1411.
Figueroa-Clarevega A, Bilder D . Malignant Drosophila tumors interrupt insulin signaling to induce cachexia-like wasting. Dev Cell 2015; 33: 47–55.
Kwon Y, Song W, Droujinine IA, Hu Y, Asara JM, Perrimon N . Systemic organ wasting induced by localized expression of the secreted insulin/IGF antagonist ImpL2. Dev Cell 2015; 33: 36–46.
Wagner EF, Petruzzelli M . Cancer metabolism: a waste of insulin interference. Nature 2015; 521: 430–431.
Lundholm K, Edstrom S, Ekman L, Karlberg I, Schersten T . Metabolism in peripheral tissues in cancer patients. Cancer Treat Rep 1981; 65 (Suppl 5): 79–83.
Baar K, Nader G, Bodine S . Resistance exercise, muscle loading/unloading and the control of muscle mass. Essays Biochem 2006; 42: 61–74.
Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001; 294: 1704–1708.
Baracos VE, DeVivo C, Hoyle DH, Goldberg AL . Activation of the ATP-ubiquitin-proteasome pathway in skeletal muscle of cachectic rats bearing a hepatoma. Am J Physiol. 1995; 268: E996–1006.
Schakman O, Kalista S, Barbe C, Loumaye A, Thissen JP . Glucocorticoid-induced skeletal muscle atrophy. Int J Biochem Cell Biol 2013; 45: 2163–2172.
Temparis S, Asensi M, Taillandier D, Aurousseau E, Larbaud D, Obled A et al. Increased ATP-ubiquitin-dependent proteolysis in skeletal muscles of tumor-bearing rats. Cancer Res 1994; 54: 5568–5573.
Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004; 117: 399–412.
Cai D, Frantz JD, Tawa NE Jr, Melendez PA, Oh BC, Lidov HG et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 2004; 119: 285–298.
Chacon-Cabrera A, Fermoselle C, Urtreger AJ, Mateu-Jimenez M, Diament MJ, de Kier Joffe ED et al. Pharmacological strategies in lung cancer-induced cachexia: effects on muscle proteolysis, autophagy, structure, and weakness. J Cell Physiol 2014; 229: 1660–1672.
Bossola M, Muscaritoli M, Costelli P, Grieco G, Bonelli G, Pacelli F et al. Increased muscle proteasome activity correlates with disease severity in gastric cancer patients. Ann Surg 2003; 237: 384–389.
Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L et al. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun 2015; 6: 6670.
Penna F, Costamagna D, Pin F, Camperi A, Fanzani A, Chiarpotto EM et al. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am J Pathol 2013; 182: 1367–1378.
McClung JM, Judge AR, Powers SK, Yan Z . p38 MAPK links oxidative stress to autophagy-related gene expression in cachectic muscle wasting. Am J Physiol Cell Physiol 2010; 298: C542–C549.
Op den Kamp CM, Langen RC, Snepvangers FJ, de Theije CC, Schellekens JM, Laugs F et al. Nuclear transcription factor kappa B activation and protein turnover adaptations in skeletal muscle of patients with progressive stages of lung cancer cachexia. Am J Clin Nutr 2013; 98: 738–748.
Tardif N, Klaude M, Lundell L, Thorell A, Rooyackers O . Autophagic-lysosomal pathway is the main proteolytic system modified in the skeletal muscle of esophageal cancer patients. Am J Clin Nutr 2013; 98: 1485–1492.
Boyer-Guittaut M, Poillet L, Liang Q, Bole-Richard E, Ouyang X, Benavides GA et al. The role of GABARAPL1/GEC1 in autophagic flux and mitochondrial quality control in MDA-MB-436 breast cancer cells. Autophagy 2014; 10: 986–1003.
Stephens NA, Skipworth RJ, Gallagher IJ, Greig CA, Guttridge DC, Ross JA et al. Evaluating potential biomarkers of cachexia and survival in skeletal muscle of upper gastrointestinal cancer patients. J Cachexia Sarcopenia Muscle 2015; 6: 53–61.
Costelli P, Reffo P, Penna F, Autelli R, Bonelli G, Baccino FM . Ca(2+)-dependent proteolysis in muscle wasting. Int J Biochem Cell Biol 2005; 37: 2134–2146.
He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, Thomas-Ahner J et al. NF-kappaB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J Clin Invest 2013; 123: 4821–4835.
He WA, Calore F, Londhe P, Canella A, Guttridge DC, Croce CM . Microvesicles containing miRNAs promote muscle cell death in cancer cachexia via TLR7. Proc Natl Acad Sci USA 2014; 111: 4525–4529.
Tzika AA, Fontes-Oliveira CC, Shestov AA, Constantinou C, Psychogios N, Righi V et al. Skeletal muscle mitochondrial uncoupling in a murine cancer cachexia model. Int J Oncol 2013; 43: 886–894.
Antunes D, Padrao AI, Maciel E, Santinha D, Oliveira P, Vitorino R et al. Molecular insights into mitochondrial dysfunction in cancer-related muscle wasting. Biochim Biophys Acta 2014; 1841: 896–905.
McLean JB, Moylan JS, Andrade FH . Mitochondria dysfunction in lung cancer-induced muscle wasting in C2C12 myotubes. Front Physiol 2014; 5: 503.
Bing C, Russell ST, Beckett EE, Collins P, Taylor S, Barraclough R et al. Expression of uncoupling proteins-1, -2 and -3 mRNA is induced by an adenocarcinoma-derived lipid-mobilizing factor. Br J Cancer 2002; 86: 612–618.
Sanchis D, Busquets S, Alvarez B, Ricquier D, Lopez-Soriano FJ, Argiles JM . Skeletal muscle UCP2 and UCP3 gene expression in a rat cancer cachexia model. FEBS Lett 1998; 436: 415–418.
Collins P, Bing C, McCulloch P, Williams G . Muscle UCP-3 mRNA levels are elevated in weight loss associated with gastrointestinal adenocarcinoma in humans. Br J Cancer 2002; 86: 372–375.
Divakaruni AS, Brand MD . The regulation and physiology of mitochondrial proton leak. Physiology (Bethesda) 2011; 26: 192–205.
Nedergaard J, Ricquier D, Kozak LP . Uncoupling proteins: current status and therapeutic prospects. EMBO Rep 2005; 6: 917–921.
Ushmorov A, Hack V, Droge W . Differential reconstitution of mitochondrial respiratory chain activity and plasma redox state by cysteine and ornithine in a model of cancer cachexia. Cancer Res 1999; 59: 3527–3534.
Barreiro E . Protein carbonylation and muscle function in COPD and other conditions. Mass Spectrom Rev 2014; 33: 219–236.
Argiles JM, Fontes-Oliveira CC, Toledo M, Lopez-Soriano FJ, Busquets S . Cachexia: a problem of energetic inefficiency. J Cachexia Sarcopenia Muscle 2014; 5: 279–286.
Zentella A, Manogue K, Cerami A . Cachectin/TNF-mediated lactate production in cultured myocytes is linked to activation of a futile substrate cycle. Cytokine 1993; 5: 436–447.
Willis MS, Bevilacqua A, Pulinilkunnil T, Kienesberger P, Tannu M, Patterson C . The role of ubiquitin ligases in cardiac disease. J Mol Cell Cardiol 2014; 71: 43–53.
Tian M, Nishijima Y, Asp ML, Stout MB, Reiser PJ, Belury MA . Cardiac alterations in cancer-induced cachexia in mice. Int J Oncol 2010; 37: 347–353.
Wysong A, Couch M, Shadfar S, Li L, Rodriguez JE, Asher S et al. NF-kappaB inhibition protects against tumor-induced cardiac atrophy in vivo. Am J Pathol 2011; 178: 1059–1068.
Mustafa I, Leverve X . Metabolic and nutritional disorders in cardiac cachexia. Nutrition 2001; 17: 756–760.
Drott C, Lundholm K . Glucose uptake and amino acid metabolism in perfused hearts from tumor-bearing rats. J Surg Res 1990; 49: 62–68.
Lieffers JR, Mourtzakis M, Hall KD, McCargar LJ, Prado CM, Baracos VE . A viscerally driven cachexia syndrome in patients with advanced colorectal cancer: contributions of organ and tumor mass to whole-body energy demands. Am J Clin Nutr 2009; 89: 1173–1179.
Tisdale MJ . Cachexia in cancer patients. Nat Rev Cancer 2002; 2: 862–871.
Julienne CM, Tardieu M, Chevalier S, Pinault M, Bougnoux P, Labarthe F et al. Cardiolipin content is involved in liver mitochondrial energy wasting associated with cancer-induced cachexia without the involvement of adenine nucleotide translocase. Biochim Biophys Acta 2014; 1842: 726–733.
Dumas JF, Goupille C, Julienne CM, Pinault M, Chevalier S, Bougnoux P et al. Efficiency of oxidative phosphorylation in liver mitochondria is decreased in a rat model of peritoneal carcinosis. J Hepatol 2011; 54: 320–327.
Holroyde CP, Skutches CL, Boden G, Reichard GA . Glucose metabolism in cachectic patients with colorectal cancer. Cancer Res 1984; 44: 5910–5913.
Tisdale MJ . Mechanisms of cancer cachexia. Physiol Rev 2009; 89: 381–410.
George A, Reichard J, Nelson I, Moury F, Norman JR, Hochella J et al. Quantitative estimation of the cori cycle in the human. J Biol Chem 1963; 238: 495–501.
Holroyde CP, Gabuzda TG, Putnam RC, Paul P, Reichard GA . Altered glucose metabolism in metastatic carcinoma. Cancer Res 1975; 35: 3710–3714.
FENNINGER LD, MIDER GB . Energy and nitrogen metabolism in cancer. Adv Cancer Res 1954; 2: 229–253.
Berriel DM, Krones-Herzig A, Metzger D, Ziegler A, Vegiopoulos A, Klingenspor M et al. Nuclear receptor cofactor receptor interacting protein 140 controls hepatic triglyceride metabolism during wasting in mice. Hepatology 2008; 48: 782–791.
Martignoni ME, Dimitriu C, Bachmann J, Krakowski-Rosen H, Ketterer K, Kinscherf R et al. Liver macrophages contribute to pancreatic cancer-related cachexia. Oncol Rep 2009; 21: 363–369.
Jones A, Friedrich K, Rohm M, Schafer M, Algire C, Kulozik P et al. TSC22D4 is a molecular output of hepatic wasting metabolism. EMBO Mol Med 2013; 5: 294–308.
Sunny NE, Parks EJ, Browning JD, Burgess SC . Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab 2011; 14: 804–810.
Patra SK, Arora S . Integrative role of neuropeptides and cytokines in cancer anorexia-cachexia syndrome. Clin Chim Acta 2012; 413: 1025–1034.
Bologa RM, Levine DM, Parker TS, Cheigh JS, Serur D, Stenzel KH et al. Interleukin-6 predicts hypoalbuminemia, hypocholesterolemia, and mortality in hemodialysis patients. Am J Kidney Dis 1998; 32: 107–114.
Brenner DA, Buck M, Feitelberg SP, Chojkier M . Tumor necrosis factor-alpha inhibits albumin gene expression in a murine model of cachexia. J Clin Invest 1990; 85: 248–255.
Hennig B, Honchel R, Goldblum SE, McClain CJ . Tumor necrosis factor-mediated hypoalbuminemia in rabbits. J Nutr 1988; 118: 1586–1590.
Fearon KC, Falconer JS, Slater C, McMillan DC, Ross JA, Preston T . Albumin synthesis rates are not decreased in hypoalbuminemic cachectic cancer patients with an ongoing acute-phase protein response. Ann Surg 1998; 227: 249–254.
Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013; 497: 633–637.
Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res 2015; 75: 544–553.
Costa G, HOLLAND JF . Effects of Krebs-2 carcinoma on the lipide metabolism of male Swiss mice. Cancer Res 1962; 22: 1081–1083.
Das SK, Eder S, Schauer S, Diwoky C, Temmel H, Guertl B et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science 2011; 333: 233–238.
Shaw JH, Wolfe RR . Fatty acid and glycerol kinetics in septic patients and in patients with gastrointestinal cancer. The response to glucose infusion and parenteral feeding. Ann Surg 1987; 205: 368–376.
Das SK, Hoefler G . The role of triglyceride lipases in cancer associated cachexia. Trends Mol Med 2013; 19: 292–301.
Russell ST, Tisdale MJ . Mechanism of attenuation of skeletal muscle atrophy by zinc-alpha2-glycoprotein. Endocrinology 2010; 151: 4696–4704.
Kir S, White JP, Kleiner S, Kazak L, Cohen P, Baracos VE et al. Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature 2014; 513: 100–104.
Petruzzelli M, Schweiger M, Schreiber R, Campos-Olivas R, Tsoli M, Allen J et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab 2014; 20: 433–447.
Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM . A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998; 92: 829–839.
Stewart AF . Clinical practice. Hypercalcemia associated with cancer. N Engl J Med 2005; 352: 373–379.
Carracedo A, Cantley LC, Pandolfi PP . Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer 2013; 13: 227–232.
DeWys WD . Changes in taste sensation and feeding behaviour in cancer patients: a review. J Hum Nutr 1978; 32: 447–453.
Martinez-Sanchez N, Alvarez CV, Ferno J, Nogueiras R, Dieguez C, Lopez M . Hypothalamic effects of thyroid hormones on metabolism. Best Pract Res Clin Endocrinol Metab 2014; 28: 703–712.
Anand BK, Pillai RV . Activity of single neurones in the hypothalamic feeding centres: effect of gastric distension. J Physiol 1967; 192: 63–77.
Wang GJ, Tomasi D, Backus W, Wang R, Telang F, Geliebter A et al. Gastric distention activates satiety circuitry in the human brain. Neuroimage 2008; 39: 1824–1831.
Moran TH, Dailey MJ . Intestinal feedback signaling and satiety. Physiol Behav 2011; 105: 77–81.
Schwartz MW, Woods SC, Porte D Jr, Seeley RJ, Baskin DG . Central nervous system control of food intake. Nature 2000; 404: 661–671.
Laviano A, Russo M, Freda F, Rossi-Fanelli F . Neurochemical mechanisms for cancer anorexia. Nutrition 2002; 18: 100–105.
Otto B, Cuntz U, Fruehauf E, Wawarta R, Folwaczny C, Riepl RL et al. Weight gain decreases elevated plasma ghrelin concentrations of patients with anorexia nervosa. Eur J Endocrinol 2001; 145: 669–673.
Chance WT, Balasubramaniam A, Borchers M, Fischer JE . Refractory hypothalamic adenylate cyclase in anorectic tumor-bearing rats: implications for NPY-induced feeding. Brain Res 1995; 691: 180–184.
Chance WT, Xiao C, Dayal R, Sheriff S . Alteration of NPY and Y1 receptor in dorsomedial and ventromedial areas of hypothalamus in anorectic tumor-bearing rats. Peptides 2007; 28: 295–301.
Laviano A, Inui A, Marks DL, Meguid MM, Pichard C, Rossi FF et al. Neural control of the anorexia-cachexia syndrome. Am J Physiol Endocrinol Metab 2008; 295: E1000–E1008.
Silva VR, Micheletti TO, Pimentel GD, Katashima CK, Lenhare L, Morari J et al. Hypothalamic S1P/S1PR1 axis controls energy homeostasis. Nat Commun 2014; 5: 4859.
Laviano A, Meguid MM, Inui A, Muscaritoli M, Rossi-Fanelli F . Therapy insight: cancer anorexia-cachexia syndrome—when all you can eat is yourself. Nat Clin Pract Oncol 2005; 2: 158–165.
Cahlin C, Gelin J, Delbro D, Lonnroth C, Doi C, Lundholm K . Effect of cyclooxygenase and nitric oxide synthase inhibitors on tumor growth in mouse tumor models with and without cancer cachexia related to prostanoids. Cancer Res 2000; 60: 1742–1749.
Davis TW, Zweifel BS, O'Neal JM, Heuvelman DM, Abegg AL, Hendrich TO et al. Inhibition of cyclooxygenase-2 by celecoxib reverses tumor-induced wasting. J Pharmacol Exp Ther 2004; 308: 929–934.
Feng P, Jyotaki M, Kim A, Chai J, Simon N, Zhou M et al. Regulation of bitter taste responses by tumor necrosis factor. Brain Behav Immun 2015; 49: 32–42.
Kluger MJ, Rothenburg BA . Fever and reduced iron: their interaction as a host defense response to bacterial infection. Science 1979; 203: 374–376.
Cahlin C, Korner A, Axelsson H, Wang W, Lundholm K, Svanberg E . Experimental cancer cachexia: the role of host-derived cytokines interleukin (IL)-6, IL-12, interferon-gamma, and tumor necrosis factor alpha evaluated in gene knockout, tumor-bearing mice on C57 Bl background and eicosanoid-dependent cachexia. Cancer Res 2000; 60: 5488–5493.
Bing C, Taylor S, Tisdale MJ, Williams G . Cachexia in MAC16 adenocarcinoma: suppression of hunger despite normal regulation of leptin, insulin and hypothalamic neuropeptide Y. J Neurochem 2001; 79: 1004–1012.
Maceyka M, Harikumar KB, Milstien S, Spiegel S . Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol 2012; 22: 50–60.
Williams KW, Elmquist JK . From neuroanatomy to behavior: central integration of peripheral signals regulating feeding behavior. Nat Neurosci 2012; 15: 1350–1355.
Grossberg AJ, Scarlett JM, Marks DL . Hypothalamic mechanisms in cachexia. Physiol Behav 2010; 100: 478–489.
Tchekmedyian NS, Tait N, Moody M, Greco FA, Aisner J . Appetite stimulation with megestrol acetate in cachectic cancer patients. Semin Oncol 1986; 13 (Suppl 4): 37–43.
Tayek JA . A review of cancer cachexia and abnormal glucose metabolism in humans with cancer. J Am Coll Nutr 1992; 11: 445–456.
Bruning PF, Bonfrer JM, van Noord PA, Hart AA, de Jong-Bakker M, Nooijen WJ . Insulin resistance and breast-cancer risk. Int J Cancer 1992; 52: 511–516.
Renehan AG, Frystyk J, Flyvbjerg A . Obesity and cancer risk: the role of the insulin-IGF axis. Trends Endocrinol Metab 2006; 17: 328–336.
G AS, Rawson RW . Diabetes and altered carbohydrate metabolism in patients with cancer. Cancer 1956; 9: 1127–1134.
Honors MA, Kinzig KP . The role of insulin resistance in the development of muscle wasting during cancer cachexia. J Cachexia Sarcopenia Muscle 2012; 3: 5–11.
Parekh N, Lin Y, Hayes RB, Albu JB, Lu-Yao GL . Longitudinal associations of blood markers of insulin and glucose metabolism and cancer mortality in the third National Health and Nutrition Examination Survey. Cancer Causes Control 2010; 21: 631–642.
Fernandes LC, Machado UF, Nogueira CR, Carpinelli AR, Curi R . Insulin secretion in Walker 256 tumor cachexia. Am J Physiol 1990; 258: E1033–E1036.
Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM . Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci USA 1994; 91: 4854–4858.
Brooks PC, Klemke RL, Schon S, Lewis JM, Schwartz MA, Cheresh DA . Insulin-like growth factor receptor cooperates with integrin alpha v beta 5 to promote tumor cell dissemination in vivo. J Clin Invest 1997; 99: 1390–1398.
Massoner P, Ladurner-Rennau M, Eder IE, Klocker H . Insulin-like growth factors and insulin control a multifunctional signalling network of significant importance in cancer. Br J Cancer 2010; 103: 1479–1484.
Cullen KJ, Yee D, Sly WS, Perdue J, Hampton B, Lippman ME et al. Insulin-like growth factor receptor expression and function in human breast cancer. Cancer Res 1990; 50: 48–53.
Maloney EK, McLaughlin JL, Dagdigian NE, Garrett LM, Connors KM, Zhou XM et al. An anti-insulin-like growth factor I receptor antibody that is a potent inhibitor of cancer cell proliferation. Cancer Res 2003; 63: 5073–5083.
Gelfand RA, Barrett EJ . Effect of physiologic hyperinsulinemia on skeletal muscle protein synthesis and breakdown in man. J Clin Invest 1987; 80: 1–6.
Lecker SH, Solomon V, Price SR, Kwon YT, Mitch WE, Goldberg AL . Ubiquitin conjugation by the N-end rule pathway and mRNAs for its components increase in muscles of diabetic rats. J Clin Invest 1999; 104: 1411–1420.
Hanson RW, Reshef L . Regulation of phosphoenolpyruvate carboxykinase (GTP) gene expression. Annu Rev Biochem 1997; 66: 581–611.
Asp ML, Tian M, Kliewer KL, Belury MA . Rosiglitazone delayed weight loss and anorexia while attenuating adipose depletion in mice with cancer cachexia. Cancer Biol Ther 2011; 12: 957–965.
Lundholm K, Korner U, Gunnebo L, Sixt-Ammilon P, Fouladiun M, Daneryd P et al. Insulin treatment in cancer cachexia: effects on survival, metabolism, and physical functioning. Clin Cancer Res 2007; 13: 2699–2706.
Bartlett DL, Charland SL, Torosian MH . Reversal of tumor-associated hyperglucagonemia as treatment for cancer cachexia. Surgery 1995; 118: 87–97.
Hartl WH, Demmelmair H, Jauch KW, Koletzko B, Schildberg FW . Effect of glucagon on protein synthesis in human rectal cancer in situ. Ann Surg 1998; 227: 390–397.
Inculet RI, Peacock JL, Gorschboth CM, Norton JA . Gluconeogenesis in the tumor-influenced rat hepatocyte: importance of tumor burden, lactate, insulin, and glucagon. J Natl Cancer Inst 1987; 79: 1039–1046.
Puppa MJ, White JP, Sato S, Cairns M, Baynes JW, Carson JA . Gut barrier dysfunction in the Apc(Min/+) mouse model of colon cancer cachexia. Biochim Biophys Acta 2011; 1812: 1601–1606.
Klein GL, Petschow BW, Shaw AL, Weaver E . Gut barrier dysfunction and microbial translocation in cancer cachexia: a new therapeutic target. Curr Opin Support Palliat Care 2013; 7: 361–367.
Bindels LB, Delzenne NM . Muscle wasting: the gut microbiota as a new therapeutic target? Int J Biochem Cell Biol 2013; 45: 2186–2190.
Cani PD . Gut microbiota and obesity: lessons from the microbiome. Brief Funct Genomics 2013; 12: 381–387.
Bindels LB, Neyrinck AM, Salazar N, Taminiau B, Druart C, Muccioli GG et al. Non digestible oligosaccharides modulate the gut microbiota to control the development of leukemia and associated cachexia in mice. PLoS ONE 2015; 10: e0131009.
Bindels LB, Porporato P, Dewulf EM, Verrax J, Neyrinck AM, Martin JC et al. Gut microbiota-derived propionate reduces cancer cell proliferation in the liver. Br J Cancer 2012; 107: 1337–1344.
Bindels LB, Beck R, Schakman O, Martin JC, De BF, Sohet FM et al. Restoring specific lactobacilli levels decreases inflammation and muscle atrophy markers in an acute leukemia mouse model. PLoS ONE 2012; 7: e37971.
Heppner KM, Piechowski CL, Muller A, Ottaway N, Sisley S, Smiley DL et al. Both acyl and des-acyl ghrelin regulate adiposity and glucose metabolism via central nervous system ghrelin receptors. Diabetes 2014; 63: 122–131.
St-Pierre DH, Karelis AD, Cianflone K, Conus F, Mignault D, Rabasa-Lhoret R et al. Relationship between ghrelin and energy expenditure in healthy young women. J Clin Endocrinol Metab 2004; 89: 5993–5997.
Porporato PE, Filigheddu N, Reano S, Ferrara M, Angelino E, Gnocchi VF et al. Acylated and unacylated ghrelin impair skeletal muscle atrophy in mice. J Clin Invest 2013; 123: 611–622.
Ruozi G, Bortolotti F, Falcione A, Dal Ferro M, Ukovich L, Macedo A et al. AAV-mediated in vivo functional selection of tissue-protective factors against ischaemia. Nat Commun 2015; 6: 7388.
Filigheddu N, Gnocchi VF, Coscia M, Cappelli M, Porporato PE, Taulli R et al. Ghrelin and des-acyl ghrelin promote differentiation and fusion of C2C12 skeletal muscle cells. Mol Biol Cell 2007; 18: 986–994.
Garcia JM, Boccia RV, Graham CD, Yan Y, Duus EM, Allen S et al. Anamorelin for patients with cancer cachexia: an integrated analysis of two phase 2, randomised, placebo-controlled, double-blind trials. Lancet Oncol 2015; 16: 108–116.
Al-Zoughbi W, Huang J, Paramasivan GS, Till H, Pichler M, Guertl-Lackner B et al. Tumor macroenvironment and metabolism. Semin Oncol 2014; 41: 281–295.
Mayers JR, Wu C, Clish CB, Kraft P, Torrence ME, Fiske BP et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat Med 2014; 20: 1193–1198.
Hanahan D, Weinberg RA . Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674.
Carmeliet P, Jain RK . Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov 2011; 10: 417–427.
Porporato PE, Dhup S, Dadhich RK, Copetti T, Sonveaux P . Anticancer targets in the glycolytic metabolism of tumors: a comprehensive review. Front Pharmacol 2011; 2: 49.
De Saedeleer CJ, Porporato PE, Copetti T, Perez-Escuredo J, Payen VL, Brisson L et al. Glucose deprivation increases monocarboxylate transporter 1 (MCT1) expression and MCT1-dependent tumor cell migration. Oncogene 2014; 33: 4060–4068.
Lewis CA, Brault C, Peck B, Bensaad K, Griffiths B, Mitter R et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 2015; 34: 5128–5140.
Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med 2011; 17: 1498–1503.
Tonjes M, Barbus S, Park YJ, Wang W, Schlotter M, Lindroth AM et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat Med 2013; 19: 901–908.
Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013; 496: 101–105.
Dang CV . MYC on the path to cancer. Cell 2012; 149: 22–35.
Maddocks OD, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E et al. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 2013; 493: 542–546.
Luo Y, Yoneda J, Ohmori H, Sasaki T, Shimbo K, Eto S et al. Cancer usurps skeletal muscle as an energy repository. Cancer Res 2014; 74: 330–340.
Taguchi A, Blood DC, del TG, Canet A, Lee DC, Qu W et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature 2000; 405: 354–360.
Yan HX, Wu HP, Zhang HL, Ashton C, Tong C, Wu H et al. p53 promotes inflammation-associated hepatocarcinogenesis by inducing HMGB1 release. J Hepatol 2013; 59: 762–768.
Tabata C, Shibata E, Tabata R, Kanemura S, Mikami K, Nogi Y et al. Serum HMGB1 as a prognostic marker for malignant pleural mesothelioma. BMC Cancer 2013; 13: 205.
Theologides A . Cancer cachexia. Cancer 1979; 43 (Suppl 5): 2004–2012.
Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 2011; 12: 489–495.
Acknowledgements
PEP is a Fonds National de la Recherche Scientifique (F.R.S.-FNRS) Postdoctoral Fellow. Work at the author’s lab is supported by a Starting Grant from the European Research Council (ERC No. 243188 TUMETABO), Interuniversity Attraction Pole (IAP) grant #UP7-03 from the Belgian Science Policy Office (Belspo), an Action de Recherche Concertée from the Communauté Française de Belgique (ARC 14/19-058), the Belgian Fonds National de la Recherche Scientifique (F.R.S.-FNRS), the Télévie, the Belgian Fondation contre le Cancer (2012–186), the Louvain Foundation and the UCL Fonds Spéciaux de la Recherche (FSR). I sincerely thank Dr. Nicoletta Filigheddu and Vincent F. Van Hée for helpful discussions. Figure 1 of this manuscript was partially adapted from Servier Medical Art.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declares no conflict of interest.
Rights and permissions
Oncogenesis is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Porporato, P. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis 5, e200 (2016). https://doi.org/10.1038/oncsis.2016.3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/oncsis.2016.3
This article is cited by
-
Virtual reality-empowered deep-learning analysis of brain cells
Nature Methods (2024)
-
Photobiomodulation therapy moderates cancer cachexia-associated muscle wasting through activating PI3K/AKT/FoxO3a pathway
Apoptosis (2024)
-
Cancer cachexia: molecular mechanisms and treatment strategies
Journal of Hematology & Oncology (2023)
-
Blood biomarkers as potential malnutrition screening alternatives among adult patients with cancer on treatment in oncology unit of jimma tertiary hospital: A cross-sectional analysis
BMC Nutrition (2023)
-
Highlighting the idea of exerkines in the management of cancer patients with cachexia: novel insights and a critical review
BMC Cancer (2023)
