
Abstract
TP53 plays essential roles in tumor initiation and progression, and is frequently mutated in cancer. However, pharmacological stabilization and reactivation of p53 have not been actively explored for targeted cancer therapies. Herein, we identify a novel Cyclophilin A (CypA) small molecule inhibitor (HL001) that induces non-small cell lung cancer (NSCLC) cell cycle arrest and apoptosis via restoring p53 expression. We find that HL001 stabilizes p53 through inhibiting the MDM2-mediated p53 ubiquitination. Further mechanistic studies reveal that the downregulation of G3BP1 and the induction of reactive oxygen species and DNA damage by HL001 contribute to p53 stabilization. Surprisingly, HL001 selectively suppresses tumor growth in p53 wild-type NSCLC harboring Arg72 homozygous alleles (p53-72R) through disrupting interaction between MDM2 and p53-72R in a CypA-dependent manner. Moreover, combining HL001 with cisplatin synergistically enhance tumor regression in orthotopic NSCLC mouse model. Collectively, this study demonstrates that pharmacologic inhibition of CypA offers a potential therapeutic strategy via specific activation of p53-72R in NSCLC.
Similar content being viewed by others


Introduction
Lung cancer is one of the most fatal malignancies worldwide, which represents about 27% of the leading cause of all cancer deaths in 2016.1 Advances in kinase inhibitors, such as gefitinib and erlotinib, have been effective in treating non-small cell lung cancer (NSCLC).2 However, patients treated with those kinase inhibitors often develop drug resistance, and their prolong survivals are typically only a few months.3, 4 In addition, most currently therapeutic agents often cause severe toxicity due to lacking of targeted specificity between cancer and normal cells.5, 6 Thus, development of new molecularly targeted therapeutic agents is very urgent to improve the clinical outcomes for cancer patients.7
Cyclophilin A (CypA), known as a peptidyl prolyl cis-trans isomerase, is overexpressed in multiple types of cancer (for example, NSCLC) and plays a critical role in tumor transformation and metastasis.8 For example, CypA stimulates cell proliferation through binding to cell surface receptor CD147 and activating ERK1/2 signaling pathways.9, 10 CypA is also able to inhibit apoptosis by sequestering cytochrome c.11 Knockdown of CypA resulted in decreased cell proliferation and increased apoptosis in NSCLC in vivo,12 suggesting that CypA is a potential therapeutic target in NSCLC.
The tumor suppressor p53, encoded by TP53, plays essential roles in cell cycle, DNA repair and apoptosis.13 TP53 is frequently inactivated by mutations or deletions in multiple cancer types.14 Recent studies demonstrated that restoration and reactivation of wild-type p53 (p53WT) function prompt effective tumor suppression.15 Hence, pharmacological restoration and activation of p53WT activity might provide a promising therapeutic strategy for the timely development of the molecularly targeted cancer therapies in clinic.14
In this study, we report a small molecule CypA inhibitor (HL001) that selectively suppresses tumor growth of NSCLC harboring p53WT Arg72 homozygous alleles (p53-72R) both in vitro and in vivo by blocking the proteasomal degradation of p53WT. Furthermore, a combination of HL001 with cisplatin synergistically inhibits tumor growth and induces tumor regression in vivo. These findings suggest that pharmacologic inhibition of CypA offer a promising avenue for the treatment of NSCLC patients with p53-72R.
Results
CypA overexpression predicts poor prognosis in lung cancer
To determine the significance of CypA in human lung cancer, we investigated the differential expression profiles of CypA-coding gene (PPIA) in lung adenocarcinoma (LUAD) data set16 from The Cancer Genome Atlas (TCGA). We found that PPIA was significantly overexpressed in LUAD (P=2.1 × 10−12, Kolmogorov–Smirnov test) compared to matched normal tissues (Figure 1a). To further validate the clinical role of PPIA expression in lung cancer, we correlated the expression of PPIA with overall survival of LUAD patients in TCGA. In the Kaplan–Meier survival analyses, we found that high PPIA expression was significantly correlated with poor prognosis in LUAD patients (P=9.4 × 10−3, Figure 1b).

The anti-proliferation activity of HL001 in vitro. (a) CypA (encoded by PPIA) differential expression analysis in LUAD using the RNA-seq (V2, read count, log2 scale) data from TCGA. The significance (P-value) of differential expression in different groups was calculated using Kolmogorov–Smirnov test. (b) Kaplan–Meier survival analysis of CypA in LUAD patients. Patients were grouped into lowly (green) and highly (red) expressed groups based on the mean expression level. All P-values of Kaplan–Meier survival analysis were performed using a log-rank test. (c) Chemical structure of HL001. (d) Graph indicates IC50 values of HL001 in various cancer cell lines. Colors demonstrate cell lines from different tissues: red, lung cancer; blue, breast cancer; yellow, colorectal cancer; pink, pancreatic cancer; aqua, liver cancer; green, prostatic cancer; orange, cervical cancer; black, neuroblastoma; purple, gastric cancer. (e) Cellular thermal shift assay showing CypA target engagement by HL001 in intact A549 cells. A549 cells were incubated with HL001 for 12 h, and cellular thermal shift assay was carried out. (f and g) A549-CypA-knockout cells are resistant to HL001. CRISPR/Cas9-mediated CypA-knockout A549 cells were analyzed by western blotting (f). The IC50 values of HL001 against CypA-knockout A549 cells represent mean±s.d. of independent biological triplicates representative of three experiments (g). (h) S cell cycle arrest induced by HL001 in A549 cells. A549 cells were treated with HL001 for 36 h at the indicated concentrations and fixed with ethanol. The cells were stained with PI to detect DNA content by FACS analysis. Representative results from three independent experiments are shown. Quantitative analysis was provided in Supplementary Figure 1h. (i) Apoptosis induced by HL001 in A549 cells. A549 cells were treated with HL001 for 72 h at the indicated concentrations, double-stained with AnnexinV-FITC and PI, and analyzed by flow cytometry. Representative results from three independent experiments are shown. Quantitative analysis was provided in Supplementary Figure 1k.
A CypA inhibitor HL001 inhibits cell viability in NSCLCs
We previously identified HL001 as a highly potent CypA peptidyl prolyl cis-trans isomerase inhibitor (Figure 1c) with an half maximal inhibitory concentration (IC50) value of 31.6 nM.17 To comprehensively investigate the anti-tumor activities of HL001 in human cancers, we evaluated a panel of lung cancer cell lines (H128, H209, H524, H460, A549, H1299, H522, H1975, H1395, H292, H358, H1650, H157, H23, H2126, H1703 and H838) as well as breast cancer (MCF7 and MDA-MB-231), colorectal cancer (HCT116, HT-29 and Ls174T), pancreas cancer (PANC-1 and SW1990), hepatocellular cancer (HepG2 and Hep3B), prostate cancer (PC3), cervical cancer (Hela and SiHa), neuroblastoma (IMR-32 and SH-SY5Y) and gastric cancer (BGC823, MGC803, SNU-1 and AGS) (Figure 1d and Supplementary Table 1). HL001 displayed anti-proliferation effects on five lung cancer cell lines (A549, H292, H460, H838 and H1650) with IC50 value less than 10 μM. HL015,17 an active analog of HL001 with an IC50 value of 159 nM in CypA enzyme assay, showed similar suppressing activities in HL001-sensitive lung cancer cells (A549 and H460), but not effective in HL001-resistant cells (H1395 and H1975). In comparison, two inactive HL001 analogs,17 HL003 and HL008, had no proliferation effect on both HL001-sensitive and -resistant cell lines (Supplementary Figure 1a and Supplementary Table 2).
HL001 inhibits the proliferation of lung cancer cells
To investigate the potential molecular mechanisms by which HL001 regulates cell viability, we first examined whether CypA was indeed targeted by HL001 in intact cells using cellular thermal shift assay on A549 cells. Figure 1e shows that HL001 stabilizes CypA in intact cells, suggesting that CypA is a binding protein of HL001. CypA stabilization following HL001 treatment was also detected in other lung cancer cells, including H460, H1395, H1975 and H1299 (Supplementary Figures 1b–e). Furthermore, knockout of CypA by CRISPR/Cas9-mediated genome editing markedly diminished effectiveness of HL001 in A549, H460 and H292 cells (Figures 1f and g and Supplementary Figures 1f–g), supporting that CypA is a major functional target of HL001. We next performed cell cycle analysis to characterize the effects of HL001. After 36 h treatment, HL001 caused a dramatic decrease in the number of cells in the G1 phase and an increase in the percentage of cells in the S phase (Figure 1h and Supplementary Figure 1h). HL001 also reduced the expression of several cell cycle-related proteins including Cyclin D1, Cyclin E, CDK2 and Cdc25A (Supplementary Figure 1i). In contrast, cellular apoptosis was increased after incubation with HL001 for 72 h, detected by the Annexin V staining assay (Figure 1i and Supplementary Figure 1j). The increased apoptosis was accompanied by an increased level of three pro-apoptotic proteins: Puma, Noxa and Bax (Supplementary Figure 1k).
G3BP1 is associated with the anti-tumor activity of HL001
We next performed two-dimensional (2D) PAGE analysis to investigate differentially expressed proteins that involves in anti-proliferation effects of HL001 in A549 cells (Supplementary Figures 2a–b). A total of 13 proteins were identified by mass spectrometry analysis (Supplementary Table 3). These identified proteins are involved in various cellular functions, including signal transduction, cell metabolism, proteolysis, cytoskeleton constituent, transcription regulation and immune regulation. We focused on Ras GTPase-activating protein-binding protein 1 (G3BP1), since it was previously known to be involved in cell proliferation and cell cycle regulation.18 As shown in Figures 2a–c and Supplementary Figures 3a–d, HL001 treatment lead to G3BP1 downregulation in a dose- and time-dependent manner in three different NSCLC cell lines (A549, H460 and H292).

Decreased expression of G3BP1 in A549 cells induced by HL001. (a) The quantitative differences of G3BP1 expression after the treatment of 10 μM HL001 in the 2-DE images through three independent experiments. Bar groups represent the mean±s.d. Student’s t-test was performed, *P<0.05. (b and c) Western blotting suggesting decreased G3BP1 induced by HL001. A549 cells were incubated with 20 μM HL001 for different time (b) or different concentrations of HL001 for 36 h (c), and cell lysates were analyzed by western blotting. (d) Kaplan–Meier survival analysis of G3BP1 in LUAD patients. (e and f) The knockdown of G3BP1 suppresses the proliferation of A549 cells. A549 cells stably expressing control shRNA or G3BP1 shRNA were lysed and subjected to western blotting (e), and the proliferation of A549-shG3BP1 cells were assessed by MTS assay at 24, 48, 72 and 96 h through three independent experiments carried out in triplicate (f). Bar groups represent the mean±s.d. Student’s t-test was performed, **P<0.01. (g–i) The overexpression of G3BP1 produces resistance to the suppression activity of HL001. A549 cells stably expressing G3BP1 were analyzed by western blotting (g), and the proliferation (h) and cell viability (i) of A549-shG3BP1 cells were assessed by MTS assay at 24, 48, 72 and 96 h in the presence of 20 μM HL001 through three independent experiments carried out in triplicate. Bar groups represent the mean±s.d. Student’s t-test was performed, **P<0.01, ***P<0.001.
To examine the relevance of G3BP1 in human lung cancer, we performed TCGA data analysis to investigate the correlation between G3BP1 expression and overall survival in LUAD patients. G3BP1 overexpression is significantly correlated with poor survival in LUAD patients (P=9.0 × 10−4, Figure 2d). Knockdown of G3BP1 in A549 cells lead to a slower proliferation compared to control cells (Figures 2e and f), consistent with a previous study.18 Moreover, resistance to HL001 was found in G3BP1-overexpressed A549 stable cell lines (Figures 2g–i). Collectively, those findings suggest that G3BP1 plays a potential role for anti-proliferation activity of HL001.
HL001 perturbs the interaction between CypA and G3BP1
We next investigated whether CypA physically interacted with G3BP1 and further contributed to anti-proliferation activity of HL001. Figure 3a and Supplementary Figure 3e reveal a potential binding between CypA and G3BP1 via co-immunoprecipitation (Co-IP) assay. We further examined the interaction between CypA and G3BP1 by pull-down assay using CypA-conjugated beads and A549 cell lysates (Supplementary Figure 3f). Figure 3b shows that CypA variant R55A (retained less than 1% catalytic activity19) abolishes the interaction with G3BP1 using the purified recombinant proteins. Additionally, incubation of CypA with increasing concentration of HL001 causes a gradual reduction of complex formation with G3BP1 (Figure 3c). Surface plasmon resonance (Biacore, GE Healthcare, Piscataway, NJ, USA) assay shows that CypA binds to G3BP1 with a KD value of 4.26 μM. Moreover, when the mixture harboring CypA and increased concentration of HL001 were passed over the biosensor surface of G3BP1, the binding capability between G3BP1 and CypA was decreased with a KD value of 64.9 μM (Figure 3d). Figure 3e further shows that CypA is colocalized with G3BP1 in cytoplasm by confocal microscopy. In addition, HL001 do not alter the intracellular localization of G3BP1, CypA and its mutant CypA R55A (Supplementary Figure 3g). Finally, we performed binding assay using truncated G3BP1 to map the region of G3BP1 required for CypA binding. Figure 3f reveals that a RNA-recognition motif domain (amino-acid residues 230–420) of G3BP1 is an essential region for CypA binding.

Downregulated G3BP1 by HL001 via inhibiting the interaction between CypA and G3BP1. (a) Co-IP to detect the interaction between CypA and G3BP1 in A549 cells. Proteins retained on the beads were analyzed by western blotting. Non-related antibodies were used as control. (b) The interaction between CypA and G3BP1 relies on the peptidyl prolyl cis-trans isomerase activity. G3BP1 was incubated with CypA WT beads or CypA R55A beads, and proteins retained on the beads were analyzed by western blotting. Bovine serum albumin-conjugated beads were used as control. (c) HL001 inhibits the interaction between CypA and G3BP1 demonstrated by pull-down assay. The lower band indicated that equal G3BP1 proteins were loaded in the pull-down assay. (d) Surface plasmon resonance binding assay for CypA and G3BP1. (e) Colocalization of G3BP1 and CypA. A549 cells were fixed and stained as indicated. (f) Region of G3BP1 necessary for binding to CypA. Different purified truncated G3BP1 was incubated with CypA beads, and proteins retained on the beads were analyzed by western blotting. Bovine serum albumin-conjugated beads were used as control. (g) A549 cells stably expressing control shRNA or CypA shRNA were analyzed by western blotting. (h) A549 cells were treated with HL001 for 36 h, and mRNA was extracted and subjected to real-time PCR. Values represent mean±s.d. of three independent experiments carried out in triplicate. (i) A549 cells were treated with 20 μM HL001 in the presence of CHX (20 μg ml−1) and collected at the indicated time points. Cell lysates were analyzed by western blotting. (j) Quantification of protein half-life analysis of G3BP1 described in i. Values represent mean±s.d. of three independent experiments carried out in triplicate.
To investigate the role of CypA in the regulation of G3BP1, we generated a stable A549 cell line bearing CypA knockdown by shRNA-mediated gene silencing. Figure 3g shows that CypA knockdown leads to the downregulation of G3BP1 level. In addition, G3BP1 mRNA level is not affected by HL001 as determined by RT–PCR assay (Figure 3h). In the presence of cycloheximide (an inhibitor of protein biosynthesis in eukaryotic organisms), G3BP1 protein level slowly decreased in a period of 12 h. However, protein half-life analysis reveals that G3BP1 is markedly degraded at 8 h by HL001 treatment (Figures 3i and j). A previous study showed that CypA regulated protein folding and assembly via its peptidyl prolyl cis-trans isomerase activity,8 suggesting that the interaction between CypA and G3BP1 might play a critical role for maintaining G3BP1 protein stability. However, HL001 causes similar extent of G3BP1 downregulation in both HL001-sensitive and -resistant cell lines (Supplementary Figure 3h). Therefore, we next examined additionally possible molecular mechanisms mediating the selectivity for anti-tumor activity of HL001.
HL001 induces p53 activation
A careful comparison of genotype between HL001-sensitive and -resistant cells revealed that cancer cells carrying p53 mutant or p53WT have different responses to HL001, suggesting that HL001 may regulate p53 function in NSCLC (Figure 1d and Supplementary Table 1). Indeed, treatment of HL001 increased steady-state p53 levels in a time- and dose-dependent manner. p21WAF1/CIP1, a key downstream regulator of p53, was also elevated upon HL001 treatment (Figures 4a and b). Knocked down p53 by shRNA-mediated gene silencing revealed that loss of p53 abrogates HL001-induced growth inhibition (Figures 4c and d and Supplementary Figures 4a–b). In addition, HL001 fails to induce S-phase arrest in p53 knockdown cells (Figure 4e and Supplementary Figure 4c), suggesting that the S-phase arrest induced by HL001 is p53 dependent. Immunofluorescence analysis further shows that HL001 induces p53 translocation into nucleus, suggesting p53 activation (Figure 4f). Consistent with the confocal microscopy studies, upregulation of p53 in nucleus is also observed by western blotting (Supplementary Figure 4d). Furthermore, gene silence of p53 target gene p21WAF1/CIP1 markedly attenuates HL001-induced S-phase arrest (Supplementary Figure 4e). Taken together, p53 activation plays a potential role in HL001-mediated tumor growth suppression.

p53 activation induced by HL001 in A549 cells. (a and b) A549 cells were incubated with 20 μM HL001 for different incubating time (a) or different concentrations of HL001 for 36 h (b), and cell lysates were analyzed by western blotting. (c and d) The knockdown of p53 produces resistance to the suppression activity of HL001. A549 cells stably expressing control shRNA or p53 shRNA were analyzed by western blotting (c), and cell viability was evaluated in the presence of 20 μM HL001 through three independent experiments carried out in triplicate (d). Bar groups represent the mean±s.d. Student’s t-test was performed, ***P<0.001. (e) A549 cells stably expressing control shRNA or p53 shRNA were treated with HL001 for 36 h at the indicated concentrations and fixed with ethanol. The cells were stained with PI to detect DNA content by FACS analysis. The number of events was plotted against the fluorescence intensity (DNA content). Representative results from three independent experiments are shown. Quantitative analysis was provided in Supplementary Figure 4c. (f) Subcellular location of p53. A549 cells were treated with 20 μM HL001 for 36 h, and were fixed and stained as indicated. (g) A549 cells were treated with 20 μM HL001. After 36 h, mRNA was extracted and subjected to real-time PCR. (h) Regulation of p53 ubiquitination by HL001. A549 cells were treated with 20 μM HL001 for 36 h and additional 4 h with 20 μM MG132 before collection. p53 was immunoblotted with monoclonal anti-p53 (DO-1) antibody. (i) The activation of p53 is CypA dependent. A549 cells stably expressing control shRNA or CypA shRNA were treated with 20 μM HL001 for 36 h, and cell lysates were analyzed by western blotting.
To explore the mechanisms underlying p53 upregulation, we sought to identify p53 regulator whose function was altered after HL001 treatment. RT–PCR analysis shows that p53 mRNA levels in A549, H460 and H292 cells were not affected by the incubation of HL001 (Figure 4g and Supplementary Figures 4f–g), suggesting that the upregulated p53 may be a result of the decreased degradation. Previous studies showed that p53 activity is mainly controlled by MDM2-mediated ubiquitination and degradation via the proteasome.20 Accordingly, we examined the effect of HL001 on p53 ubiquitylation in NSCLC cells. In the presence of proteasome inhibitor MG132, a ladder of slowly migrating ubiquitylated forms was observed. Addition of HL001 inhibits ubiquitination of p53 in three different NSCLC cell lines (Figure 4h and Supplementary Figures 4h–i). We reasoned that CypA might participate in p53 ubiquitination and degradation. To test this hypothesis, CypA was knocked down in A549 cells via shRNA-mediated gene silencing. Loss of CypA reduced HL001-induced elevation of p53 (Figure 4i), suggesting that HL001-mediated p53 activation in A549 cells by targeting CypA. Collectively, these observations suggest that HL001 activates p53 function by reducing its ubiquitination and degradation.
A recent study suggested that the decreased G3BP1 also contributed to p53 stabilization by diminishing the redistribution of p53 from nucleus to cytoplasm.21 To investigate the relationship between G3BP1 downregulation and p53 activation induced by HL001, shG3BP1 stable knockdown A549 cells were treated with HL001, and p53 activation appeared at an earlier time point (6 h) (Supplementary Figure 5a), compared to the control cells at 8 h (Figure 4a). In addition, G3BP1 cannot interact with MDM2 and regulate the function of MDM2 directly (Supplementary Figure 5b), and has no effect on p53–MDM2 interaction (Supplementary Figure 5c), consistent with previous data.21 In summary, although G3BP1 plays a potential role for anti-proliferation activity of HL001 (Figure 2), the downregulation of G3BP1 by HL001 may partially contribute to p53 stabilization via inhibiting p53 redistribution from nucleus to cytoplasm.
HL001 induces oxidative stress and subsequent DNA damage
As p53 is known to be activated by cellular stress, such as endoplasmic reticulum stress, oxidative stress and DNA damage,22 we examine whether HL001-induced cellular stress. Supplementary Figures 6a–c show that HL001 induces reactive oxygen species (ROS) generation and γ-H2AX phosphorylation. However, HL001 has minor effects on two endoplasmic reticulum stress markers: GRP78 and XBP-1.
We further used the antioxidant N-acetyl-L-cysteine (NAC) to abrogate the oxidative stress to examine the role of ROS in HL001-induced p53 activation. Supplementary Figure 6d shows that NAC (5 mM) greatly suppresses ROS generation and DNA damage induced by HL001. Of note, in the presence of NAC (5 mM), HL001 still activates p53 in a time- and dose-dependent manner (Supplementary Figures 6e–f). Interestingly, the steady activation of p53 is delayed to 10 h (Supplementary Figure 6e) in the presence of NAC (5 mM), compared to the p53 activation of control cells at 8 h (Figure 4a). Furthermore, the S-phase arrest induced by HL001 becomes weaker (S-phase arrest was decreased from 23.52 to 15.25%) (Supplementary Figure 6g), and the IC50 value increases from 7.22 to 10.35 μM in the presence of NAC at 5 mM (Supplementary Table 4).
We next investigated the effects of HL001 on the phosphorylation status of key components in p53 signaling pathway, including MDM2, p53, Cdc25C, CHK1, CHK2 and CDK1 (Supplementary Figures 6h–i). HL001 induces p53 phosphorylation at S15 and S392, but has no effects on S20, S46 and T18. Moreover, HL001 has no effect on the phosphorylation of Cdc25C (S216), CHK1 (S345), CHK2 (T68) and p53 (S6), while the DNA damage agent topotecan induces the elevated levels of phosphorylated Cdc25C (S216), CHK1 (S345), CHK2 (T68) and p53 (S6).
We further examined the activities of two ROS-inducing agents (elesclomol and β-lapachone) and a DNA damage agent (topotecan) against both HL001-sensitive cells (A549, H460 and H838) and HL001-resistant cells (H1395, H1975, H1299, H157, Hela, HCT116, MCF7, MDA-MB-231, PANC-1, H2126, Hep3B and IMR-32) across various cancer types. Those three anti-tumor agents do not show similar selectivity as HL001 (Supplementary Table 5). In addition, HL001 induces similar ROS and DNA damage in both HL001-sensitive (A549 and H460) and HL001-resistant lung cancer cells (H1395 and H1975) (Supplementary Figures 6j–k). Taken together, these results suggest that the induction of ROS generation partially contributes to HL001-induced p53 activation and anti-proliferation activity of HL001.
Selective tumor suppression activity of HL001 in p53WT NSCLC
TP53 is often mutated in approximate 50% cancer patients, whose somatic alterations are associated with tumor progression, adverse prognosis and the development of drug resistance.14, 23 We examined the role of CypA-coding gene PPIA in human lung cancer based on different p53 genotypic statuses. We collected p53 nonsynonymous mutations and copy number variant data from TCGA.16 Interestingly, we found that high PPIA expression was significantly correlated with poor survival in p53WT LUAD patients (P=5.0 × 10−4, Figure 5a). However, high PPIA expression is not significantly correlated with poor survival in p53 mutant patients (P=0.8556, Figure 5a). Furthermore, high PPIA expression is significantly correlated with poor survival rate for patients (P=0.0177) whose tumors are p53 natural diploid. In contrast, high PPIA expression is not significantly correlated with poor survival rate for LUAD patients whose tumors have p53 deletions (P=0.1186) or gains (P=0.8615) in Figure 5b. These findings suggest that CypA inhibitors may improve potential clinical benefits for a subset of lung cancer patients whose tumors are p53WT or natural diploid.

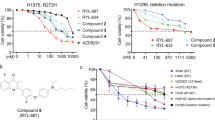
Therapeutic profiles of HL001 on inducing p53 activation and suppressing cell proliferation in cells with p53-72R than p53-72P. (a and b) Kaplan–Meier survival analysis for CypA in LUAD patients grouped by TP53 genotypes: (a) TP53 DNA copy number variants and (b) TP53 mutated (Mut) versus WT. TP53 DNA copy number variant data are grouped based on GISTIC 2.0 values: gain (values=1 or 2), natural diploid (values=0), loss (values=−1 or −2). Patients are categorized into lowly (green) and highly (red) expressed groups based on the mean expression level. All P-values of Kaplan–Meier survival analysis were performed using a log-rank test. (c) Colony formation assay of lung cancer cells (A549, H292, H460, H1395, H1299 and H1975) treated with 1 μM HL001 for 7 days. The cells were fixed and stained with crystal violet. Quantifications of three independent colony formation assays were shown in the right panel. Bar groups represent the mean±s.d. Student’s t-tests were performed, ***P<0.001. (d) HL001 inhibits the proliferation of p53-72R-transfected H1299 cells. H1299 cells transfected with p53-72P or p53-72R were treated with 20 μM HL001 for 72 h, and cell viability was evaluated by three independent MTT assays carried out in triplicate. Bar groups represent the mean±s.d. Student’s t-test was performed, **P<0.01. (e) HL001 stabilizes p53 in p53-72R-transfected H1299 cells. H1299 cells transfected with p53-72P or p53-72R (200 ng) were treated with 20 μM HL001 for 36 h, and cell lysates were analyzed by western blotting. (f) HL001 inhibits the interaction between p53-72R and MDM2. H1299 cells transfected with p53-72P or p53-72R were treated with HL001 for 24 h, and cell lysates were immunoprecipitated with p53 antibodies DO-1. Proteins retained on the beads were analyzed by western blotting. Non-related antibodies were used as control. (g) HL001 selectively activates p53 in cells with p53-72R. H292 (p53-72R/R), H460 (p53-72R/R), H1395 (p53-72P/P) and H128 (p53-72P/P) cells were treated with 20 μM HL001 for 36 h, and cell lysates were analyzed by western blotting.
We next searched in International Agency for Research on Cancer p53 database to investigate the p53 mutation status across different cancer cell lines used in this study. Consistently with TCGA analysis (Figures 5a and b), HL001 is not effective in cancer cells harboring p53 mutations or deletion, but exhibits remarkable suppressing activities in several p53WT cell lines (Supplementary Table 1). We performed the colony formation assay to verify HL001 selectivity in lung cancer cell lines with different p53 status in a broader range of NSCLC cell lines. HL001 at a concentration of 1 μM notably suppresses the colony formation of A549, H460 and H292 cells (p53WT) after 7 days’ incubation, while the growth of H1299 (p53 null) and H1975 (p53 mutant) cells are less affected at the same concentration (Figure 5c). Collectively, p53 genetic status may represent a potential biomarker for characterizing the selectivity of anti-proliferation activity of HL001 in lung cancer.
HL001 specifically activates p53-72R function
Inconsistent responses to HL001 observed in several different p53WT lung cancer cell lines suggest that other genetic profiles may mediate selectivity of HL001 activity in lung cancer (Supplementary Table 1). The codon 72 in p53, a unique polymorphism in human, encodes either proline or arginine. A previous study reported that CypA interacted with the proline-rich region of p53 (amino acids 64–91), and the proline 72 played a critical role in mediating the interaction.24 We next processed DNA sequencing data in both lung and non-lung cancer cells on the exon 4 of p53 and examined whether the codon 72 polymorphism affects selective tumor suppression activity of HL001. Surprisingly, HL001 is more effective (IC50 <10 μM) in cells expressed p53WT with homozygous R/R at the codon 72 (A549, H292, H460, H838 and H1650) than that of homozygous P/P across 17 different lung cancer cell lines (Supplementary Table 1). For instance, the activities of HL001 in lung cancer cell lines expressing p53WT with homozygous P/P (H128, H209 and H1395) are 20 times weaker than cells with R/R at the codon 72.
We expressed p53 with either a proline or an arginine at the codon 72 in H1299 (p53-null lung carcinoma cell line) and examined the cell viability of H1299 cells treated with HL001. After 48 h incubation, the cell viability of H1299 cells transfected with p53-72R is decreased more notably than that of p53-72P (Figure 5d). Moreover, p53-72R level is increased in p53-transfected H1299 cells after the treatment with HL001 for 24 h, while the expression of p53-72P is not affected (Figure 5e).
We further performed Co-IP assay to investigate the selective activation of p53 polymorphism at the codon 72 by HL001. As shown in Figure 5f, the binding of MDM2 to p53-72R in H1299 cell lysates is suppressed by the incubation of HL001, while the interaction between MDM2 and p53-72P is not affected. Pull-down assay using MDM2-conjugated beads and H1299-transfected cell lysates show similar trends (Supplementary Figure 7a). Furthermore, the interaction between MDM2 and p53-72R is not interrupted by HL001 using purified recombinant proteins (Supplementary Figure 7b). Collectively, HL001 is not a direct inhibitor for MDM2–p53-72R interaction.
To identify the specific p53 activation induced by HL001, we investigated whether p53 is activated by HL001 in different lung cancer cells containing p53-72R homozygote (H292 and H460) and p53-72P homozygote (H128 and H1395). We found that p53 was strongly activated by incubation of 20 μM HL001 in H292 and H460 cells expressing p53-72R (Figure 5g). However, p53 expression is not affected in H1395 and H128 cells encoding p53-72P. Furthermore, HL001 induces S-phase arrest in H292 and H460 cells (similar with A549 cells) with p53-72R, while the cell cycle progress of H1395 (p53-72P), H1299 (p53 null) and H1975 (p53 mutant) are not affected significantly (Supplementary Figure 7c). In summary, these observations suggest that HL001 selectively targets p53WT lung cancer cells harboring Arg72 homozygote.
Synergistic effects of HL001 in combination with cisplatin
Cisplatin is a widely used cytotoxic chemotherapeutic agent for the treatment of NSCLC in clinic. The combination of cytotoxic chemotherapy drugs and targeted therapy drugs is an effective approach in lung cancer therapy by minimizing toxicity and acquired resistance.25 Accordingly, we examined the potential efficacy of combining HL001 with cisplatin in A549 cells harboring p53-72R. As shown in Figure 6a, the combination of HL001 with cisplatin displays a dramatically synergistic anti-proliferative effect with a combination index of 0.385 in A549 cells. Furthermore, the colony formation assay confirms that this drug combination triggers dramatically synergistic inhibitory effect against cell growth at a low concentration (Figure 6b). Interestingly, the HL001-cisplatin combination is also effective in HL001-sensitive H460 cells (p53-72R), while it is invalid in HL001-resistant H1395 (p53-72P) and H1975 (p53 mutant) cells (Supplementary Figures 8a–c).

Effects of HL001 in combination with cisplatin on the growth of A549 cells in vitro and in vivo. (a) Evaluation of effect of HL001 in combination with cisplatin on cell viability. A549 cells were treated with several ratios combination of HL001-cisplatin for 48 h, and cell viability was assessed by MTT assay. The combination index was calculated as described in ‘Materials and Methods’ section. Synergism of HL001-cisplatin is shown by combination index <1. (b) Synergistic effects of HL001-cisplatin combination. A549 cells were treated with indicated single drug (HL001, 0.5 μM; cisplatin, 4 μM) or drug combination for 7 days, and the cells were fixed and stained with crystal violet. Quantifications of three independent colony formation assays were shown in the right panel. Bar groups represent the mean±s.d. Student’s t-tests were performed, ***P<0.001. (c) HL001 suppresses A549-generated tumor growth in vivo. Nude mice with tumor xenografts formed by A549 cells were treated daily for 40 days by oral gavage with vehicle or HL001 (20, 60 and 120 mg kg−1). Tumor measurements are mean±s.d. Student’s t-tests were performed, **P<0.01, ***P<0.001, n=6 per group. (d and e) HL001 induces increased p53 and decreased G3BP1 in A549 xenograft tumors. Tumor tissues from different treatment groups were lysed and analyzed by western blotting (d) or immunohistochemical stained by antibodies against p53 or G3BP1 (e). (f and g) In vivo effects of HL001 and cisplatin combination therapy in an orthotopic lung tumor model. Mice with established tumors (n=6 per group) were treated for 30 days with vehicle, HL001 (20 mg kg−1 in 0.5% CMC-Na by daily gavage), cisplatin (1 mg kg−1 in normal saline by intraperitoneal injection once a week) or drug combination of HL001 and cisplatin (HL001, 20 mg kg−1 in 0.5% CMC-Na by daily gavage. Cisplatin, 1 mg kg−1 in normal saline by intraperitoneal injection once a week). The representative bioluminescent 2D images (f) or three-dimensional modeling (g) from each group were shown. (h) Four representative macroscopic observations of lungs in nude mice at day 30.
We then tested the therapeutic effects of HL001 on both xenograft models of p53-72R/R and p53-72P/P tumors by subcutaneous injection of A549 and H1395 cells, respectively. Mice receiving HL001 show a significant reduction in in vivo tumor growth of A549 (p53-72R/R) (P<0.001, Figure 6c), but not for H1395 (p53-72P/P) (Supplementary Figure 8d), consistent with the in vitro observations. In addition, HL001 shows minor effects on A549-CypA R55A cells-derived xenograft model and its inactive analog HL003 fails to impair the in vivo tumor growth of A549-derived xenograft model (Supplementary Figures 8e–f), suggesting that CypA plays important roles for HL001’s anti-cancer activities.
To further evaluate the mechanism of tumor suppression by HL001, we examined the expression of G3BP1, p53 and CypA in the tumor tissues by western blot analyses and immunohistochemistry assays. HL001 upregulates the expression of p53 and downregulates the expression of G3BP1 compared with vehicle group (Figures 6d and e). Taken together, HL001 suppresses lung cancer growth by upregulation of p53 level in vivo.
We further investigated the potency of combinational therapy using an orthotopic lung cancer mouse model in vivo. After 30 days treatment, tumors in the control group grow fast and distribute diffusely in lung tissue. The single-agent-treated group shows a limited anti-tumor activity at the given dose and tumor spread is observed. In contrast, combining HL001 with cisplatin exhibits significantly coordinative anti-tumor activity (Figure 6f and Supplementary Figure 8g). In addition, three-dimensional co-registration also shows that tumors grow and distribute diffusely in the entire lung in the control group or the single-agent group (Figure 6g). In contrast, tumor growth is significantly suppressed and only a small lightspot is found in the HL001 and cisplatin combination group (Figure 6g and Supplementary Movie 1). Finally, macroscopic lung tumors from different groups suggest the synergistic effect of HL001 in combination with cisplatin on lung tumor suppression, consistently with the 2D image and the three-dimensional reconstruction data (Figure 6h). Furthermore, no markedly difference in mouse body weight is observed, indicating low toxicity in the HL001-cisplatin combination (Supplementary Figure 8h). In summary, these results suggest that HL001 offers a promising preliminary lead agent to treat lung cancer by rational combinational therapy with cisplatin for human lung cancers.
Discussion
The development of precision oncology provides novel strategies for prevention and treatment of cancers.26 Genetic profiles like single nucleotide polymorphisms can be exploited to predict risk of cancer or evaluate outcome and/or response to clinical treatment. However, identifying new drugs targeting proteins with specific single nucleotide polymorphism is still in the early stage in precision oncology.27 Over 200 natural p53 sequence variations are reported and one of the most common p53 polymorphisms is at codon 72, encoding either Pro or Arg.28 Comparative genomic studies suggest that p53-72P is the ancestral form, in which p53-72R is found with over 50% frequency in some populations.29 In this study, we found that HL001, a small molecule CypA inhibitor, selectively restores p53-72R homozygote expression and reactivates p53-dependent cancer cell growth suppression. Moreover, HL001-cisplatin drug combination is highly effective in WT p53-72R cells (A549 and H460), but not in WT p53-72P (H1395 cells) and mutant p53-72R (H1975 cells). To the best of our knowledge, this is the first study that p53-72R can be selectively activated by a small molecule, providing a potential biomarker for the development of targeted cancer therapies. Further studies are needed to elucidate the causative pathways and molecular mechanisms involved in highly selective anti-tumor activities of HL001 in p53-72R lung cancer. HL001 and its analog would provide a useful small molecule probe for understanding the functional roles and mechanisms of p53 polymorphism at codon 72.
Previous studies have suggested that p53-72P interacted more readily with inhibitory protein iASPP than p53-72R,30 and iASPP was reported to inhibit p53 activity.31 In addition, the Arg72 variant is associated with greater binding and ubiquitination of p53 by MDM2.32 Moreover, CypA interacts with the proline-rich region of p53 (amino acids 64–91), and the proline 72 plays a critical role in mediating the interaction.24 In this study, we showed that HL001 disrupted the interaction between p53-72R and MDM2 in the presence of CypA using the Co-IP and pull-down assays (Figure 5f). In addition, p53-72R is more stable than p53-72P after treatment with HL001, which result in selectively suppressing growth of the p53-72R lung cancer cells. Further studies will be needed to determine precisely how HL001 specifically regulates p53-72R in clinic.
Alterations in p53 signaling pathways are believed to be needed for the development of most cancers, and various accumulated evidences indicate that the restoration and reactivation of p53 function have the marked therapeutic benefits.33 Although a variety of compounds that restore p53 function have been reported, the biological consequences and clinical benefits of these compounds remain unclear.34, 35 For instance, p53 is activated by various cellular stresses and MDM2 is a main negative regulator of p53. Herein, we showed that HL001 selectively upregulates p53-72R through disrupting interaction between MDM2 and p53-72R in a CypA-dependent manner.
Alternatively, CypA interferes intracellular ROS generation and its inhibitor Cyclosporin A significantly increases intracellular ROS level.36, 37 As a potent CypA inhibitor, HL001 indeed induces ROS generation and DNA damage in a time-dependent manner. DNA damage may be the secondary effect of the oxidative stress induced by HL001, as γ-H2AX phosphorylation is not observed in the presence of NAC. We also found that HL001 still activated p53 even in the presence of antioxidant NAC, although a delayed steady activation of p53. In addition, HL001 induces downregulation of G3BP1 through blocking the interaction between CypA and G3BP1. The downregulation of G3BP1 facilitates early p53 activation, but does not affect HL001-induced steady state of p53 activation. The p53 activation is observed in a panel of NSCLC cell lines by using western blotting and immunofluorescence. However, p53 is lost in 2D PAGE analysis. One possible explanation is that the limited detection sensitivity of 2D PAGE may not cope with the actual dynamic range of p53 protein concentration in cell extracts owing to a well-known instability of p53.38 Collectively, these data suggest that the oxidative stress and G3BP1 downregulation induced by HL001 partially contribute to p53 activation.
Overexpression of CypA plays an important role in the resistance to cisplatin in cancer cells.36 A well-known CypA inhibitor Cyclosporin A significantly and selectively increases the sensitivity of cisplatin-resistant cells to the cytolysis by cisplatin.39 However, several clinical trials utilizing cisplatin/carboplatin in combination with Cyclosporin A yielded unsatisfactory results due to its strong immunosuppressive activity.40 As a potent inhibitor of CypA, HL001 exhibits weak activity on concanavalin A-simulated mouse spleen cell proliferation (data not shown), indicating a safer therapeutic option for cisplatin resistance. Furthermore, we described markedly synergistic effects of HL001 in combination with cisplatin on tumor growth suppression in NSCLC both in vitro and in vivo, providing an achievable targeted therapeutic strategy for individual patient according to their p53 codon 72 polymorphism, although the detailed mechanism of synergistic effects needed to be studied further.
In summary, our preclinical study demonstrated that HL001, a small molecule inhibitor of CypA, selectively activated p53-72R and subsequently inhibited growth of human lung cancer cells. Furthermore, mouse models indicate that in vivo administration of HL001 alone or co-treatment with cisplatin promote significant tumor suppression in NSCLC. The preclinical results presented here demonstrate that CypA inhibitors may serve as a new pharmacologic approach to the targeted therapy for NSCLC.
Materials and methods
Please see the Supplementary Methods for more details.
Cellular protein thermal shift assay
Protein thermal shift assay in intact cells was described previously.41 Briefly, cells were incubated with dimethylsulfoxide as control or HL001 at a final concentration of 20 μM for 12 h. After treatment, the cells were digested with trypsin and resuspended in phosphate-buffered saline (PBS) and the cell suspension was divided into eight equal PCR tubes. The tubes were heated to 44, 46, 48, 50, 52, 54, 56 or 58 °C for 3 min and followed by cooling at room temperature for 3 min. Then the cells were lysed by three cycles of freeze-thawing using liquid nitrogen. The lysates were centrifuged at 20 000 g for 20 min to separate the soluble proteins from precipitates. The supernatants were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) followed by western blot using CypA antibody.
Flow cytometry analysis
Flow cytometry analysis was performed as described previously.42 After reaching 70–80% confluence, cells were treated with different concentrations of HL001 for 36 h. For cell cycle analysis, cells were collected and washed in PBS. The cell pellet was resuspended in PBS and fixed in 70% ice-cold ethanol at 4 °C overnight. The fixed cells were centrifuged and resuspended in 1 ml PBS. RNase A (0.05 mg ml−1) was added and the samples were incubated at 37 °C for 30 min. With the addition of 0.05 mg ml−1 propidium iodide (PI), the incubation was carried out at 4 °C in the dark for 20 min. Cell cycle profiles were analyzed on BD FACS Calibur flow cytometer (Franklin Lakes, NJ, USA).
For cell apoptosis assay, cells were treated with various concentration of HL001 for 72 h. After treatment, cells were collected, washed three times with PBS and resuspended with 500 μl binding buffer at a concentration of 1 × 106 cells per ml. Then 5 μl Annexin V-FITC and 5 μl PI were added and the solution was incubated at room temperature for 20 min. Analysis was carried out using BD FACS Calibur flow cytometer.
Co-IP assay
Co-IP assay was carried out as described previously with modifications.43 H1299 cells were transfected with different cDNA expressing p53-72P, p53-72R or MDM2 for 24 h, and treated with HL001 (20 μM) for 36 h. Cells were washed with cold PBS and lysed in lysis buffer (50 mM Tris-HCl, pH=7.0, 150 mM NaCl, 1 mM EDTA and 1% NP-40 supplemented with protease inhibitor cocktail). The whole-cell lysates were incubated with 2 μg p53 antibody DO-1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and 20 μl protein A/G Sepharose beads (Santa Cruz Biotechnology) overnight at 4 °C. The immunocomplexes were washed with lysis buffer for three times and separated by SDS–PAGE followed by western blotting analysis. As p53 protein band is overlapping with IgG bands, IgG light chain was further detected by using specific anti-IgG light chain secondary antibody from (Jackson immunoResearch, West Grove, PA, USA).
Confocal microscopy
Confocal microscopy assay was performed as described previously with modifications.44 A549 cells were transferred to confocal dishes and treated with 20 μM HL001 for 36 h or not. After washed three times with PBS, the cells were fixed with 4% paraformaldehyde in PBS for 30 min at 4 °C, permeabilized with 0.1% Triton X-100 for 15 min and blocked with 3% bovine serum albumin for 1 h at room temperature. Then the cells were incubated with primary antibody at 4 °C overnight. p53 and G3BP1 were visualized using Alexa Fluor 555-labeled goat anti-mouse secondary antibody, and CypA was visualized using Alexa Fluor 488-labeled goat anti-mouse secondary antibody. The cells were finally incubated with Hoechst 33342 for 30 min at room temperature to stain nucleus. The samples were imaged using Zeiss Axioplan 2 confocal microscopy (Carl Zeiss, Inc., Thornwood, NY, USA).
p53 Codon 72 genotyping
Genotyping of the p53-72 polymorphism was carried out on cancer cells with WT p53 reported by the International Agency for Research on Cancer p53 database. The exon 4 of p53 was amplified from genomic DNA of the cell lines using the forward primer 5′-AAGGGAGTTGGGAATAGG-3′ and reverse primer 5′-GAAGCCAAAGGGTGAAGA-3′. The PCR products were purified and sequenced directly.
Animal studies
The mouse experiments were conducted according to the guidelines of the Institution Animal Care and Use Committee of Shanghai and the National Research Council Guide for Care and Use of Laboratory Animals.
Statistical analysis
Data were represented as the mean±s.d. with at least three independent experiments with triplicate. Comparisons between two groups were analyzed for statistical significance using two tailed Student’s t-test. For multiple comparisons, one-way ANOVAs were applied. The levels of significance were set at P<0.05 (*), P<0.01 (**) and P<0.001 (***).
References
Siegel RL, Miller KD, Jemal A . Cancer statistics, 2016. CA Cancer J Clin 2016; 66: 7–30.
Cohen P, Alessi DR . Kinase drug discovery—what's next in the field? ACS Chem Biol 2012; 8: 96–104.
Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014; 346: 1480–1486.
Knight ZA, Lin H, Shokat KM . Targeting the cancer kinome through polypharmacology. Nat Rev Cancer 2010; 10: 130–137.
Bai JP, Abernethy DR . Systems pharmacology to predict drug toxicity: integration across levels of biological organization. Annu Rev Pharmacol Toxicol 2013; 53: 451–473.
Cheng F, Loscalzo J . Autoimmune cardiotoxicity of cancer immunotherapy. Trends Immunol 2017; 38: 77–78.
Rosell R, Bivona TG, Karachaliou N . Genetics and biomarkers in personalisation of lung cancer treatment. Lancet 2013; 382: 720–731.
Nigro P, Pompilio G, Capogrossi MC . Cyclophilin A: a key player for human disease. Cell Death Dis 2013; 4: e888.
Zhu D, Wang Z, Zhao JJ, Calimeri T, Meng J, Hideshima T et al. The Cyclophilin A-CD147 complex promotes the proliferation and homing of multiple myeloma cells. Nat Med 2015; 21: 572–580.
Yang H, Chen J, Yang J, Qiao S, Zhao S, Yu L . Cyclophilin A is upregulated in small cell lung cancer and activates ERK1/2 signal. Biochem Biophys Res Commun 2007; 361: 763–767.
Bonfils C, Bec N, Larroque C, Del Rio M, Gongora C, Pugnire M et al. Cyclophilin A as negative regulator of apoptosis by sequestering cytochrome c. Biochem Biophys Res Commun 2010; 393: 325–330.
Howard BA, Furumai R, Campa MJ, Rabbani ZN, Vujaskovic Z, Wang XF et al. Stable RNA interference–mediated suppression of cyclophilin A diminishes non-small-cell lung tumor growth in vivo. Cancer Res 2005; 65: 8853–8860.
Kruse J-P, Gu W . Modes of p53 regulation. Cell 2009; 137: 609–622.
Mandinova A, Lee SW . The p53 pathway as a target in cancer therapeutics: obstacles and promise. Sci Transl Med 2011; 3: 64rv61.
Junttila MR, Karnezis AN, Garcia D, Madriles F, Kortlever RM, Rostker F et al. Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature 2010; 468: 567–571.
Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014; 511: 543–550.
Ni S, Yuan Y, Huang J, Mao X, Lv M, Zhu J et al. Discovering potent small molecule inhibitors of cyclophilin A using de novo drug design approach. J Med Chem 2009; 52: 5295–5298.
Shim JH, Su ZY, Chae JI, Kim DJ, Zhu F, Ma WY et al. Epigallocatechin gallate suppresses lung cancer cell growth through Ras-GTPase-activating protein SH3 domain-binding protein 1. Cancer Prev Res 2010; 3: 670.
Zydowsky LD, Etzkorn FA, Chang HY, Ferguson SB, Stolz LA, Ho SI et al. Active site mutants of human cyclophilin A separate peptidyl-prolyl isomerase activity from cyclosporin A binding and calcineurin inhibition. Protein Sci 1992; 1: 1092–1099.
Pant V, Lozano G . Limiting the power of p53 through the ubiquitin proteasome pathway. Genes Dev 2014; 28: 1739–1751.
Kim MM, Wiederschain D, Kennedy D, Hansen E, Yuan ZM . Modulation of p53 and MDM2 activity by novel interaction with Ras-GAP binding proteins (G3BP). Oncogene 2007; 26: 4209–4215.
Junttila MR, Evan GI . p53—a Jack of all trades but master of none. Nat Rev Cancer 2009; 9: 821–829.
Leroy B, Girard L, Hollestelle A, Minna JD, Gazdar AF, Soussi T . Analysis of TP53 mutation status in human cancer cell lines: a reassessment. Hum Mutat 2014; 35: 756–765.
Baum N, Schiene-Fischer C, Frost M, Schumann M, Sabapathy K, Ohlenschlager O et al. The prolyl cis/trans isomerase cyclophilin 18 interacts with the tumor suppressor p53 and modifies its functions in cell cycle regulation and apoptosis. Oncogene 2009; 28: 3915–3925.
Camidge DR, Pao W, Sequist LV . Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol 2014; 11: 473–481.
Arnedos M, Vicier C, Loi S, Lefebvre C, Michiels S, Bonnefoi H et al. Precision medicine for metastatic breast cancer-limitations and solutions. Nat Rev Clin Oncol 2015; 12: 693–704.
Wood AJ, Evans WE, McLeod HL . Pharmacogenomics—drug disposition, drug targets, and side effects. N Engl J Med 2003; 348: 538–549.
Whibley C, Pharoah PD, Hollstein M . p53 polymorphisms: cancer implications. Nat Rev Cancer 2009; 9: 95–107.
Puente XS, Velasco G, Gutierrez-Fernandez A, Bertranpetit J, King MC, Lopez-Otin C . Comparative analysis of cancer genes in the human and chimpanzee genomes. BMC Genomics 2006; 7: 15.
Bergamaschi D, Samuels Y, Sullivan A, Zvelebil M, Breyssens H, Bisso A et al. iASPP preferentially binds p53 proline-rich region and modulates apoptotic function of codon 72–polymorphic p53. Nat Genet 2006; 38: 1133–1141.
Bergamaschi D, Samuels Y, O'Neil NJ, Trigiante G, Crook T, Hsieh JK et al. iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat Genet 2003; 33: 162–167.
Dumont P, Leu JI, Della Pietra AC 3rd, George DL, Murphy M . The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet 2003; 33: 357–365.
Muller PA, Vousden KH . Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 2014; 25: 304–317.
Boeckler FM, Joerger AC, Jaggi G, Rutherford TJ, Veprintsev DB, Fersht AR . Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Pro Natl Acad Sci USA 2008; 105: 10360–10365.
Bykov VJ, Issaeva N, Zache N, Shilov A, Hultcrantz M, Bergman J et al. Reactivation of mutant p53 and induction of apoptosis in human tumor cells by maleimide analogs. J Biol Chem 2005; 280: 30384–30391.
Choi KJ, Piao YJ, Lim MJ, Kim JH, Ha J, Choe W et al. Overexpressed cyclophilin A in cancer cells renders resistance to hypoxia- and cisplatin-induced cell death. Cancer Res 2007; 67: 3654–3662.
Hong F, Lee J, Song J-W, Lee SJ, Ahn H, Cho JJ et al. Cyclosporin A blocks muscle differentiation by inducing oxidative stress and inhibiting the peptidyl-prolyl-cis-trans isomerase activity of cyclophilin A: cyclophilin A protects myoblasts from cyclosporin A-induced cytotoxicity. FASEB J 2002; 16: 1633–1635.
Rajagopalan S, Andreeva A, Teufel DP, Freund SM, Fersht AR . Interaction between the transactivation domain of p53 and PC4 exemplifies acidic activation domains as single-stranded DNA mimics. J Biol Chem 2009; 284: 21728–21737.
Mutch DG, Herzog TJ, Chen CA, Collins JL . The effects of cyclosporin A on the lysis of ovarian cancer cells by cisplatin or adriamycin. Gynecol Oncol 1992; 47: 28–33.
Hamilton G . Cyclophilin A as a target of cisplatin chemosensitizers. Curr Cancer Drug Targets 2014; 14: 46–58.
Molina DM, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013; 341: 84–87.
Klein DK, Hoffmann S, Ahlskog JK, O’Hanlon K, Quaas M, Larsen BD et al. Cyclin F suppresses B-Myb activity to promote cell cycle checkpoint control. Nat Commun 2015; 6: 5800.
Choudhury R, Roy SG, Tsai YS, Tripathy A, Graves LM, Wang Z . The splicing activator DAZAP1 integrates splicing control into MEK/Erk-regulated cell proliferation and migration. Nat Commun 2014; 5: 3078.
Abraham S, Scarcia M, Bagshaw RD, McMahon K, Grant G, Harvey T et al. A Rac/Cdc42 exchange factor complex promotes formation of lateral filopodia and blood vessel lumen morphogenesis. Nat Commun 2015; 6: 7286.
Acknowledgements
We are grateful to Mr Lianbiao Cheng, Miss Jiaqian Pu and Miss Ningning Dong (all from Jin Huang lab) for material and intellectual help with the design and execution of some of these experiments. The authors thank Professor Jose M Cuezva (Centro de Biología Molecular Severo Ochoa, Universidad Autónoma de Madrid, Madrid, Spain) for providing G3BP1 plasmid; Professor Xiangshi Tan (Department of Chemistry, Fudan University, Shanghai, China) for providing the pMAL-c2x-derived MBPHT-Cherry2 vector; Professor Yi Yang (Shanghai Key Laboratory of New Drug Design, School of Pharmacy, East China University of Science and Technology, Shanghai, China) for providing H23, H838, H1703 and H2126 lung cancer cell lines. The work is supported by the National Natural Science Foundation of China (21222211, 31371485, 81402482, 91313303 and 81573020), CAS Key Laboratory of Receptor Research, the Shanghai Committee of Science and Technology (14ZR1411100 and 15431902000), China Postdoctoral Science Foundation Grant (2014M551361 and 2015T80415).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Lu, W., Cheng, F., Yan, W. et al. Selective targeting p53WT lung cancer cells harboring homozygous p53 Arg72 by an inhibitor of CypA. Oncogene 36, 4719–4731 (2017). https://doi.org/10.1038/onc.2017.41
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2017.41
This article is cited by
-
The effects of CypA on apoptosis: potential target for the treatment of diseases
Applied Microbiology and Biotechnology (2024)
-
Regulated cell death (RCD) in cancer: key pathways and targeted therapies
Signal Transduction and Targeted Therapy (2022)
-
Tracking the multiple-step formation of an iron(III) complex and its application in photodynamic therapy for breast cancer
Science China Chemistry (2019)
