
Abstract
One characteristic of cancer cells is the abnormally high rate of cell metabolism to sustain their enhanced proliferation. However, the behind mechanism of this phenomenon is still elusive. Here we find that enhanced precursor 45s ribosomal RNA (pre-45s rRNA) is one of the core mechanisms in promoting the pathogenesis of colorectal cancer (CRC). Pre-45s rRNA expression is significantly higher in primary CRC tumor tissues samples and cancer cell lines compared with the non-tumorous colon tissues, and is associated with tumor sizes. Knockdown of pre-45s rRNA inhibits G1/S cell-cycle transition by stabilizing p53 through inducing murine double minute 2 (MDM2) and ribosomal protein L11 (RpL11) interaction. In addition, we revealed that high rate of cancer cell metabolism triggers the passive release of calcium ion from endoplasmic reticulum to the cytoplasm. The elevated calcium ion in the cytoplasm activates the signaling cascade of calcium/calmodulin-dependent protein kinase II, ribosomal S6 kinase (S6K) and ribosomal S6K (CaMKII-S6K-UBF). The activated UBF promotes the transcription of rDNA, which therefore increases pre-45s rRNA. Disruption of CaMKII-S6K-UBF axis by either RNAi or pharmaceutical approaches leads to reduction of pre-45s rRNA expression, which subsequently suppresses cell proliferation in colon cancer cells by causing cell-cycle arrest. Knockdown of APC activates CaMKII-S6K-UBF cascade and thus enhances pre-45s rRNA expression. Moreover, the high expression level of pre-45s rRNA is associated with poor survival of CRC patients in two independent cohorts. Our study identifies a novel mechanism in CRC pathogenesis mediated by pre-45s rRNA and a prognostic factor of pre-45s rRNA in CRC patients.
Similar content being viewed by others


Introduction
Colorectal cancer (CRC) is the third most common cancer worldwide.1 One million new CRC cases are reported annually, and the disease-specific mortality rate is nearly 33%.2 With the changes of living and dietary habits, the incidence of CRC is rapidly increasing in Asian in the past decade. However, the molecular mechanisms that drive the development of CRC remain elusive.
Altered nuclear morphologies such as enlargement and increased numbers of nucleoli are observed in many types of cancer cells, including CRC. Nucleoli are sites where rDNA transcription and ribosome biogenesis occur.3 To produce a functional ribosome, the precursor rRNA precursor 45s ribosomal RNA (pre-45s rRNA) (also known as 47S rRNA) has to be transcribed from rDNA and processed to generate 18 S, 5.8 S and 28 S rRNAs.4 To support the growth of tumor cells, the rate of ribosomal biogenesis is increased to produce ribosome that is composed of rRNAs and proteins, resulting in a prominent increase in nucleolus size.5, 6, 7 Enhanced proliferation in colon cancer cells should be supported by elevated metabolic mechanisms. To synthesize effectively enough cellular components for cell division, the efficiency of protein synthesis should be enhanced. Increase in the number of ribosomes is one way to support it. As rRNAs are the essential component of ribosome, it is expected that the level of primary rRNA, pre-45s rRNA, should be high so as to maintain the efficiency of protein synthesis. In this study, we detected the high expression status of pre-45s rRNA in two independent cohorts of CRC patients. High expression of pre-45s rRNA could be prognostic factor of poor outcome of CRC patients. We explored the underlying mechanism of pre-45s rRNA in promoting CRC carcinogenesis and characterized its clinical implication in CRC patients.
Results
Pre-45s rRNA levels are significantly elevated in two cohorts of CRC patients
We first compared the expression level of pre-45s rRNA in paired tumor and adjacent normal tissues in two cohorts of CRC patients. We found that 49 out of 52 (P<0.0001) cases from Hong Kong cohort showed higher level of pre-45s rRNA in tumor tissues compared with their adjacent non-tumor tissues (Figure 1a); the mean value of relative expression level of pre-45s rRNA was also significantly elevated in tumor tissues compared with the adjacent non-tumor tissues (P<0.0001, Figure 1b); however, the expression of pre-45s rRNA was not correlated with tumor, node or metastasis (TNM) stages (Figure 1c). The enhanced expression level of pre-45s rRNA in tumor tissues was confirmed in the second cohort of 28 pairs of CRC patients from Zhejiang (P<0.0001); the mean expression level of pre-45s rRNA was slightly higher in TNM stages III/IV compared with stages I/II (Figures 1d–f). Altogether, we confirmed that the expression level of pre-45s rRNA in primary tumor tissues was higher compared with that in non-tumor tissues.

Upregulation of pre-45s rRNA in CRC patients. (a) Pre-45s rRNA was upregulated in tumor tissues obtained from CRC patients in Hong Kong cohort (P<0.0001, n=52). (b) The average relative expression of pre-45s rRNA in both adjacent normal (0.375±0.079) and tumor tissues (6.602±0.563). (c) The expression levels of pre-45s rRNA in tumor tissues from the patients at TNM I and II were compared with that from patients at TNM III and IV (P=0.867). (d) Pre-45s rRNA was upregulated in tumor tissues obtained from CRC patients in Zhejiang cohort. The expression level of pre-45s rRNA in paired adjacent normal and tumor tissues was compared (P<0.0001, n=28). (e) The average relative expression of pre-45s rRNA in adjacent normal (1.151±0.068) and tumor tissues (6.591±0.465). (f) The expression levels of pre-45s rRNA in tumor tissues from the patients at TNM I and II were compared with that from patients at TNM III and IV (P=0.037). β-Actin was used as an internal control for real-time PCR. Data are expressed as mean±s.d.
Expression level of pre-45s rRNA is essential for the growth of colon cancer cells
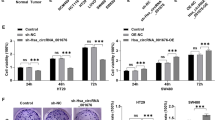
We evaluated the expression status of pre-45s rRNA in nine different colon cancer cell lines (Caco-2, DLD-1, HCT116, HT29, LOVO, LS180, SW480, SW620 and SW1116) and a normal colon epithelial cell line NCM460. All nine colon cancer cell lines showed higher expression of pre-45s rRNA relative to NCM460 (Figure 2a). We therefore investigated the functional significance of pre-45s rRNA in colon cancer. Downregulation of pre-45s rRNA by pre-45s rRNA siRNA in three colon cancer cell lines HCT116, HT29 and Caco-2 was confirmed by quantitative reverse transcription–polymerase chain reaction (Supplementary Figures 1A–C). Knockdown of pre-45s rRNA significantly suppressed cell growth in all these cell lines (Figure 2b), but not in normal epithelial cells (NCM460; Supplementary Figures 2A–C), suggesting that pre-45s rRNA is essential in colon tumorigenesis.

Pre-45s rRNA was essential for supporting the growth of CRC tumor cells. (a) Expression levels of pre-45s rRNA in colon cancer cell and normal colon epithelial cell were evaluated by real-time PCR. Expression of pre-45s rRNA was compared with commercial normal colon tissue. Knockdown of pre-45s rRNA suppressed cell growth. (b) MTT assay was performed in HCT116, HT29 and Caco-2. Colony formation assay was performed in HCT116, HT29 and Caco-2. Represented images were shown. Flow cytometry was used to evaluate the effect of pre-45s rRNA downregulation on cell-cycle arrest in (c) HCT116, (d) HT29 and (e) Caco-2. Representive graphs were shown. (f) Knockdown of pre-45s rRNA favored the interaction between MDM2 and RpL11. Co-immunoprecipitation was used to determine the interaction between MDM2 and RpL11. RpL11 was immunoprecipiated; immunoblot was used to determine the presence of RpL11 and MDM2 in the immunoprecipitant. siRNA of 5 pmol against pre-45s rRNA was used to knockdown the rRNA in cells. β-Actin was used as an internal control for real-time PCR. All data points are expressed as mean±s.d.
Knockdown of pre-45s rRNA inhibits G1/S transition by inducing MDM2/RpL11 interaction and therefore p53 stabilization
To determine whether knockdown of pre-45s rRNA decreased colon cancer cell growth by altering cell cycle, we investigated the effect of pre-45s rRNA knockdown on cell-cycle distribution. Downregulation of pre-45s rRNA suppressed G1/S transition in colon cancer cells HCT116, HT29 and Caco-2 (Figures 2c–e and Supplementary Figure 2D). Bromodeoxyuridine labeling assay confirmed that knockdown of pre-45s rRNA can reduce the portion of cells in the S phase in colon cancer cells HCT116, HT29 and Caco-2 (Supplementary Figure 3). It is well established that cell-cycle arrest can be induced by the overexpression of p53.8 Ribosomal proteins can interact with E3 ubiquitin-protein ligase murine double minute 2 (MDM2), suppressing its function on p53 degradation. Among the ribosomal proteins, RpL11 has a significant role in p53 regulation.9 We found that downregulation of pre-45s rRNA favored the interaction of MDM2 and RpL11 in HCT116 and HT29 cells (Figure 2f) as determined by co-immunoprecipitation, which subsequently resulted in p53 stabilization and upregulation of p53 downstream effectors p21 and p27 (Supplementary Figure 4).
Intracellular Ca2+ level mediates upregulation of pre-45s rRNA in colon cancer cells
To identify a messenger molecule involved in pre-45s rRNA upregulation in colon cancer cells, we tested if cyclic adenosine 3′, 5′-monophosphate (cAMP), nitric oxide or calcium ion (Ca2+) would be involved. Our data showed that only treatment with EGTA-AM (a chelator of Ca2+), but not H89 (a protein kinase A inhibitor) and l-NMMA (an inhibitor of all NOS isoforms), could reduce the level of pre-45s rRNA in HCT116 and HT29 colon cancer cell lines (Figure 3). In keeping with this, the concentration of Ca2+ was higher in all colon cancer cells compared with normal colon cell line NCM460 (Figure 4a). The feature suggested that the higher concentration of Ca2+ might be responsible for mediating the high level of pre-45s rRNA in these cells. To verify the relation between Ca2+ and pre-45s rRNA, we treated nine colon cancer cell lines with another Ca2+ chelator, named 1,2-bis(o-aminophenoxy) ethane-N,N,N',N'-tetraacetic acid (BAPTA-AM), at an optimal dosage of 5 μm (Supplementary Figures 5A and B). BAPTA-AM significantly reduced the expression of pre-45s rRNA in all nine colon cancer cell lines (Figure 4b). However, when these cells were treated with cell-impermeable BAPTA, pre-45s rRNA expression did not change (Figure 4b). To further confirm this result, we used Northern blot to determine the expression levels of pre-45s rRNA transcript in HCT116 colon cancer cell line. BAPTA-AM treatment reduced the level of pre-45s rRNA transcript, whereas BAPTA treatment did not change pre-45s rRNA transcript in HCT116 cells by Northern blot (Supplementary Figure 6A). Therefore, intracellular Ca2+ was essential for mediating the upregulation of pre-45s rRNA.

The effect of different chemical on pre-45s rRNA level. (a) Cells were treated with 0.1 μM of H89. (b) Cells were treated with 2 μM of l-NMMA. (c) Cells were treated with 1 mM of EGTA-AM. Real-time PCR was performed to test the expression of pre-45s rRNA . β-Actin was used as an internal control for real-time PCR. Data are expressed as mean±s.d.

Ca2+ used CaMKII-S6K-UBF signaling cascade to mediate high expression level of pre-45s rRNA in colon cancer cells. (a) The concentration of free Ca2+ in the cytoplasm was determined by fluorescence-based method in normal colon epithelial and colon cancer cells. (b) Treatment of Ca2+ chelator BATPA-AM could suppress the expression level of pre-45s rRNA in all colon cancer cells. Real-time PCR was used to determine the expression level of pre-45s rRNA. (c) Immunoblot analysis on the levels of indicated candidates. (d) Treatment of KN93 suppressed the expression level of pre-45s rRNA in colon cancer cells. KN93 is a small molecule that can inhibit the activity of CaMKII. Real-time PCR was performed to determine the level of pre-45s rRNA. (e) The effect of KN93 on cell growth in NCM460, HCT116 and HT29 was revealed by MTT. The effect of KN93 on cell growth in NCM460, HCT116 and HT29 was revealed by colony formation assays. Represented images were shown. Five micro molar of KN93 was used to treat cells. (f) Immunoblot analysis on the levels of indicated candidates and real-time PCR analysis on pre-45s rRNA levels in paired normal adjacent (N) and tumor (T) tissues from seven patients. Corresponding siRNA of 5 pmol was used. BAPTA-AM/BAPTA of 5 μm was used. GAPDH was used as a loading control in western blot. β-Actin was used as an internal control for real-time PCR. All data points are expressed as mean±s.d.
CaMKII-S6K-UBF axis mediates the upregulation of pre-45s rRNA
It is known that CaMKII can be activated by Ca2+ in nanomolar level.10 Activation of CaMKII by Ca2+ induces the activity of S6K. The activated S6K subsequently activates upstream binding factor (UBF),11 enhancing the activity of UBF to induce rRNA transcription.12 With these involved connecting components, Ca2+ links to rDNA transcription. We thus determined the levels of p-CaMKII, p-S6K and p-UBF in colon cancer cells and found that protein expression of p-CaMKII, p-S6K and p-UBF were higher in colon cancer cell lines (HCT116 and HT29) than in normal colon epithelial cell NCM460 (Figure 4c). The expression level of pre-45s rRNA was significantly reduced by a CaMKII inhibitor KN93 in colon cancer cell lines HCT116 and HT29, but not in normal colon epithelial cell NCM460 (Figure 4d). This was confirmed by Northern blot that KN93 could significantly reduce the level of pre-45s rRNA level in HCT116 cells (Supplementary Figure 6B). KN93 treatment reduced the levels of p-CaMKII, p-S6K and p-UBF in HCT116 cells (Supplementary Figure 7A), suggesting that KN93 inhibits CaMKII-S6K-UBF axis. Since the level of pre-45s rRNA was essential for the growth of cancer cells, both cell viability and colony formation assays confirmed this growth-suppressive effect by KN93 treatment on HCT116 and HT29 cells, but not in NCM460 (Figure 4e). Moreover, KN93 could increase the level of p53, p21Cip/Waf1and p27Kip1 in HCT116 (Supplementary Figure 7B). To further confirm the involvement of CaMKII-S6K-UBF axis in the mediation of pre-45s rRNA in colon cancer, we used small interfering RNA (siRNA) to knockdown UBF in HCT116, HT29 and Caco-2. The knockdown efficiency was shown in Supplementary Figure 8A. Knockdown of UBF in these colon cancer cells could reduce the expression level of pre-45s rRNA (Supplementary Figure 8B). Clonogenicity assay showed that knockdown of UBF could suppress cell proliferation in HCT116, HT29 and Caco-2 cells (Supplementary Figure 8C). Subsequently, we evaluated the activity of this signaling cascade in primary tumor tissues from CRC patients. The results showed that the signaling cascade was more active in tumor tissues in all cases compared with their adjacent non-tumor tissues, which was consistent with the upregulated pre-45s rRNA levels in these tumor tissues (Figure 4f). Collectively, our findings suggested that CaMKII-S6K-UBF axis mediates the upregulation of pre-45s rRNA in colon cancer cells.
Loss of function of APC enhances pre-45s rRNA expression via inducing CaMKII-S6K-UBF axis
One well known cancer driver in CRC is adenomatous polyposis coli (APC) loss of function. CaMKII-S6K-UBF axis mediated the upregulation of pre-45s rRNA would be a general mechanism to support colon tumorigenesis. Therefore, we evaluated the effect of APC knockdown on HCT116 and HT29. HCT116 bears wild type and full length of APC, whereas HT29 bears truncated APC, which is still functional. Knockdown of APC enhanced protein synthesis in these cell lines (Figure 5a). Also, we found that knockdown of APC enhanced the level of free Ca2+ in these cell lines (Figure 5b). The increased Ca2+ by knockdown of APC could enhance the activity of CaMKII-S6K-UBF axis, whereas the enhanced activity of this signaling cascade could be blocked by BAPTA-AM in both HCT116 and HT29 (Figure 5c). As a result, knockdown of APC led to the elevation of pre-45s rRNA level, whereas such an elevation could be abolished by BAPTA-AM (Figure 5d). Moreover, the enhanced cell growth induced by APC knockdown could be diminished by knockdown of pre-45s rRNA in both colon cancer cells (Figures 5e and f). However, there is no significant difference of pre-45s rRNA levels in HCT116 p53+/+ and HCT116 p53−/− cells (Supplementary Figure 9). These data indicated that pre-45s rRNA is important in supporting tumorigenesis mediated by certain CRC drivers such as APC.

Pre-45s rRNA was essential for supporting the cell growth in colon cancer cells with APC loss of function. (a) Knockdown of APC enhanced the rate of protein synthesis in colon cell lines. (b) Knockdown of APC led to elevation of free Ca2+ in the cytoplasm in colon cells. Real-time PCR was used to determine the expression level of pre-45s rRNA. (c) Activation of CaMKII-S6K-UBF signaling cascade in APC knocked down cells depended on free Ca2+. GAPDH was used as a loading control. Cells were treated with 5 μM of BATPA-AM. (d) Upregulation of pre-45s rRNA mediated by APC knockdown was abolished by BATPA-AM treatment in colon cancer cells. Expression of pre-45s rRNA was compared with untreated NCM460, HCT116 and HT29. Knockdown of pre-45s rRNA suppressed cell growth in colon cancer cells. (e) HCT116 and (f) HT29 with APC knocked down. Cell growth was evaluated by MTT assay. siRNA of 5 pmol against pre-45s rRNA was used to knockdown the rRNA in cells. GAPDH was used as a loading control in western blot. β-Atin was used as internal control for real-time PCR. All data points are represented as mean±s.d. *P<0.05, **P<0.01 and ***P<0.001.
High expression of pre-45s rRNA is an independent predictor of poor outcome in CRC patients
The association between clinicopathologic features and pre-45s rRNA expression in human CRCs was evaluated. We categorized the patients into two groups, based on the expression level of pre-45s rRNA, high expression (⩾mean in the study cohort) and low expression (Pre-45s rRNA expression only associated with tumor length and recurrence in Hong Kong cohort (Table 1). We found a positive and linear relationship (R2=0.8232) between these tumor lengths and pre-45s rRNA level with Spearman's coefficient equaled to 0.9163 (Figure 6a). Therefore, our data implicated that pre-45s rRNA might be one of factors to support CRC carcinogenesis. We next examined that correlation between the expression level of pre-45s rRNA and survival outcome in CRC patients. In both Hong Kong cohort and Zhejiang cohort, the survival outcomes were significantly different between high expression and low expression groups as shown in the Kaplan–Meier survival curves (P<0.0001; Figures 6b and c). We further used univariate and multivariate Cox regression analyses to evaluate the disease-free hazard ratio (HR) of different parameters in the two cohorts. We found that the survival outcomes were significantly different between high pre-45s rRNA expression and low pre-45s rRNA expression groups (univariate: HR=5.941; 95% confidence interval (CI)=2.924–12.070; multivariate: HR=7.385; 95% CI=3.504–15.568; Table 2). Therefore, high expression level of pre-45s rRNA was an independent prognostic factor for poor outcome of CRC patients.

Clinical significance of pre-45s rRNA in CRC patients. (a) Tumor size positively correlated with the expression level of pre-45s rRNA. Only samples from Hong Kong were included in this analysis. Red line represents 95% CI. High expression level of pre-45s rRNA correlated with poor survival outcome of patients from (b) Hong Kong and (c) Zhejiang. Expression level of pre-45s rRNA was an independent prognostic marker for CRC patients. (d) Schematic illustration of the molecular mechanism of pre-45s rRNA in colon cancer. The high metabolism in the colon cancer cells triggers the passive release of Ca2+ from endoplasmic reticulum to the cytoplasm. The elevated Ca2+ in the cytoplasm activates the signaling cascade composed of CaMKII-S6K-UBF. The activated UBF promotes the transcription of rDNA by different mechanisms, which therefore increases the level of precursor of ribosomes, pre-45s rRNA. The enhanced pre-45s rRNA promotes G1/S cell-cycle transition by reducing the inhibitory factors on MDM2 and therefore decreasing the level of p53, which contributes to colon tumorigenesis.
Discussion
In this study, we identified that pre-45s rRNA was markedly upregulated in primary tumors of CRC and colon cancer cell lines. A series of in vitro functional experiments revealed that pre-45s rRNA possessed a strong tumorigenic function in CRC. Pre-45s rRNA enhanced tumor cell proliferation, which is attributable to its G1/S cell-cycle-promoting ability. The mechanism of the upregulation of pre-45s rRNA in colon cancer cells was investigated and we found that higher concentration of Ca2+ was involved in mediating the increased level of pre-45s rRNA in CRC. Changing the cytosolic concentrations of Ca2+ leads to a broad range of cellular events, including those important in tumorigenesis mediated by specific Ca2+ channels and pumps.13, 14, 15 The high level of pre-45s rRNA induced by higher concentration of Ca2+ is at least one of the molecular mechanisms contributing to the tumorigenesis of CRC (Figure 6d).
Pre-45s rRNA can be processed to 18 s and 28 s rRNA for assembly of large and small ribosome subunits. Ribosomal proteins of RpL5, RpL11 and RpL23 are essential elements of ribosome subunits. It was reported that inhibition of rRNA by nucleolar stress led to the accumulation of free ribosomal proteins including RpL5, RpL11 and RpL23 RpL23.16, 17, 18 The unassembled free RpL11 could interact with MDM2 and led to a marked accumulation of p53.19 In this study, we found that knockdown of pre-45s rRNA enhanced MDM2/RpL11 interaction (Figure 2f). Collectively, these findings suggested that depletion of pre-45s rRNA enhances MDM2/RpL11 interaction through the release of free RpL11, thereby inhibiting colon cancer cell proliferation.
We further extend the knowledge on roles played by non-ATP-dependent channels in colon cancer cells. We found that high metabolism could lead to increase in cytosolic Ca2+ via translocons. We confirmed this notion by treating cells with protein synthesis inhibitors cyclohexamer and anisomycin. Through gene knockdown and pharmaceutical intervention (CaMKII inhibitor KN93) approaches, we uncovered CaMKII-S6K-UBF axis for mediating the upregulation of pre-45s rRNA in colon cancer cells (Figure 6d). It was reported that inhibition of Ca2+ by an N-myristoylated EF-hand Ca2+-binding protein, calcineurin homologous protein 1, suppresses rRNA transcription by interacting with UBF, which is a component of the RNA polymerase I complex.20 Inhibition of RNA polymerase I promotes the cancer-specific activation of p53 and subsequent suppression of tumor growth.21 On the other hand, the interaction between calcineurin homologous protein 1 and UBF can be abolished by the presence of Ca2+, thereby promoting rRNA transcription.20, 22 These findings collectively suggested that the effect of CaMKII-S6K-UBF on the regulation of pre-45s rRNA transcription in CRC is potentially mediated by RNA polymerase I complex.
Mutations on APC activate Wnt signaling pathway,23, 24, 25 resulting in enhanced protein synthesis.6, 26 In noncanonical Wnt β-catenin-independent signaling pathway, activation of Wnt signaling can induce the elevation of Ca2+ in the cytoplasm and subsequent activation of CaMKII.27 The binding of Wnt to Frizzled receptor triggers the production of inositol 1,4,5-triphosphate and 1,2 diacylglycerol.28 Both inositol 1,4,5-triphosphate and 1,2 diacylglycerol can induce the release of Ca2+ through the interaction with the calcium channels present on the endoplasmic reticulum membrane.27 Sustained activation of Wnt signaling maintains a relative high intracellular Ca2+, which induces the activation of CaMKII-S6K-UBF axis and eventually leading to high expression of pre-45s rRNA. In addition, we found that knockdown of APC enhanced pre-45s rRNA levels, and consequently promoted cell growth. On the other hand, downregulation of pre-45s rRNA abolished the promoting effect of APC knockdown on cell growth. These findings suggest that CRC driver mutation such as APC also contributed to the upregulation of pre-45s rRNA to sustain their oncogenic function in CRC. Thus, pre-45s rRNA is a central hub of CRC carcinogenesis.
Recognizing the biological functions of pre-45s rRNA in CRC, its enhanced expression would favor tumor progression and a worse outcome in the patients. In this regard, the clinical significance of pre-45s rRNA expression with patient’s outcome was evaluated in two independent cohorts of 80 primary CRC patients. Our results indicated that higher pre-45s rRNA expression was significantly associated with poor survival in cohort I (Hong Kong) CRC patients independent of patient characteristics (P<0.0001). This finding was supported by that of cohort II (Zhejiang; P<0.0001). Our data supported an adverse effect of pre-45s rRNA on the survival of CRC patients, providing an additional evidence for the promoting role of pre-45s rRNA in the development of CRC.
In conclusion, we demonstrated that pre-45s rRNA is upregulated in tumor tissues and such an upregulation is mediated by signaling cascade composed of CaMKII-S6K-UBF mediated by Ca2+. The high level of pre-45s rRNA level promotes the development of CRC by inducing G1/S cell-cycle transition through suppressing MDM2/RpL11 interaction and therefore reducing p53 stabilization (Figure 6d). P re-45s rRNA is associated with the poor survival of CRC patients.
Materials and methods
Clinical samples collection and study
Eighty pairs of CRC tumor and their adjacent non-tumor tissues were collected from 1999 to 2009 who underwent surgery at The Prince of Wales Hospital, Hong Kong (n=52) and The Second Affiliated Hospital, Zhejiang (n=28), and were histologically confirmed in a blinded manner by two pathologists. The adjacent normal tissue is composed of normal colonic mucosa located ~10 cm away from the cancer tissue. All subjects provided informed consent before specimen collection. The study protocols were approved by the Ethics Committee of the Chinese University of Hong Kong and Human Subject Research Ethics Committee of the Second Affiliated Hospital, School of Medicine, Zhejiang University.
Cell cultures
Colon cell lines (Caco-2, DLD-1, HCT116, HT29, LOVO, LS180, SW480, SW620 and SW1116) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). NCM460 was obtained from In Cell (San Antonio, TX, USA). HCT116 p53−/− was provided by Dr B Vogelstein (Johns Hopkins University, Baltimore, MD, USA). Dulbecco's modified Eagle's medium (Gibco BRL, Rockville, MD, USA) supplemented with 10% of fetal bovine serum and 1% of penicillin and streptomycin was used.
RNA extraction and real-time PCR analyses
Total RNA was extracted from cells, tissues and serum using TRIzol Reagent (Molecular Research Center Inc., Cincinnati, OH, USA). cDNA was synthesized from 2 μg of total RNA using Transcriptor Reverse Transcriptase (Roche, Indianapolis, IN, USA). β-Actin was used as internal control; the primer sequences for β-actin were as follows: forward, 5′-AAAAGCCACCCCACTTCTCT-3′ and reverse, 5′-CTCAAGTTGGGGGACAAAAA-3′. The primer sequences of pre-45s rRNA were as follows: forward, 5′-CCGTCCGTCCGTCGTCCTCCTCGC-3′ and reverse, 5′-TGTACCGGCCGTGCG TACTTAGAC-3′. Real-time PCR was performed using the SYBR Green master mixture (Roche, Indianapolis, IN, USA) on the ABI7500 Instrument (Thermo Fisher Scientific, Grand Island, NY, USA). Each sample was tested in triplicate. ΔΔCT method was used to determine the fold change in gene expression level. ΔCT method was used to determine the relative expression levels of corresponding genes.
RNA interference and transfection
Knockdown of pre-45s rRNA expression in was performed by siRNA targeting pre-45s rRNA (5'-UCAACCCAAGGACACACGA[dT][dT]-3' (Sigma-Aldrich, St Louis, MO, USA). HCT116, HT29 and NCM460 cells were transfected with 50 nM APC siRNA (s1434; Life Technologies, Carlsbad, CA, USA), CaMKII siRNA (L-004536; Dharmacon, Lafayette, CO, USA), ribosomal S6K siRNA (L-003616; Dharmacon), UBF siRNA (sc-29514, Santa Cruz Biotechnology, Dallas, TX, USA) or control siRNA (AM4611) using Lipofectamine 2000 (Life Technologies).
Chemicals
KN93 (Santa Cruz Biotechnology), H89 (Sigma-Aldrich), l-NMMA (Cayman Chemical Company, Ann Arbor, MI, USA), EGTA-AM (Life Technologies), BAPTA-AM (Sigma-Aldrich) and BAPTA (Sigma-Aldrich) were purchased. These chemicals were dissolved in dimethyl sulfoxide or double-distilled water.
Western blot analysis
Total protein was separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The proteins in sodium dodecyl sulfate–polyacrylamide gel electrophoresis were transferred onto nitrocellulose membranes (GE Healthcare, Piscataway, NJ, USA). The membrane was incubated with primary antibodies overnight, and then with secondary antibody at room temperature for 1 h. Proteins of interest were visualized using ECL Plus Western Blotting Detection Reagents (GE Healthcare). The antibodies used and their dilutions were listed in Supplementary Table S1.
Northern blot
DNA probe against pre-45s rRNA 5′-CTGACACGCTGTCCTCTGGCGA-3′ and the probe against actin 5′-TAGGATGGCAAGGGACTTCCTG-3′ were used. The probes were biotin-labeled. The RNA sample was denatured by heating at 65 °C for 15 min in 50% of formamide. Ten micrograms of denatured RNA was resolved by electrophoresis in 4% of denaturing polyacrylamide gel. RNA in the gel was then transfered to Nylon membrane Hybond-N+ (GE Healthcare). RNA was crosslinked to the membrane by UV illumination. Signal was generated by using North2South Chemiluminescent Hybridization and Detection Kit (Life Technologies). X-ray film was used to show the signal.
Cell viability assay
Cell viability was examined using the Vybrant MTT Cell viability Assay Kit (Life Technologies) according to the manufacturer’s instructions. All experiments were conducted three times in triplicates. Bromodeoxyuridine (B23151) and Bromodeoxyuridine Monoclonal Antibody Alexa Fluor 488 (B35130) were purchased from Life Technologies. The labeling and detection were performed according to the manufacturer’s protocol.
Protein synthesis measurement
Click-iT HPG Alexa Fluor 488 Protein Synthesis Assay Kit was purchased (Life Technologies). Experiments were performed according to manufacturer’s instructions. All experiments were conducted three times in triplicates.
Colony formation assay
Cells (2 × 105/well) were plated in a 12-well plate. After 24 h, cells were treated with indicated treatment. After culturing for 4 to 12 days, cells were fixed with 70% ethanol and stained with 0.5% crystal violet solution. Colonies with more than 50 cells per colony were counted. All experiments were conducted three times in triplicates.
Cell-cycle analysis
The cells (HCT116, HT29, Caco-2 and NCM460) were fixed in 70% ethanol-phosphate-buffered saline for 24 h. The cells were then labeled with 50 μg/ml of propidium iodide (BD Pharmingen, Franklin Lakes, NJ, USA). The cells were sorted by FACSCalibur (BD Biosciences, San Diego, CA, USA). Cell-cycle profiles were analyzed by the ModFit 3.0 Software (BD Biosciences). All experiments were conducted three times in triplicates.
Ca2+ measurement
Cells were treated with 4 μm of Fura-2-AM (Life Technologies) for 10 min. Fluorescence intensity was measured by Perkin-Elmer Fluorescence Spectrometer (Perkin-Elmer, Waltham, MA, USA). Standard curve was plotted for calculating the concentration of Ca2+. All experiments were conducted three times in triplicates.
Statistical analysis
The difference in pre-45s rRNA levels between paired tissue samples was determined by the Wilcoxon's matched-pair test. Correlations between tumor size and pre-45s rRNA expression level were determined by the Spearman's correlation test. Disease-free HR of survival associated with pre-45s rRNA level and other predictor variables were first estimated using Univariate Cox proportional hazards regression model. Multivariate Cox model was constructed to estimate the adjusted HR for high expression of pre-45s rRNA. P<0.05 was taken as statistical significance. All the tests were performed by the Graphpad Prism 5.0 (GraphPad Software Inc., La Jolla, CA, USA) or SPSS18 (IBM Corporation, Armonk, NY, USA).
Abbreviations
- ATP:
-
adenosine triphosphate
- APC:
-
adenomatous polyposis coli
- BAPTA:
-
1,2-bis(o-aminophenoxy) ethane-N,N,N′,N′-tetraacetic acid
- cAMP,:
-
cyclic adenosine 3′, 5′-monophosphate
- Ca2+:
-
calcium ion
- CaMKII:
-
calcium/calmodulin-dependent protein kinase II
- CRC:
-
colorectal cancer
- HR:
-
hazard ratio
- MDM2:
-
mouse double minute 2 homolog
- pre-45s rRNA:
-
precursor 45s ribosomal RNA
- RpL11:
-
ribosomal protein L11
- S6K:
-
ribosomal S6 kinase
- UBF:
-
upstream binding factor.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D . Global cancer statistics. CA Cancer J Clin 2011; 61: 69–90.
Fujita T . Colorectal cancer. Lancet 2010; 376: 331–331.
Grummt I . Life on a planet of its own: regulation of RNA polymerase I transcription in the nucleolus. Gene Dev 2003; 17: 1691–1702.
Mullineux ST, Lafontaine DLJ . Mapping the cleavage sites on mammalian pre-rRNAs: Where do we stand? Biochimie 2012; 94: 1521–1532.
Rodnina MV, Wintermeyer W . Recent mechanistic insights into eukaryotic ribosomes. Curr Opin Cell Biol 2009; 21: 435–443.
van Riggelen J, Yetil A, Felsher DW . MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer 2010; 10: 301–309.
Ruggero D, Pandolfi PP . Does the ribosome translate cancer? Nat Rev Cancer 2003; 3: 179–192.
Khoo KH, Verma CS, Lane DP . Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov 2014; 13: 217.
Kim TH, Leslie P, Zhang YP . Ribosomal proteins as unrevealed caretakers for cellular stress and genomic instability. Oncotarget 2014; 5: 860–871.
Shifman JM, Choi MH, Mihalas S, Mayo SL, Kennedy MB . Ca2+/calmodulin-dependent protein kinase II (CaMKII) is activated by calmodulin with two bound calciums. Proc Natl Acad Sci USA 2006; 103: 13968–13973.
Hannan KM, Brandenburger Y, Jenkins A, Sharkey K, Cavanaugh A, Rothblum L et al. mTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Mol Cell Biol 2003; 23: 8862–8877.
Kihm AJ, Hershey JC, Haystead TAJ, Madsen CS, Owens GK . Phosphorylation of the rRNA transcription factor upstream binding factor promotes its association with TATA binding protein. Proc Natl Acad Sci USA 1998; 95: 14816–14820.
Monteith GR, McAndrew D, Faddy HM, Roberts-Thomson SJ . Calcium and cancer: targeting Ca2+ transport. Nat Rev Cancer 2007; 7: 519–530.
Prevarskaya N, Ouadid-Ahidouch H, Skryma R, Shuba Y . Remodelling of Ca2+ transport in cancer: how it contributes to cancer hallmarks? Philos Trans R Soc Ser B 2014; 369: 20130097.
Panner A, Wurster RD . T-type calcium channels and tumor proliferation. Cell Calcium 2006; 40: 253–259.
Sasaki M, Kawahara K, Nishio M, Mimori K, Kogo R, Hamada K et al. Regulation of the MDM2-P53 pathway and tumor growth by PICT1 via nucleolar RPL11. Nat Med 2011; 17: 944–U153.
Dai MS, Lu H . Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J Biol Chem 2004; 279: 44475.
Dai MS, Zeng SX, Jin Y, Sun XX, David L, Lu H . Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol Cell Biol 2004; 24: 7654–7668.
Zhang YP, Wolf GW, Bhat K, Jin A, Allio T, Burkhart WA et al. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol Cell Biol 2003; 23: 8902–8912.
Jimenez-Vidal M, Srivastava J, Putney LK, Barber DL . Nuclear-localized Calcineurin homologous protein CHP1 interacts with upstream binding factor and inhibits ribosomal RNA synthesis. J Biol Chem 2010; 285: 36260–36266.
Bywater MJ, Poortinga G, Sanij E, Hein N, Peck A, Cullinane C et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell 2012; 22: 51–65.
Panov KI, Friedrich JK, Russell J, Zomerdijk JCBM UBF . activates RNA polymerase I transcription by stimulating promoter escape. EMBO J 2006; 25: 3310–3322.
Fearnhead NS, Britton MP, Bodmer WF . The ABC of APC. Hum Mol Genet 2001; 10: 721–733.
Fodde R, Smits R, Clevers H . APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer 2001; 1: 55–67.
Schneikert J, Behrens J . The canonical Wnt signalling pathway and its APC partner in colon cancer development. Gut 2007; 56: 417–425.
Grzmil M, Hemmings BA . Translation regulation as a therapeutic target in cancer. Cancer Res 2012; 72: 3891–3900.
De A . Wnt/Ca2+ signaling pathway: a brief overview. Acta Biochim Biophys Sin 2011; 43: 745–756.
Kestler HA, Kuhl M . Generating a Wnt switch: it's all about the right dosage. J Cell Biol 2011; 193: 431–433.
Acknowledgements
This project was supported by RGC-GRF Hong Kong (766613, 14106145, 14111216) HMRF Hong Kong (1196728), The National Key Technology R&D Program (2014BAI09B05), 135 program project (2016YFC1303200), and Shenzhen Virtual University Park Support Scheme to CUHK-Shenzhen Research Institute.
Author contributions
HT designed the study, performed the experiments, analyzed data and drafted the manuscript. KCL, YD, CKL and JZ performed experiments. SCN, SSMN, YXC, SZ and JF collected samples. XZ analyzed data and revised the manuscript. JY designed, supervised the studies and wrote the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Tsoi, H., Lam, K., Dong, Y. et al. Pre-45s rRNA promotes colon cancer and is associated with poor survival of CRC patients. Oncogene 36, 6109–6118 (2017). https://doi.org/10.1038/onc.2017.86
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2017.86
This article is cited by
-
RRP15 deficiency induces ribosome stress to inhibit colorectal cancer proliferation and metastasis via LZTS2-mediated β-catenin suppression
Cell Death & Disease (2023)
-
UBTF tandem duplications define a distinct subtype of adult de novo acute myeloid leukemia
Leukemia (2023)
-
UBTF tandem duplications are rare but recurrent alterations in adult AML and associated with younger age, myelodysplasia, and inferior outcome
Blood Cancer Journal (2023)
-
Amelioration for an ignored pitfall in reference gene selection by considering the mean expression and standard deviation of target genes
Scientific Reports (2022)
-
UBTF facilitates melanoma progression via modulating MEK1/2-ERK1/2 signalling pathways by promoting GIT1 transcription
Cancer Cell International (2021)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}