
Abstract
Melanoma is curable when it is at an early phase but is lethal once it becomes metastatic. The recent development of BRAFV600E inhibitors (BIs) showed great promise in treating metastatic melanoma, but resistance developed quickly in the treated patients, and these inhibitors are not effective on melanomas that express wild-type BRAF. Alternative therapeutic strategies for metastatic melanoma are urgently needed. Here we report that ERBB3, a member of the epidermal growth factor receptor family, is required for the formation of lung metastasis from both the BI-sensitive melanoma cell line, MA-2, and the BI-resistant melanoma cell line, 451Lu-R. Further analyses revealed that ERBB3 does not affect the initial seeding of melanoma cells in lung but is required for their further development into overt metastases, indicating that ERBB3 might be essential for the survival of melanoma cells after they reach the lung. Consistent with this, the ERBB3 ligand, NRG1, is highly expressed in mouse lungs and induces ERBB3-depdnent phosphorylation of AKT in both MA-2 and 451Lu-R cells in vitro. These findings suggest that ERBB3 may serve as a target for treating metastatic melanomas that are resistant to BIs. In support of this, administration of the pan-ERBB inhibitor, canertinib, significantly suppresses the metastasis formation of BI-resistant melanoma cell lines.
Similar content being viewed by others


Introduction
Melanoma is one of the most aggressive cancer types. It accounts for <5% of all skin cancer incidences but results in >75% of skin cancer-related deaths.1 Melanoma arises from melanocytes, the pigment-producing cells residing in the basal layer of the epidermis. If detected at early stages, melanoma is curable with a 5-year survival rate of >97%. Once it becomes metastatic, however, it is highly aggressive, and the 5-year survival rate of patients drops to ∼15%.1 The regulation of melanoma metastasis is poorly understood.
Almost 60% of melanomas express a constitutively active mutant BRAF (mostly V600E),2 which leads to persistent activation of the MAPK/ERK (mitogen-activated protein kinase/extracellular signal–regulated kinase) pathway. Recently an FDA (Food and Drug Administation)-approved inhibitor of BRAFV600E (BI), vemurafenib, showed impressive benefits for late-stage melanoma patients.3 Nevertheless, regression typically lasted only a few months and was quickly overtaken by relapse owing to the development of drug resistance.4 Furthermore, these inhibitors are not effective on melanomas that express wild-type BRAF (BRAFWT). Alternative or combinatory therapeutic strategies have been proposed, and some are in clinical trials,5, 6 but their efficacies are far from being satisfactory. A greater understanding of melanoma biology and metastasis will significantly accelerate the advancement of this front.
Metastasis is the spread of cancer cells to distant organs and accounts for >90% of death in cancer patients.7 Cancer cells must complete at least five steps to successfully metastasize: (1) their detachment from the primary tumor, (2) intravasation into the circulation, (3) survive the circulation, (4) extravasation (exit) from the circulation, and (5) survival/growth in a distant organ. The last step is considered one of the rate-limiting steps during metastasis. Clinically detectable metastases are relatively rare compared with the large number of tumor cells circulating in cancer patients.8 Moreover, it is known that metastatic cells can remain dormant in distant organs for decades before giving rise to detectable metastases.9 Consequently, understanding how this step is regulated may provide opportunities for effective control and treatment of metastasis.
A widely used method to study the survival and growth of metastatic cancer cells is the experimental metastasis assay. In this assay, metastatic cancer cells are directly injected into the circulation of immunodeficient mice and form metastases in various tissues, most frequently the lung.10, 11 Cells with increased metastatic potentials could then be selected, and genes differentially regulated in these cells could be identified via gene expression profiling. We used this method to study the mechanisms of metastasis in melanoma. A series of highly metastatic melanoma cell lines were derived from a poorly metastatic human parental line, A375P.12, 13 Bioinformatic analyses revealed a gene signature whose high expression levels correlate with lower survival among melanoma patients carrying metastases.13 A significant number of genes in this signature encode either transmembrane receptors or extracellular proteins, which are more accessible to therapeutic agents than cytosolic proteins and thus have a higher therapeutic potential.
We analyzed the function of one of these secreted proteins, ERBB3, in melanoma metastasis. ERBB3 is an epidermal growth factor (EGF) receptor (EGFR) family member and binds to EGF-like growth factors, neuregulins (NRGs), with high affinity.14 It does not have a functional kinase domain and needs to form heterodimers with other EGFR family members (EGFR, ERBB2 or ERBB4) to transmit signals.15 Distinct from ERBB1 and ERBB2, ERBB3 (and ERBB4) directly binds to the p85 subunit of phosphatidylinositol 3′-kinase (PI3K) and is thought to signal predominantly via the PI3K-AKT pathway to promote cell survival.16, 17, 18 It has been implicated in promoting the progression of multiple cancer types, and the ERBB3 inhibitors showed tumor-inhibitory effects in animal models.19, 20, 21, 22 In melanoma, upregulation of ERBB3 correlated with melanoma malignancy.20 Its downregulation by short hairpin RNA (shRNA) or inhibition by small inhibitors led to a reduction in melanoma cell proliferation or subcutaneous melanoma growth.22, 23, 24, 25, 26 Nevertheless, how ERBB3 regulates melanoma metastasis and whether its inhibition could serve as a therapeutic approach for treating metastatic melanoma are not clear.
We report here that high expression levels of ERBB3 correlate significantly with a poor survival of melanoma patients carrying metastases. Furthermore, knocking down of ERBB3 led to a reduced metastasis formation not only from the BI-sensitive human melanoma cell line, MA-2, but also from the BI-resistant human melanoma cell line, 451Lu-R. To understand the mechanisms by which ERBB3 regulates melanoma metastasis, we performed time course analyses and found that ERBB3 did not affect the initial seeding of the melanoma cells in the lung but was essential for their subsequent survival and proliferation to form overt metastases. We also investigated whether the function of ERBB3 in melanoma metastasis is mediated by its ligand, NRG1. We found that the Nrg1 mRNA was highly expressed in the mouse lungs and the NRG1 protein induced ERBB3-dependent phosphorylation of AKT in vitro in the MA-2 and 451Lu-R cell lines, as well as in three other melanoma cell lines. Taken together, we propose that the NRG1/ERBB3 signaling axis is essential for metastasis formation of melanoma cells in the lung and may be targeted alone or in combination with BIs to treat patients with metastatic melanoma. Consistent with this, administration of a pan-ERBB inhibitor, canertinib, led to a significant reduction in metastasis formation from the BI-resistant melanoma cell lines, 451Lu-R and MeWo.
Results
Expression levels of ERBB3 mRNA correlate with a poor survival in patients carrying melanoma metastases
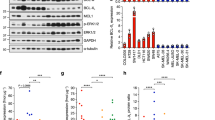
ERBB3 was found upregulated in melanoma cells with high metastatic potentials, but whether this upregulation affects patient survival has not been studied. To address this, we divided the metastases-carrying melanoma patients in our previous study5 into two groups: group 1 expresses the ERBB3 mRNA at levels higher than the median, and group 2 expresses the ERBB3 mRNA at levels lower than the median. Kaplan–Meier survival analyses were performed and showed a significant decrease in the survival of patients from group 1 (Figure 1), indicating that high levels of ERBB3 expression correlate positively to melanoma malignancy.

ERBB3 contributes to poor survival of melanoma patients with metastases. Fifty human metastatic melanomas were separated into two groups based on their ERBB3 expression levels. The survival probability of patients expressing high levels of ERBB3 (medium survival=5 months) was significantly lower than those expressing low levels of ERBB3 (medium survival=28 months).
Knocking down of ERBB3 led to a reduction in metastasis formation from MA-2 and 451Lu-R melanoma cell lines
The effects of ERBB3 on melanoma metastasis were examined in two melanoma cell lines, MA-2 and 451Lu-R. The MA-2 cell line is a metastatic derivative of A375P,27 which was reported to express BRAFV600E and is sensitive to inhibition by BRAFV600E inhibitors (BIs).28 The 451Lu-R cell line was also reported to express BRAFV600E, but it was selected in vitro in the presence of BIs and has gained BI resistance.29 We confirmed the BRAFV600E mutation status of the two cell lines by sequence analyses (Table 1). The 451Lu-R cells were cultured routinely in the presence of SB 590885, a BRAFV600E inhibitor, to maintain their BI resistance.
ERBB3 was knocked down in both cell lines via lentiviral expression of shRNAs. The knockdown cells express significantly lower levels of ERBB3 mRNA and protein (Figures 2a and c, 5a and b). These cells, along with control cells expressing the shRNA against green fluorescent protein (GFP), were injected intravenously into the immunodeficient NSG mice. Lungs were harvested, and metastases counted. The knockdowns of ERBB3 resulted in a significant reduction in the number of lung metastases from both cell lines (Figures 2b and d), suggesting that ERBB3 is required for the metastasis formation of human melanoma cells and this effect may be independent of the sensitivity of melanoma cells to BIs.

ERBB3 is required for the metastasis formation of melanoma cell lines in lung. (a, c). ERBB3 was knocked down by two different shRNAs in MA-2 cells (a) or 451Lu-R cells (c). The efficiency of knockdown was measured by qRT–PCR. (b, d). ERBB3 knockdown led to a significant decrease in the number of lung metastases from MA-2 cells (b) or 451Lu-R cells (d). Student’s t-test, ***P<0.001.
ERBB3 does not affect the initial seeding of MA-2 and 451Lu-R cells in the mouse lungs but is essential for their persistence at later stages
The formation of overt metastases from circulating tumor cells is thought to go through several steps (Figure 3a): they need to seed the target tissue (for example, lung), survive and then proliferate into overt metastases. We asked at which step ERBB3 may exert its effects on the metastasis formation of melanoma cells. MA-2 cells expressing control or ERBB3 shRNAs were labeled with 5-chloromethylfluorescein diacetate (CMFDA), an intense green fluorescent Cell Tracker dye, before intravenous injections into NSG mice. Lungs were harvested shortly (3, 6 or 12 h) after injections, sectioned and visualized under microscope (see Materials and methods). Green fluorescent cells were readily detectable in the lungs at all time points (Figure 3b, white arrows). No difference in the cell number was observed between control- and ERBB3-knockdown MA-2 cells (Figure 3c), suggesting that ERBB3 does not affect the initial seeding of MA-2 cells in the lung (Figure 3a; step 1). We then examined whether ERBB3 affects the survival of MA-2 cells in the lung at later time points. Lungs were harvested 24 h after the injections, and the number of green fluorescent cells on each lung section was counted (Figure 3b). A significant reduction of labeled cells was observed when ERBB3 was knocked down (Figure 3d), suggesting that ERBB3 might be required for the survival of MA-2 cells after their seeding in the lung.

ERBB3 affects the survival of MA-2 cells in the lung at 24 h after injections. (a) Schematic presentation of the predicted processes for CTCs (circulating tumor cells) to form overt metastases. (b) Representative images of the mouse lungs at 3, 6, 12 or 24 h after intravenous injections of MA-2(shGFP), MA-2(shERBB3#1) or MA-2(shERBB3#2) cells. Green: metastatic melanoma cells (white arrows). Red: autofluorescence of the lung. Blue: DAPI for nuclei. (c) ERBB3 knockdown did not affect the number of fluorescent cells in the lung at 3, 6 or 12 h after tail vein injections. In all, 2–3 mice were used under each condition. NS: not significant. (d) Twenty-four hours after injections, the number of MA-2 cells in the lung was significantly reduced upon ERBB3 knockdown. **P<0.01; ***P<0.001.
We performed similar analyses in 451Lu-R cells, except that the 451Lu-R cells were not labeled with CMFDA before injections. Mouse lungs were collected at different time points after injections, sectioned and stained with a human-specific anti-vimentin antibody to detect the human melanoma cells (Figure 4a, black arrows; see Materials and methods). In contrast to the MA-2 cells, knocking down of ERBB3 did not lead to any reduction of 451Lu-R cells in the mouse lung 24 h after injections (Figure 4b), arguing that ERBB3 does not affect the seeding of 451Lu-R cells and their initial survival in the lung. The metastasis formation from these cells was then traced over a course of 3 weeks. No difference in metastasis formation was observed at the 1-week time point (Figure 4b), but at the 3-week time point, a significant reduction of metastases was observed in ERBB3-knockdown cells (Figure 4b). These findings suggest that, although ERBB3 might not be required for the survival of 451Lu-R cells immediately after their seeding in the lung, it is required for their persistent survival and/or proliferation later on to form overt metastases (Figure 3a; steps 2 and 3).

ERBB3 affects the formation of 451Lu-R metastases in the lung at later time points. ERBB3 knockdown did not reduce the number of 451Lu-R cells in the lungs at 24 h or 1 week after injections but led to a significant reduction in lung metastases at the 3-week time point. (a) Representative images of lung sections at 24 h, 1 week or 3 weeks after intravenous injections of 451Lu-R(shGFP), 451Lu-R(shERBB3#1) and 451Lu-R(shERBB3#2) cells. (b) Quantitation of lung metastases at each time point. NS: not significant. *P<0.05. **P<0.01.
The ERBB3 ligand, NRG1, is highly expressed in the mouse lungs and induces ERBB3-dependent AKT phosphorylation in MA-2 and 451Lu-R cells
As mentioned earlier, ERBB3 is thought to predominantly signal through the PI3K/AKT pathway to regulate cancer cell survival.16, 17, 18 A high-affinity ligand for ERBB3 is neuregulin, NRG1.30 It activates ERBB3 to induce AKT phosphorylation and has been shown to promote melanoma growth.31 We predicted that ERBB3 promotes melanoma metastasis formation via the NRG1-ERBB3-AKT survival pathway. To test this, serum-starved MA-2 cells expressing control shRNA or ERBB3 shRNA were stimulated with 500 ng/ml NRG1 for 15, 30 or 60 min. Robust induction of phospho-ERBB3 and phospho-AKT was observed in the control cells but not in the ERBB3-knockdown cells (Figure 5a). Similar impairment of ERBB3 and AKT phosphorylation was observed in 451Lu-BR cells expressing ERBB3 shRNAs (Figure 5b). These results demonstrate that NRG1 induces ERBB3-dependent AKT activation in MA-2 and 451Lu-R cells in vitro.

NRG1 induces ERBB3-dependent AKT phosphorylation in MA-2 and 451Lu-R cells in vitro. (a, b) MA-2 (a) and 451Lu-R (b) cells were stimulated with NRG1 for 0, 15, 30, 60 or 120 min. Cells were lysed and probed for phospho-ERBB3, total ERBB3, phospho-AKT or total AKT. γ-tubulin was used as a loading control. (c) qRT–PCR analyses of Nrg1 mRNA in the mouse lungs and in MA-2 or 451Lu-R cells. Three independent experiments were performed and graphed. (d) A panel of melanoma cell lines were stimulated with NRG1 for 0, 15 or 30 min. Cells were lysed and probed for phospho-ERBB3, total ERBB3, phospho-AKT or total AKT (top panel). γ-tubulin was used as a loading control. Band intensities were quantified by the ImageJ software (bottom panel).
Both lung tissues and melanoma cells have been reported to express NRG1,31, 32 so the NRG1/ERBB3 signaling could regulate melanoma metastasis through paracrine and/or autocrine mechanisms. To investigate which mechanism might apply to metastasis of MA-2 and 451Lu-R cells, quantitative reverse transciptase–PCR (qRT–PCR) was performed to determine the relative abundance of NRG1 mRNA in the mouse lungs and in the human melanoma cell lines. As shown in Figure 5c, NRG1 mRNA was indeed expressed in the mouse lungs and in both melanoma cell lines, but its level in the mouse lungs was approximately 40-fold higher than that in either melanoma cell line. Assuming that the differential expression of NRG1 mRNA results in a similar differential expression of NRG1 protein, the majority of NRG1 protein to support metastasis formation of MA-2 and 451Lu-R cells would be provided by the lung parenchyma, and, as such, would regulate their survival and growth via a paracrine mechanism. In the absence of ERBB3, this paracrine mechanism is disrupted, leading to a failure of the MA-2 and 451Lu-R cells to form lung metastases.
The high levels of NRG1 in the lung and responses of both MA-2 and 451Lu-R cells to NRG1 stimulation (Figures 5a–c) suggest that NRG1-ERBB3 signaling may be a general mechanism for lung metastasis of melanoma cells. To test this, we investigated the expression levels of ERBB3 and its activity upon NRG1 stimulation in a panel of metastatic melanoma cell lines, including MA-2, WM266-4, 451Lu-R, SK-MEL-2 and MeWo (Table 1). These cell lines carry either the constitutively active BRAF mutant (MA-2, WM266-4, 451Lu-R) or BRAFWT (SK-MEL-2 and MeWo)29, 33, 34 and are sensitive (MA-2 and WM266-4) or resistant (451Lu-R, SK-MEL-2 and MeWo) to BI inhibition28(Table 1). The levels of total ERBB3 vary slightly among the cell lines (Figure 5d), with 451Lu-R expressing the highest and MeWo expressing the lowest (Figure 5d, bottom panel). Upon stimulation by 500 ng/ml NRG1 for 15 or 30 min, phosphorylation of ERBB3 was induced in all the cell lines (Figure 5d, top panel), but the induction at 15 min (relative to the basal level at 0 min) was significantly higher in SK-MEL-2 and MeWo cell lines, the two cell lines that express BRAFWT, and the lowest in 451Lu-R cells (Figure 5d, bottom panel). Phosphorylation of AKT was also induced in all the cell lines upon NRG1 stimulation (Figure 5d, top panel), but the induction at 15 min (relative to the basal level at 0 min) in SK-MEL-2 and MeWo cell lines did not differ significantly from the MA-2 cells and was slightly lower than that of the WM266-4 cells (Figure 5d, bottom panel). Perhaps ERBB3 is more responsive to NRG1 stimulation in the BRAFWT-expressing melanoma cell lines than those expressing BRAFV600E, but only a portion of this elevated ERBB3 activity is translated into AKT activation. ERBB3 activates the MAPK/ERK pathway in addition to the PI3K/AKT pathway. This input of ERK activation from ERBB3 may be more essential and abundant for cell lines expressing BRAFWT than for those expressing BRAFV600E, which activates ERK constitutively. The induction of phospho-AKT in 451Lu-R cells was the lowest (Figure 5d, bottom panel), similar to the induction of phospho-ERBB3, suggesting that ERBB3 activation may be reduced in these cells. This is in contrast to the recent report that ERBB3 was upregulated and hyperactivated in melanoma cells treated with BIs.26 In that report, BI treatment was on cells that are sensitive to BIs and was for short term.26 The 451Lu-R cells used in this study have established resistance to BIs and were cultured routinely in the presence of BI. Perhaps, upon short-term BI treatment, ERBB3 is upregulated in the BI-sensitive cells to overcome apoptosis, but this is no longer needed when cells have already established BI resistance.
The pan-ERBB inhibitor, canertinib, suppresses metastasis formation of BI-resistant 451Lu-R and MeWo cells in vivo
Our above findings imply that ERBB3 might be a therapeutic target for melanoma metastasis, particularly those resistant to BIs. ERBB3 does not have a functional kinase domain at its C-terminus, so its signaling relies on dimerization with other ERBB members. To maximize the effects on ERBB3 inhibition, we obtained the irreversible pan-ERBB inhibitor, canertinib,35, 36 for our study. Two BI-resistant melanoma cell lines, 451Lu-R and MeWo, were injected intravenously into NSG mice. Once the metastases were established, the injected mice were treated daily with canertinib, or vehicle, for a few days and then left off drug for an additional period of time before being euthanized for analyses (see Materials and methods). Their lungs were harvested, sectioned and stained with the anti-vimentin antibody to score metastases. Canertinib treatment led to a significant reduction in metastasis formation from both 451Lu-R and MeWo cells (Figures 6a and b). We also assessed the potential of canertinib on prolonging the survival of metastasis-bearing mice. A small group of mice carrying 451Lu-R metastases were kept for 64 days after the start of treatment. The vehicle-treated mice (n=3) died on day 46, 49 and 64, whereas two of the three canertinib-treated mice were still alive on day 64 and one died on day 58. Although the sample size in each group is too small to allow a reliable statistical calculation, canertinib-treated mice appeared to have prolonged survival relative to the vehicle-treated mice (Figure 6c), suggesting that pan-ERBB inhibitors may have therapeutic effects on treating BI-resistant metastatic melanoma.

The pan-ERBB inhibitor, canertinib, inhibits metastasis formation of BI-resistant melanoma cells. (a, b) Canertinib treatment led to a reduction in metastases from 451Lu-R cells (a) and MeWo cells (b). Left: human-specific vimentin staining of the mouse lung sections treated with vehicle or canertinib. Metastases are in red and indicated by black arrows. Right: the number of lung metastases from canertinib- or vehicle-treated mice. *P<0.05; **P<0.01. (c) Survival probability of 451Lu-R metastases-bearing mice, treated with canertinib or vehicle.
Discussion
The roles of ERBB3 in cancer are extensively studied and have been shown in many cancer types to regulate cell survival and proliferation through the AKT and ERK pathways.19, 20, 21, 22, 30 Anti-ERBB3 therapies have shown beneficial effects on cancer patients.37, 38 Nevertheless, these studies have mostly focused on the effects of ERBB3 on the growth of primary tumors. How ERBB3 regulates metastasis is poorly understood. We show here that ERBB3 is required for the metastasis formation of two different melanoma cell lines, MA-2 and 451Lu-R. Both cell lines express the BRAFV600E mutant, but only the MA-2 line is sensitive to BIs; the 451Lu-R line has gained resistance via in vitro selections. The fact that ERBB3 is required for the formation of metastasis from both lines indicates that it might serve as a more versatile therapeutic target for treating melanomas. Our proof-of-principle study using the irreversible pan-ERBB inhibitor supports this notion.
The observed effects of ERBB3 on metastasis formation of melanoma cells are consistent with its known function in regulating cell survival and proliferation. It receives signals from NRG1, one of the high-affinity ligands for ERBB3 that has been shown to promote melanoma growth,14, 31 and activate AKT and induce downstream survival signals.31, 39 The effects of NRG1 are likely through a paracrine mechanism, as the mouse lungs express a significant higher level of NRG1 mRNA than either the MA-2 or the 451Lu-R cell line. This predicted dominance of paracrine regulation over the autocrine regulation by NRG1/ERBB3 signaling may explain the lack of effects of ERBB3 on lung seeding of MA-2 and 451Lu-R cells. Successful lung seeding requires the survival of melanoma cells in circulation, but little paracrine NRG1 would be available during this process, and as such, the contribution from NRG1/ERBB3 signaling would be minimal.
The kinetics of ERBB3 effects during the course of metastasis formation appears to differ between the two cell lines. In the case of MA-2 cells, ERBB3 was required for their survival almost immediately after seeding in the lung (within 24 h after injections). In the case of 451Lu-R cells, however, the apparent effects of ERBB3 were delayed (>1 week after seeding). Perhaps other survival mechanisms exist in 451lu-R cells to compensate for the loss of ERBB3 at the early stage of metastasis, but these mechanisms are absent in MA-2 cells. It was reported that the IGF-1R (insulin-like growth factor 1 receptor)/PI3K/AKT signaling is elevated in 451lu-R cells, relative to the BI-sensitive parental line.29 It is possible that this elevated IGF-1R/PI3K/AKT signaling and/or other anti-apoptosis mechanisms enable the short-term survival of 451Lu-R cells after they reach the lung, even in the absence of ERBB3. Their long-term survival in the lung, however, requires persistent signaling through ERBB3, resulting in the reduction of lung metastases at later time points when ERBB3 is knocked down.
ERBB3 does not have a functional kinase domain at its cytoplasmic tail and so relies on dimerization with other ERBB members to signal.15 In melanomas and other cancer types, ERBB3 is thought to preferentially dimerize with ERBB2 to promote cell survival, migration and proliferation.23, 40 Consequently, both ERBB3-specific and pan-ERBB inhibitors were shown to effectively curb the progression of melanoma.25, 37, 38 Most of these studies investigate the effects of ERBB3 on primary tumor growth but not metastasis. Our results suggest that ERBB3 inhibition may block the metastatic growth of melanoma cells, and this suppressive role appears to be effective regardless of their sensitivity to BIs. Consequently, combining ERBB3 inhibitors and BIs might have improved efficacy in treating metastatic melanomas, compared with BI treatment only. In support of this, activation of ERBB3 has been shown to be an early response of melanoma cells to Raf/MEK inhibition24 and knockdowns of ERBB3 resulted in a more pronounced reduction in subcutaneous melanoma growth in response to PLX4302 (vemurafenib), the BI currently in use in clinic for treating metastatic melanoma.26
Materials and methods
Cell lines and reagents
The 451Lu-R and WM266-4 cell lines were received from Dr Meenhard Herlyn (Wistar Institute, Philadelphia, PA, USA). The MeWo and SK-MEL-2 cell lines were purchased from ATCC (HTB-65 and HTB-68, Manassas, VA, USA). 451Lu-R cells29 were cultured in Dulbecco’s Modified Eagle Medium (DMEM) with 5% fetal bovine serum, glutamax and penicillin/streptomycin and 1 μM SB 590885 (Tocris Bioscience, Bristol, UK). WM266-4, MeWo and SK-MEL-2 cells were cultured in Minimum Essential Medium with Earles Salt, 10% fetal bovine serum, non-essential amino acid, sodium pyruvate, glutamax and penicillin/streptomycin. The MA-2 cell line was derived from A375 cells (ATCC No. CRL-1619)13 and cultured in DMEM with 10% fetal bovine serum, glutamax and penicillin/streptamycin. The V600E mutation in the BRAF gene in MA-2, WM266-4, 451Lu-R and SK-MEL-2 cells was confirmed by analyses using PCR and hybridization with an allele-specific fluorescently labeled oligodeoxyribonucleotide probe. Canertinib dihydrochloride salt was purchased from LC Labs (C-1201, Woburn, MA, USA). NRG1 (Heregulin-B2 Human Recombinant) was purchased from ProSpec (CYT-407, Rehovot, Israel). Rabbit anti-phospho-ERBB3, rabbit anti-pan AKT and rabbit anti-phospho-AKT antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Rabbit anti-total ERBB3 antibody was from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA) Mouse anti-γ-tubulin antibody was purchased from ThermoFisher Scientific (Waltham, MA, USA).
Survival analyses
The Kaplan–Meier survival curves were generated using the GenePattern software (http://www.broadinstitute.org/cancer/software/genepattern/). The expression levels of ERBB3 mRNA were extracted from the microarray data generated in our previous study.13 The values above the medium were included in the first group and those below the medium were included in the second group. The survival probability of each group of patients was calculated and graphed.
Knocking down ERBB3
Lentiviruses expressing shRNAs against human ERBB3 were purchased (ThermoFisher Scientific) and used to infect melanoma cell lines. The efficiency of knockdown was measured by qRT–PCR. Two shRNAs resulted in significant knockdowns of ERBB3. They target the following sequences: 5′-AATTCTCTACTCTACCATTG-3′ (shERBB3 #1) and 5′-CCAGAGCTTCAAGACTGTTTA3′ (shERBB3 #2). The shRNA that targets green fluorescent protein (5′-CAAGCTGACCCTGAAGTTC-3′) was used as a control. Infected cells were selected under puromycin. Positive clones were amplified and cultured for further analyses.
qRT–PCR
Total RNA was extracted from the cultured cells or mouse lungs using the Qiagen RNeasy Mini Kit (Qiagen, Valencia, CA, USA). cDNA was synthesized from 1 μg RNA using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA) and amplified by quantitative PCR using the following primers: ERBB3-forward: 5′-GGACAGTACGGGAGATCACAG-3′, ERBB3-reverse: 5′-GCACTAATTTCCTTCAGGGATCG-3′, NRG1-forward: 5′-CTGTGTGAATGGAGGCGAGT-3′, NRG1-reverse: 5′-GTAGGCCACCACACACATGA-3′, mouse-ß ACTIN-forward: 5′-AGGAGTACGATGAGTCCGGC-3′, mouse-ß ACTIN-reverse: 5′-AAAACGCAGCTCAGTAACAGTC-3′, human-ß ACTIN-forward: 5′-CGCCTTTGCCGATCCGCCG-3′, and human-ß ACTIN-reverse 5′-CGACGAGCGCGGCGATATCAT-3′. Qunatitative PCR was performed in Bio-rad MyiQ2 Two Color Real-Time PCR Detection System, and data were analyzed using the iQ5 Optical System Software 2.1 (Bio-Rad), according to the manufacturer’s instructions.
Experimental metastasis assay and time course analyses
Cells were injected intravenously into NSG mice (NOD/Cg-Prkdscid Il2rgtmlWfl/SzJ, The Jackson Laboratory, Maine, MA, USA) at 20 000 cells/mouse for MA2 and 100 000 cells/mouse for 451Lu-R. Lungs were harvested 4 weeks later, and metastases were counted. For the lung-seeding assays of MA-2 cells, cells were labeled with Cell Tracker Green CMFDA (Life Technologies, Grand Island, NY, USA) for 15 min and then injected intravenously into NSG mice. Lungs were harvested at the indicated time points and frozen in Optimal Cutting Temperature Compound. Four non-continuous 6-μm cryosections were cut for each lung, with ∼10 sections skipped in between. They were fixed with 4% paraformaldehyde and stained with DAPI (4,6-diamidino-2-phenylindole). Images were taken from five random fields on each section, using the Zeiss Axio Imager M2m (Zeiss International, Jena, Germany) and the AcioVision Software 4.8.2 (Zeiss International) and Adobe Photoshop software (Adobe Systems Incorporated, San Jose, CA, USA). The number of green fluorescent cells in each of five fields was counted and added together as the number of fluorescent cells per section. For time course analyses of 451Lu-R cells, cells were not labeled with CMFDA before injections into NSG mice. Lungs were harvested at the indicated time points, fixed in formalin, sectioned and were stained with the human-specific anti-vimentin antibody for immunohistochemical analyses.
Immunohistochemical analyses
Mouse lungs were fixed in 10% formalin overnight, processed, embedded and sectioned. Immunohistochemical analyses were performed according to the standard protocols. Briefly, sections were rehydrated through ethanol series and subjected to antigen retrieval in citrate buffer (pH 6.0). After treatment with H2O2 and blockage in 5% normal donkey serum, sections were incubated with the antibody against human-specific vimentin (1:400; DAKO, Carpinteria, CA, USA) at 4 °C overnight. The signals were detected by biotinylated donkey anti-mouse secondary antibody and amplified with the ABC Kit (Vector Laboratory, Burlingame, CA, USA). Colorimetric development was performed using the VIP Substrate Kit (Vector Laboratory), with hematoxylin as the counterstain. Images were taken from five random fields on each section, using the AxioCam ICc1 (Zeiss International) camera. The number of micrometastases in each of the five fields was counted and added together as the number of metastases per section.
NRG1 stimulation and western blotting
Melanoma cell lines were plated in a six-well plate at 500 000 cells per well in the complete media. The next day, cells were switched into serum-free medium and cultured for 24 h before the addition of 500 ng/ml of NRG1 for the specified time intervals. The treated cells were then lysed in the lysis buffer (150 mM Tris pH 7.4, 100 mM NaF, 120 mM NaCl, 5% NP-40, 100 uM Sodium Vanadate, protease inhibitors), sonicated and spun down for 10 min at 4 °C. 30–50 μg of protein in each lysate was loaded onto an sodium dodecyl sulfate–polyacrylamide gel and probed with the rabbit anti-phospho-ERBB3, rabbit anti-total ERBB3, rabbit anti-pan AKT, rabbit anti-phospho-AKT or the mouse anti-γ-tubulin antibodies. The signals were detected by corresponding secondary antibodies and visualized with enhanced chemical luminescence (ThermoFisher Scientific). Band intensities were quantified with the ImageJ software (NIH, Bethesda, MD, USA).
Canertinib treatment
A total of 250 000 451Lu-R or MeWo cells were injected intravenously into NSG mice (n=14 for 451Lu-R and n=15 for MeWo). One week later, 1.2 mg canertinib (in 10% DMSO, 0.15% NaCl), or vehicle control, was injected intraperitoneally into each mouse daily, based on the published protocols.36 The treatment lasted 10 weekdays for 451Lu-R injected mice and 8 contiguous days for MeWo-injected mice. The mice were left off drug for another week before being examined for lung metastases. Lungs were fixed in formalin, sectioned and stained with the human-specific anti-vimentin antibody. 4–6 non-continuous sections of each lung were stained, and vimentin-positive metastases were counted and pooled. Six of the 451Lu-R injected mice (three vehicle treated and three canertinib treated) were left off drug after treatment until day 64 for survival analyses.
Data analyses and statistics
The difference between the survival curves was evaluated by the log-rank test in the GenePattern software http://www.broadinstitute.org/cancer/software/genepattern/. For the rest of data analyses, Student’s t-test was performed using the software in http://www.physics.csbsju.edu/stats/.
References
Tsao H, Atkins MB, Sober AJ . Management of cutaneous melanoma. New Engl J Med 2004; 351: 998–1012.
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417: 949–954.
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. New Engl J Med 2011; 364: 2507–2516.
Hersey P, Smalley KS, Weeraratna A, Bosenberg M, Zhang XD, Haass NK et al. Meeting report from the 7th International Melanoma Congress, Sydney, November, 2010. Pigment Cell Melanoma Res 2010; 24: e1–15.
Tsao H, Chin L, Garraway LA, Fisher DE . Melanoma: from mutations to medicine. Genes Dev 2012; 26: 1131–1155.
Sullivan RJ, Lorusso PM, Flaherty KT . The intersection of immune-directed and molecularly targeted therapy in advanced melanoma: where we have been, are, and will be. Clin Cancer Res 2013; 19: 5283–5291.
Nguyen DX, Bos PD, Massague J . Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer 2009; 9: 274–284.
Xu L, Hynes RO . GPR56 and TG2: possible roles in suppression of tumor growth by the microenvironment. Cell Cycle 2007; 6: 160–165.
Klein CA . Framework models of tumor dormancy from patient-derived observations. Curr Opin Genet Dev 2011; 21: 42–49.
Mohanty S, Xu L . Experimental metastasis assay. J Vis Exp 2010; pii: 1942.
Kozlowski JM, Hart IR, Fidler IJ, Hanna N . A human melanoma line heterogeneous with respect to metastatic capacity in athymic nude mice. J Natl Cancer Inst 1984; 72: 913–917.
Xu L, Begum S, Hearn JD, Hynes RO . GPR56, an atypical G protein-coupled receptor, binds tissue transglutaminase, TG2, and inhibits melanoma tumor growth and metastasis. Proc Natl Acad Sci USA 2006; 103: 9023–9028.
Xu L, Shen SS, Hoshida Y, Subramanian A, Ross K, Brunet JP et al. Gene expression changes in an animal melanoma model correlate with aggressiveness of human melanoma metastases. Mol Cancer Res 2008; 6: 760–769.
Sibilia M, Kroismayr R, Lichtenberger BM, Natarajan A, Hecking M, Holcmann M . The epidermal growth factor receptor: from development to tumorigenesis. Differentiation 2007; 75: 770–787.
Pinkas-Kramarski R, Soussan L, Waterman H, Levkowitz G, Alroy I, Klapper L et al. Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO J 1996; 15: 2452–2467.
Prigent SA, Gullick WJ . Identification of c-erbB-3 binding sites for phosphatidylinositol 3'-kinase and SHC using an EGF receptor/c-erbB-3 chimera. EMBO J 1994; 13: 2831–2841.
Soltoff SP, Carraway KL 3rd, Prigent SA, Gullick WG, Cantley LC . ErbB3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth factor. Mol Cell Biol 1994; 14: 3550–3558.
Hellyer NJ, Cheng K, Koland JG . ErbB3 (HER3) interaction with the p85 regulatory subunit of phosphoinositide 3-kinase. Biochem J 1998; 333: Pt 3 757–763.
Rajkumar T, Stamp GW, Hughes CM, Gullick WJ . c-erbB3 protein expression in ovarian cancer. Clin Mol Pathol 1996; 49: M199–M202.
Gyorffy B, Lage H . A Web-based data warehouse on gene expression in human malignant melanoma. J Invest Dermatol 2007; 127: 394–399.
Tovey SM, Witton CJ, Bartlett JM, Stanton PD, Reeves JR, Cooke TG . Outcome and human epidermal growth factor receptor (HER) 1-4 status in invasive breast carcinomas with proliferation indices evaluated by bromodeoxyuridine labelling. Breast Cancer Res 2004; 6: R246–R251.
Ueno Y, Sakurai H, Tsunoda S, Choo MK, Matsuo M, Koizumi K et al. Heregulin-induced activation of ErbB3 by EGFR tyrosine kinase activity promotes tumor growth and metastasis in melanoma cells. Int J Cancer 2008; 123: 340–347.
Zhang K, Wong P, Duan J, Jacobs B, Borden EC, Bedogni B . An ERBB3/ERBB2 oncogenic unit plays a key role in NRG1 signaling and melanoma cell growth and survival. Pigment Cell Melanoma Res 2013; 26: 408–414.
Fattore L, Marra E, Pisanu ME, Noto A, Belleudi F, de Vitis C et al. Activation of an early feedback survival loop involving phospho-ErbB3 is a general response of melanoma cells to RAF/MEK inhibition and is abrogated by anti-ErbB3 antibodies. J Transl Med 2013; 11: 180.
Belleudi F, Marra E, Mazzetta F, Fattore L, Giovagnoli MR, Mancini R et al. Monoclonal antibody-induced ErbB3 receptor internalization and degradation inhibits growth and migration of human melanoma cells. Cell Cycle 2012; 11: 1455–1467.
Abel EV, Basile KJ, Kugel CH 3rd, Witkiewicz AK, Le K, Amaravadi RK et al. Melanoma adapts to RAF/MEK inhibitors through FOXD3-mediated upregulation of ERBB3. J Clin Invest 2013; 123: 2155–2168.
Xu L . GPR56 interacts with extracellular matrix and regulates cancer progression. Adv Exp Med Biol 2010; 706: 98–108.
Joseph EW, Pratilas CA, Poulikakos PI, Tadi M, Wang W, Taylor BS et al. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc Natl Acad Sci USA 2010; 107: 14903–14908.
Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010; 18: 683–695.
Hynes NE, MacDonald G . ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol 2009; 21: 177–184.
Zhang K, Wong P, Zhang L, Jacobs B, Borden EC, Aster JC et al. A Notch1-neuregulin1 autocrine signaling loop contributes to melanoma growth. Oncogene 2012; 31: 4609–4618.
Liu J, Kern JA . Neuregulin-1 activates the JAK-STAT pathway and regulates lung epithelial cell proliferation. Am J Respir Cell Mol Biol 2002; 27: 306–313.
Gao L, Feng Y, Bowers R, Becker-Hapak M, Gardner J, Council L et al. Ras-associated protein-1 regulates extracellular signal-regulated kinase activation and migration in melanoma cells: two processes important to melanoma tumorigenesis and metastasis. Cancer Res 2006; 66: 7880–7888.
Kaufmann WK, Nevis KR, Qu P, Ibrahim JG, Zhou T, Zhou Y et al. Defective cell cycle checkpoint functions in melanoma are associated with altered patterns of gene expression. J Invest Dermatol 2008; 128: 175–187.
Smaill JB, Rewcastle GW, Loo JA, Greis KD, Chan OH, Reyner EL et al. Tyrosine kinase inhibitors. 17. Irreversible inhibitors of the epidermal growth factor receptor: 4-(phenylamino)quinazoline- and 4-(phenylamino)pyrido. J Med Chem 2000; 43: 3199.
Djerf Severinsson EA, Trinks C, Green H, Abdiu A, Hallbeck AL, Stal O et al. The pan-ErbB receptor tyrosine kinase inhibitor canertinib promotes apoptosis of malignant melanoma in vitro and displays anti-tumor activity in vivo. Biochem Biophys Res Commun. 2011; 414: 563–568.
Baselga J, Swain SM . Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer 2009; 9: 463–475.
Schoeberl B, Faber AC, Li D, Liang MC, Crosby K, Onsum M et al. An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation. Cancer Res 2010; 70: 2485–2494.
Buac K, Xu M, Cronin J, Weeraratna AT, Hewitt SM, Pavan WJ . NRG1 / ERBB3 signaling in melanocyte development and melanoma: inhibition of differentiation and promotion of proliferation. Pigment Cell Melanoma Res 2009; 22: 773–784.
Gordon-Thomson C, Jones J, Mason RS, Moore GP . ErbB receptors mediate both migratory and proliferative activities in human melanocytes and melanoma cells. Melanoma Res 2005; 15: 21–28.
Acknowledgements
We thank Dr Liquan Yang (Emory University) for useful suggestions and Dr Meenhard Herlyn (Wistar Institute) for providing the 451Lu-R cell line. This work was supported by the start-up fund from University of Rochester (to LX), the JWCC/RPCI Collaborative Pilot Grant (to NZ and LX) and the NYSTEM IDEA AWARD (no. N08G-480; to LX).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
Oncogenesis is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Tiwary, S., Preziosi, M., Rothberg, P. et al. ERBB3 is required for metastasis formation of melanoma cells. Oncogenesis 3, e110 (2014). https://doi.org/10.1038/oncsis.2014.23
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/oncsis.2014.23
This article is cited by
-
ERBB3 binding protein 1 promotes the progression of malignant melanoma through activation of the Wnt/ β-catenin signaling pathway
Cancer Cell International (2022)
-
Effects of RNA methylation N6-methyladenosine regulators on malignant progression and prognosis of melanoma
Cancer Cell International (2021)
-
Combining ERBB family and MET inhibitors is an effective therapeutic strategy in cutaneous malignant melanoma independent of BRAF/NRAS mutation status
Cell Death & Disease (2019)
-
Development and validation of a plasma-based melanoma biomarker suitable for clinical use
British Journal of Cancer (2018)
-
Characterising the phenotypic evolution of circulating tumour cells during treatment
Nature Communications (2018)
