
Abstract
TEA domain (TEAD) transcription factors are key components of the Hippo–YAP1 signaling pathway, but their functional role and regulatory mechanisms remain unclear. This study aims to comprehensively explore the expression pattern and functional role of TEAD family in gastric carcinogenesis and investigate its regulation by microRNAs (miRNAs). The mRNA and protein expression of TEAD family were examined by quantitative reverse transcription–PCR (qRT–PCR) and western blot. Their functional roles were determined by in vitro and in vivo studies. The clinicopathological association of TEAD4 in gastric cancer (GC) was studied using immunohistochemistry on tissue microarray. The prediction of miRNAs, which potentially target TEAD1/4, was performed by TargetScan and miRDB. The regulation of TEAD1/4 by miRNAs was confirmed by qRT–PCR, western blot and luciferase assays. TEAD1/4 were overexpressed in GC cell lines and primary GC tissues. Knockdown of TEAD1/4 induced a significant anticancer effect in vitro and in vivo. TEAD1 was confirmed to be a direct target of miR-377-3p and miR-4269, while TEAD4 was negatively regulated by miR-1343-3p and miR-4269. Among them, miR-4269 was the most effective inhibitor of TEAD1/4. Ectopic expression of these miRNAs substantiated their tumor-suppressive effects. In primary GC tumors, downregulation of miR-4269 was associated with poor disease-specific survival and showed a negative correlation with TEAD4. TEAD1 and TEAD4 are oncogenic factors, whose aberrant activation are, in part, mediated by the silence of miR-377-3p, miR-1343-3p and miR-4269. For the first time, the nuclear accumulated TEAD4 and downregulated miR-4269 are proposed to serve as novel prognostic biomarkers in GC.
Similar content being viewed by others


Introduction
Gastric cancer (GC) is the fourth most common cancer and the third leading cause of cancer-related deaths worldwide.1 In spite of its declining incidence and mortality within the recent decades, GC is still an emergent public health problem, particularly in Eastern Asia. Multiple risk factors have been reported to contribute to this cancer, such as Helicobacter pylori or Epstein–Barr virus infection, high-salt and low-vegetable diet, smoking, chronic gastritis with intestinal metaplasia.2 Approximately 95% of GC are adenocarcinomas that are histologically classified as intestinal-, diffuse- or mixed type according to Lauren’s characterization.3 Even though patients who suffer from intestinal GC live longer than those with diffuse type, the overall survival remains poor, as most patients are diagnosed at an advanced stage4 and there is a lack of effective therapies for them. Thus, there is an urgent need to investigate the molecular mechanisms underlying GC, to offer some clues for clinical instructions and to identify better biomarkers that predict prognosis.
Several dysregulated signaling pathways are involved in GC development, and the Hippo–YAP1 pathway has been shown to have a role in gastric carcinogenesis.5, 6 TEA domain (TEAD) transcription factors, also known as transcriptional enhancer factor, are crucial parts of Hippo–YAP1 signaling. In mammals, there are four members (TEAD1–4) with highly conserved domains. All of them contain a TEA domain for binding with DNA elements and a transactivation domain for interaction with transcription co-activators such as YAP1/TAZ. By binding with co-activators, TEADs function as key mediators in tumorigenesis, including liver cancer,7, 8, 9 ovarian cancer,10 breast cancer11 and prostate cancer.12 Three groups of co-activators have been identified and classified by Pobbati et al.,13 including YAP1/TAZ, Vgll proteins and p160 family nuclear receptor co-activators.
However, the underlying mechanisms of TEAD transcription factors in gastric tumorigenesis stay poorly understood. Apart from their biological roles, regulation of TEADs by microRNA (miRNA) has not been investigated either. In this study, we aim to comprehensively reveal the expression pattern and functional role of TEAD family and perform an in-depth investigation to elucidate the miRNA deregulation mechanisms contributing to TEAD activation in GC.
Results
TEAD1 and TEAD4 are overexpressed in GC
Through analyzing the expression microarray data from GENT data set, we found that both TEAD1 and TEAD4 were highly expressed in 311 GC tissues compared with the corresponding normal gastric tissues (P<0.001; Figure 1a).14 In GC cell lines, the expression levels of TEAD1 and TEAD4 were significantly higher than that of TEAD2 and TEAD3 (Figure 1b). In another published GC data set NCBI/GEO/GSE63089, TEAD1 and TEAD4 demonstrated elevated expression in GC samples compared with adjacent normal tissues (n=45, P<0.001; upper panel of Supplementary Figure S1a).15 Nonetheless, only TEAD4 exhibited a concordant trend in The Cancer Genome Atlas (TCGA) cohort (n=32, P<0.001; lower panel of Supplementary Figure S1a). Opposite to the expression pattern of TEAD2/3, both mRNA and protein of TEAD1 and TEAD4 were highly expressed in most of the GC cell lines in contrast with immortalized gastric epithelium cell line GES-1 (Figures 1c and d; Supplementary Figure S1b). Overexpression of TEAD1 and TEAD4 was associated with poor progression-free survival in primary GCs according to GSE14210, GSE15459, GSE22377, GSE29272, GSE51105 and GSE62254 (P<0.001; Figure 1e) by the analysis of Kaplan–Meier plotter (www.kmplot.com).16 Similarly, the abundance of TEAD2/3 predicted unfavorable outcomes as well (P<0.001; Supplementary Figure S1c). However, due to the upregulation in primary GC samples as well as in GC cell lines and concordant results between the expression patterns and survival situations, we only included TEAD1 and TEAD4 for further study. To elucidate the correlation of TEAD1/4 expression with the molecular classification of GC, TCGA cohort was analyzed.17, 18 TEAD1 upregulation was mostly found in genomically stable (GS) subtype, while high TEAD4 expression was strongly associated with Epstein–Barr virus-positive and microsatellite instability subtypes (Figure 1f).

TEAD1 and TEAD4 are upregulated and predict poor prognosis in GC. (a) TEAD1 and TEAD4 displayed high expression in 311 GC samples compared with normal gastric tissues (http://medical-genome.kribb.re.kr/GENT/; *P<0.05; **P<0.001; NS, not significant; unpaired t-test). (b) TEAD1 and TEAD4 were upregulated in GC cell lines compared with TEAD2/3; meanwhile, expression of YAP1 is significantly higher than that of TAZ (**P<0.001; unpaired t-test). (c) mRNA expression of TEAD1 and TEAD4 in 11 GC cell lines compared with immortalized gastric epithelium cell line GES-1. (d) High protein level of TEAD1 and TEAD4 was detected in most of the GC cell lines (upper panel). And the corresponding quantification by densitometry was shown at nether panel. (e) Overexpressed TEAD1 and TEAD4 were associated with poor progression-free survival in primary GCs (P<0.001) from Kaplan–Meier plotter (NCBI/GEO/GSE14210, GSE15459, GSE22377, GSE29272, GSE51105 and GSE62254). (f) Distribution of TEAD1 and TEAD4 mRNA expression in four molecular subtypes of GC (*P<0.05; **P<0.001; TCGA cohort; unpaired t-test). (g) Immunohistochemistry images of TEAD4 in GC tissue microarray (Scale bars are 200 μm). TEAD4 exhibited negative or cytoplasmic expression in normal epithelium cells, but it was localized in the nucleus of the cancer cells. (h) High TEAD4 nuclear accumulation was associated with poor disease-specific survival (overall cases, P=0.020; advanced-stage cases, P=0.030; log-rank (Mantel–Cox) test).
We next performed immunohistochemistry to investigate TEAD4 protein expression in GC tissue microarray. In non-tumorous gastric epithelium, TEAD4 either displayed negative expression or was mainly localized in the cytoplasm of the epithelial cells. However, in both intestinal- and diffuse type of GC, TEAD4 was found to be localized in the nucleus of GC cells, marked by deep brown staining (Figure 1g). Accordingly, when using 5% positive GC cells as the cutoff for low- and high expression (Supplementary Figure S1d), TEAD4 nuclear accumulation was associated with poor disease-specific survival (overall cases, P=0.020; advanced-stage cases, P=0.030; Figure 1h). Supplementary Table S1 summarized the correlation of TEAD4 with other clinicopathologic parameters in 128 GC samples. It turned out that TEAD4 expression was not correlated with any of these parameters. By univariate Cox regression analysis, elder age, diffuse type, high grade, advanced T, N and M stage, lymph node metastasis and high expression of TEAD4 were related to poor outcome, respectively. More importantly, through multivariate analysis, upregulated TEAD4 was found to be still associated with poor prognosis (P=0.049), together with elder age and advanced T, N and M stage. These data suggested that TEAD4 served as an independent prognostic biomarker for GC (Supplementary Table S2).
TEAD1/4 knockdown exerts anti-oncogenic effects in vitro and in vivo
As TEAD1/4 was upregulated in GC, small interfering RNA-mediated knockdown was used to investigate the functional role of TEAD1/4 in MGC-803 and SGC-7901 cells. TEAD1/4 were markedly decreased after knockdown by specific siRNAs in GC cell lines (P<0.001; Figure 2a), whereas YAP1 expression was not affected by siTEAD1-1/2 or siTEAD4-1/2 treatment. TEAD1/4 knockdown suppressed cell proliferation in a 5-day MTT assay in MGC-803 and SGC-7901 cells (P<0.001; Figure 2b). The cell growth-inhibitory effect was further confirmed by monolayer colony formation assay (P<0.001; Figure 2c; Supplementary Figure S2a). Moreover, cell invasion ability was significantly inhibited by siTEAD1-1/2 or siTEAD4-1/2 (P<0.001; Figure 2d; Supplementary Figure S2b).

Downregulation of TEAD1 or TEAD4 exerts an anti-oncogenic role in GC. (a) Protein expression of TEAD1, TEAD4 and YAP1 after the siRNA-mediated knockdown in GC cell lines. (b) TEAD1/4 knockdown suppressed cell proliferation in a 5-day MTT assay in MGC-803 and SGC-7901 cells (**P<0.001; unpaired t-test). (c) Knocking down TEAD1 or TEAD4 reduced monolayer colony formation (**P<0.001; unpaired t-test). (d) Cell invasion ability was significantly inhibited by siTEAD1-1/2 or siTEAD4-1/2 (**P<0.001; unpaired t-test). (e) Accumulation of G0/G1 cells was found in siTEAD1-1/2 or siTEAD4-1/2 transfectants compared with the mock control and scramble siRNA controls 24 h after transfection. (f) Both siTEAD1-1/2 and siTEAD4-1/2 induced senescence by the β-galactosidase staining in a 3-day transfection assay (**P<0.001; unpaired t-test). (g) Western blot analysis of related cell cycle regulators and apoptotic markers. Cyclin D1, cyclin D3, CDK6 and p-Rb showed decreased expression, whereas p21 and p27 were uniformly upregulated in TEAD1- and TEAD4-depleted cells. (h) Both TEAD1 and TEAD4 knockdown in MGC-803 cells inhibited xenograft formation in vivo (**P<0.001; unpaired t-test). (i) Enrichment plots of gene expression signatures for cell proliferation (P=0.002) and survival (P<0.001) according to TEAD4 mRNA expression levels. The barcode plot indicated the position of the genes in each gene set; red and blue colors represented the high and low expression of TEAD4, respectively. ES, enrichment score; NES, normalized enrichment score. (j) Correlation between TEAD1/4 mRNA expression and extents of methylation of promoter CpG sites in TCGA cohort (TEAD1: n=330, P=0.003; TEAD4: n=372, P<0.001; Pearson’s correlation).
As growth-inhibitory effects were observed in siTEAD1-1/2- or siTEAD4-1/2-transfected cells, we analyzed cell cycle parameters by flow cytometry. Accumulation of G0/G1 cells and a decreased percentage of S phase cells were found in siTEAD1-1/2 or siTEAD4-1/2 transfectants compared with mock control or scramble siRNA controls (Figure 2e). In addition, cell senescence was determined by β-galactosidase staining in a 3-day transfection assay. Both siTEAD1-1/2 and siTEAD4-1/2 significantly induced cell senescence in three GC cell lines (Figure 2f), which was concordant with G0/G1-phase cell cycle arrest. The related cell cycle regulators and apoptosis markers were also examined by western blot (Figure 2g). Cyclin D1, cyclin D3, CDK6 and their downstream effector, p-Rb, demonstrated decreased expression, whereas p21 and p27 were uniformly upregulated in TEAD1- and TEAD4-silenced cells. The oncogenic role of TEAD1 and TEAD4 in gastric tumorigenesis was confirmed in vivo. Both TEAD1 and TEAD4 knockdown markedly inhibited the growth of tumor xenografts in nude mice (P<0.001; Figure 2h). Consistent with the growth-promoting effect of TEAD4 in vitro and in vivo, gene set enrichment analysis19, 20, 21, 22, 23 using a published GC data set NCBI/GEO/GSE57303 (Figure 2i) revealed that the whole set of cell proliferation-related genes were significantly enriched in TEAD4-upregulated cases (P=0.002). In summary, TEAD1/4 is critical for GC development through inducing cell proliferation and preventing cell senescence. Moreover, TEAD4 abundance was negatively associated with longer cancer-related survival (P<0.001).
To further illustrate the mechanism that underlies the overexpression of TEAD1/4 in GC, we analyzed copy number changes, somatic mutation and mRNA upregulation of TEAD1 and TEAD4 in the TCGA cohort. From the TCGA cohort analyzed by cBioPortal, 13% cases (34/258) have at least one alteration in TEAD1 or TEAD4 (Supplementary Figure S1e). Copy number changes of TEAD1 and TEAD4 were significantly correlated with mRNA expression (P<0.05; Supplementary Figure S1f). Given that the proportion of this change only accounted for 13% of the total cases, genomic amplification was merely one of the multiple reasons for high TEAD1/4 mRNA expression in GC. Thereby, we checked the promoter methylation status of TEAD1/4 in GC. A range of 1500 bp in front of the first exon was included, where the promoter region was likely to be located in. In consequence, we found multiple methylated CpG islands (TEAD1: cg09113513 and cg22694703; TEAD4: cg13508391, cg14742305, cg25710178 and cg21637033). Withal the lower methylated level, the higher level of TEAD1 and TEAD4 mRNA was detected (Figures 2j; P=0.003 and P<0.001, respectively, TCGA cohort). Nevertheless, the correlation coefficients were rather low, which implied that apart from genomic and epigenetic regulation, post-transcriptional regulation might act as another mechanism on TEAD1/4 expression.
Regulation of TEAD1 and TEAD4 by miR-377-3p, miR-1343-3p and miR-4269
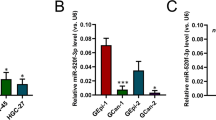
By TargetScan (www.targetscan.org), TEAD1 3′-untranslated region (UTR) was found to be potentially targeted by miR-873-5p (context score value: −0.47), miR-377-3p (−0.17), miR-4269 (−0.38), miR-340 (−0.12) and miR-124 (−0.82). Meanwhile, miR-1343-3p (−0.57), miR-4269 (−0.63) and miR-375 (−0.11) might target TEAD4. All the putative binding sites were also predicted by miRDB (http://mirdb.org/miRDB/). In the first-round screening by quantitative reverse transcription–PCR, we found miR-377-3p, miR-1343-3p, miR-4269, miR-375 and miR-124 might regulate the expression of TEAD1 or TEAD4 (Supplementary Figure S3a). As miR-375 and miR-124 were well characterized in GC,24, 25, 26, 27 we chose three novel miRNAs, miR-377-3p, miR-1343-3p and miR-4269, for further investigation. The putative binding sites in 3′-UTR of TEAD1 or TEAD4 for these miRNAs were listed in Figure 3a. In both mRNA (Figure 3b) and protein (Figure 3c) levels, TEAD1 was decreased after overexpression of miR-377-3p or miR-4269 precursors, while ectopic expression of either miR-1343-3p or miR-4269 precursors inhibited the expression of TEAD4.

TEAD1 and TEAD4 are direct targets of miR-377-3p, miR-1343-3p and miR-4269. (a) Putative binding sites in 3′-UTR of TEAD1 or TEAD4 for the related miRNA binding. (b) mRNA expression of TEAD1 and TEAD4 in MGC-803 and SGC-7901 cells after ectopic expression of miR-377-3p, miR-1343-3p and miR-4269 (*P<0.05; **P<0.001; unpaired t-test). (c) Western blot analysis of TEAD1 and TEAD4 when overexpression of miR-377-3p, miR-1343-3p and miR-4269 in MGC-803 and SGC-7901 cells. (d) Both miR-377-3p and miR-4269 suppressed the relative luciferase activity of constructs encompassing the binding sites in TEAD1 3′-UTR. Meanwhile, after ectopic expression of miR-1343-3p or miR-4269, the luciferase activities were also inhibited in the constructs containing the wild-type binding sites in TEAD4 3′-UTR (**P<0.001; unpaired t-test).
Next, we performed luciferase reporter assays to investigate the direct binding affinity between these miRNAs with the 3′-UTRs of TEAD1 and TEAD4. As shown in Figure 3d, both miR-377-3p and miR-4269 suppressed the relative luciferase activities of constructs encompassing the binding sites in TEAD1 3′-UTR. Similarly, luciferase activities were inhibited in the constructs containing wild-type binding sites in TEAD4 3′-UTR after ectopic expression of miR-1343-3p or miR-4269 (P<0.001; Figure 3d). These results indicated that miR-377-3p and miR-4269 directly recognized binding sites in TEAD1 3′-UTR, and TEAD4 3′-UTR was directly targeted by miR-1343-3p and miR-4269 in GC cells.
miR-377-3p, miR-1343-3p and miR-4269 are tumor-suppressive miRNAs
To investigate the biological function of miR-377-3p, miR-1343-3p and miR-4269 in GC, their precursors were transfected into MGC-803 and SGC-7901 cells. Overexpression of these three miRNAs impaired cell proliferation in a 4-day MTT assay (Figure 4a). Consistently, miR-377-3p, miR-1343-39 and miR-4269 overexpression reduced colony size and number in monolayer colony formation assay, as compared with scramble miRNA (P<0.001; Figure 4b; Supplementary Figure S3b). Ectopic expression of these three miRNAs also suppressed cell invasive ability of MGC-803 and SGC-7901 cells (P<0.001; Figure 4c; Supplementary Figure S3c). Given that cell growth was inhibited by these miRNAs, cell cycle analysis was also performed. miR-1343-3p and miR-4269 overexpression increased the proportion of G0/G1-phase cells (Figure 4d; Supplementary Figure S3d). To investigate whether these three miRNAs exerts tumor-suppressive effects by inducing apoptosis, we performed fluorescence-activated cell sorting analysis of cells double-stained with Annexin V and 7-AAD, and found miR-377-3p and miR-4269 promoted late apoptosis (Figure 4e; Supplementary Figure S3e). Among these miRNAs, miR-4269 was the only one that was capable of inducing G0/G1 cell cycle arrest and late apoptosis, presenting its crucial role in gastric carcinogenesis. Furthermore, western blot of cell cycle regulators and apoptosis markers showed good concordance with the flow cytometry results (Figure 4f), further confirming these miRNAs’ function as tumor suppressors.

miR-377-3p, miR-1343-3p and miR-4269 are tumor-suppressive miRNAs. (a) Overexpression of miR-377-3p, miR-1343-3p and miR-4269 suppressed cell proliferation in a 4-day MTT assay (**P<0.05; **P<0.001; unpaired t-test). (b) Smaller colony size and reduced colony number were observed in miR-377-3p, miR-1343-3p and miR-4269 transfectants compared with negative control (**P<0.001; unpaired t-test). (c) Cell invasion abilities were impaired by ectopic miR-377-3p, miR-1343-3p and miR-4269 expression in MGC-803 and SGC-7901 cells (**P<0.001; unpaired t-test). (d) Cell cycle analysis by flow cytometry revealed that miR-1343-3p and miR-4269 overexpression increased the proportion of G0/G1-phase cells (**P<0.001; unpaired t-test). (e) miR-377-3p and miR-4269 promoted late apoptosis in GC cells (**P<0.001; unpaired t-test). (f) Western blot analysis of cell cycle regulators and apoptosis-related markers, validating the flow cytometry results.
To confirm whether the epigenetic modification was responsible for miR-377-3p, miR-1343-3p and miR-4269 downregulation in GC, we examined the expression of the three miRNAs after treating AGS, MKN1, NCI-N87 and MGC-803 cells with 5-Aza-2'-Deoxycytidine (5-Aza), Trichostatin A (TSA) or a combination of both. Expression of miR-377-3p was restored after 5-Aza or TSA treatment. Furthermore, the drug combination of 5-Aza and TSA was more effective for restoring miR-377-3p expression. For miR-1343-3p or miR-4269, the elevated expression was only observed in NCI-N87 or AGS, respectively, after treatment (Supplementary Figure S3f).
miR-4269 is involved in Hippo–YAP1 signaling by targeting TEAD1 and TEAD4
To evaluate whether all three miRNAs were able to regulate Hippo–YAP1 signaling by targeting TEAD1 or TEAD4, the expression of its downstream was examined after ectopic expression of miR-377-3p, miR-1343-3p or miR-4269 in GC cell lines. Interestingly, only miR-4269 effectively inhibited both mRNA and protein expression of connective tissue growth factor (CTGF), Cyr61 and c-Myc (P<0.001; Figures 5a and b). The downstream effectors (CTGF, Cyr61 and c-Myc) of Hippo–YAP1 signaling were meanwhile downregulated by direct siRNA knockdown of TEAD1 or TEAD4, suggesting that miR-4269 regulated the downstream expression through targeting TEAD1 and TEAD4 (Supplementary Figure S4a). Similarly, after TEAD1/4 depletion, miR-4269 was incapable of regulating CTGF expression, which was revealed by luciferase activity assays (Supplementary Figure S4b). Decreased expression of miR-4269 was found in seven out of eleven GC cell lines compared with GES-1 cells (Figure 5c). To confirm whether TEAD1 and TEAD4 are the functional targets of miR-4269, rescue experiments were conducted in MGC-803 and SGC-7901 cells. Western blot was applied to validate that TEAD1 and TEAD4 were successfully re-expressed in GC cells, respectively (Figure 5d). Re-expression of TEAD1 or TEAD4 partly diminished the tumor-suppressive effect of miR-4269 in MTT proliferation assays (P<0.05; Figure 5e) and monolayer colony formation (P<0.001; Figure 5f). Cell invasive ability (P<0.001; Figure 5g) was also restored in part after re-overexpression of TEAD1 or TEAD4 in miR-4269-treated cells. On the other hand, when TEAD1 and TEAD4 were silenced by siRNAs, miR-4269 failed to exert its growth-inhibitory effect on cell proliferation (Supplementary Figure S4c), indicating that miR-4269 has a tumor-suppressive role mainly through targeting TEAD1 and TEAD4. Furthermore, both siTEAD1-1/siTEAD4-1 and miR-4269 promoted anticancer drug (5-fluorouracil and cisplatin) sensitivity in a dose-dependent manner of GC cells (*P<0.05; **P<0.001; Figure 5h). All these results suggested that TEAD1 and TEAD4 are main functional targets of miR-4269 in GC.

Only miR-4269 regulates the downstream expression of Hippo–YAP1 signaling by targeting TEAD1 and TEAD4. (a) mRNA expression of CTGF and CYR61 after overexpression of miR-377-3p, miR-1343-3p or miR-4269 in MGC-803 and SGC-7901 cell lines (**P<0.001; unpaired t-test). (b) Western blot analysis of CTGF, CYR61 and c-Myc upon ectopic expression of miR-4269. (c) miR-4269 presented decreased expression in seven out eleven GC cell lines compared with immortalized gastric epithelium cell GES-1. (d) Re-expression of both TEAD1 and TEAD4 was confirmed by western blot in the rescue experiments. (e) Re-overexpression of TEAD1 or TEAD4 partly diminished the tumor-suppressive effect of miR-4269 revealed by MTT proliferation assays (*P<0.05; unpaired t-test). (f) Colony formation ability was partly restored in miR-4269-treated GC cells after re-expression of TEAD1 or TEAD4 (**P<0.001; unpaired t-test). (g) TEAD1 or TEAD4 re-expression also revived cell invasive ability, which was previously impaired by miR-4269 (**P<0.001; unpaired t-test). (h) Both siTEAD1-1/siTEAD4-1 and miR-4269 enhanced anticancer drug sensitivity of GC cells (*P<0.05; **P<0.001; unpaired t-test).
Downregulation of miR-4269 correlates with poor survival and shows negative correlation with TEAD4 in GC
The expression of miR-4269 was measured in 41 paired primary GC and adjacent normal samples. miR-4269 expression in GC displayed descending expression compared with adjacent non-tumorous tissues (P=0.006; Figure 6a). Accordingly, our cohort (n=76) was stratified into two groups (37 high- and 39 low-miR-4269 expression cases) based on the receiver-operating characteristic curve. The low-expression group showed a poor disease-specific survival compared with high-expression group (P=0.003; Figure 6b). In addition, downregulation of miR-4269 was only marginally correlated with advanced stage by clinical correlation analysis (P=0.050; Supplementary Table S3). By univariate analysis, female patients, diffuse type, advanced TNM stage, lymph node metastasis and low expression of miR-4269 were associated with shorter patient survival. However, by multivariate analysis, the only parameter that mattered in disease-specific survival appeared to be advanced stage (Supplementary Table S4).

miR-4269 downregulation correlates with poor survival and is negatively associated with TEAD4 expression in GC. (a) miR-4269 showed decreased expression in primary gastric tumors compared with paired adjacent non-tumorous tissues (n=41, P=0.006; paired t-test). (b) Downregulation of miR-4269 predicted a shorter disease-specific survival in primary GC samples (P=0.003). (c) Expression correlation of TEAD1 (P=0.535; Pearson’s correlation) or TEAD4 (P=0.031; Pearson’s correlation) mRNA with miR-4269 in 28 primary tumors. (d) Expression correlation of miR-4269 with TEAD1 or TEAD4 protein expression in 11 GC cell lines. TEAD4 (P=0.017; Pearson’s correlation), instead of TEAD1 (P=0.688; Pearson’s correlation), showed negative correlation with miR-4269. (e) HDAC4 expression in paired primary GCs (n=32, P=0.045; paired t-test). (f) Correlation between HDAC4 mRNA expression and extents of methylation in related CpG sites (n=372, P<0.001; Pearson’s correlation). (g) Expression correlation between HDAC4 and TEAD4 in TCGA cohort (n=415, P=0.05; Spearman’s correlation).
To validate the regulatory effect of miR-4269 on TEAD1 and TEAD4 in primary samples, the expression of TEAD1, TEAD4 and miR-4269 in 28 primary tumors were measured. TEAD4 mRNA (P=0.031), instead of TEAD1 mRNA (P=0.535), was negatively correlated with miR-4269 expression in primary GC (Figure 6c). In GC cell lines, we quantified the protein expression of TEAD1 and TEAD4 by ImageJ densitometry and detected a similar result: expression of miR-4269 showed a negative correlation with TEAD4 (P=0.017), rather than TEAD1 (P=0.688; Figure 6d). Meanwhile, we checked the expression of histone deacetylase 4 (HDAC4), the host gene of miR-4269, and found that HDAC4 exhibited decreased expression in cancer tissues in contrast with adjacent non-tumorous tissues (n=32, P=0.045; Figure 6e) in TCGA cohort. To simply explain this diminution, we again looked into the methylation status of the CpG islands, which were located within 1500 bp before the encoding region of HDAC4. As a result, two sites (cg00360072 and cg02457900) were uncovered to be severely methylated and their extents of hypermethylation were related to the decreased level of HDAC4 (r=−0.190, P<0.001, TCGA cohort; Figure 6f). Thereby, as a product of this genomic locus, low-miR-4269 expression in some GC samples might be explained by hypermethylation as we described in Supplementary Figure S3f. Besides, expression of HDAC4 showed a marginally negative correlation with TEAD4 (r=−0.100, n=415, P=0.050; Figure 6g). All these findings suggested that miR-4269 downregulation was partly responsible for the aberrant TEAD4 activation in GC cells.
Schematic summary of miRNA regulation on TEAD1/4 in GC
In normal gastric epithelium, TEAD1 and TEAD4 were negatively modulated, which, in a way, was attributed to the normal expression of miR-377-3p, miR-1343-3p and miR-4269. However, the expression of these three miRNAs was downregulated or silenced in GC, thereby they lost the inhibitory effect on TEAD1/4. In addition, upregulated TEAD1/4, in turn, interacted with YAP1. As the central component of Hippo–YAP1 axis, YAP1 was translocated into the nucleus in cancer cells. As transcription factors, TEAD1 and TEAD4 exerted their oncogenic effect by activating the expression of downstream oncogenic factors CTGF, Cyr61 and c-Myc (Figure 7).

Schematic presentation of TEAD1/4 regulation by miR-377-3p, miR-1343-3p and miR-4269 in GC. TEAD1/4 showed low expression in normal gastric epithelium cells, which was partially inhibited by miR-377-3p, miR-1343-3p and miR-4269. However, the related miRNA showed decreased expression in GC, thus they loss the inhibition on TEAD1 and TEAD4. Via MST–YAP1 axis, YAP1–TEAD1/4 complex was translocated into the nucleus to promote gastric tumorigenesis by regulating the downstream expression.
Discussion
It has been well identified that TEAD transcription factor family is involved in the development of several types of cancer28 and is overexpressed in breast cancer,29, 30 fallopian tube carcinoma,31 germ cell tumor,32 renal cell carcinoma,33 medulloblastoma34 and liver cancer.35 In this study, TEAD1 and TEAD4, instead of TEAD2 and TEAD3, were found to be abundantly expressed, which means their function is predominant in this family and functional studies demonstrated their oncogenic role during gastric carcinogenesis. Moreover, we unraveled downregulation of multiple tumor-suppressive miRNAs as a novel mechanism by which, partially, TEAD1 and TEAD4 were overexpressed in GC, which in turn activated Hippo–YAP1 signaling pathway. Finally, TEAD4 and its repressor miR-4269 were identified as prognostic markers for GC patients, in keeping with previous reports.36, 37
TEAD1 and TEAD4 are upregulated in GC according to our cohort and other publically available data sets. Functional investigations explicated that TEAD1 and TEAD4 were essential oncogenic factors in gastric carcinogenesis by promoting cell growth. It has been suggested that the aberrant activation of YAP1–TEAD complex contributes to the pathogenesis of various cancers. This complex drives cell growth and transformation through several target genes, including CTGF, Cyr6138, 39 and c-Myc.40, 41 In GC, Vgll4 was believed to be a suppressor for YAP1 activity through competing with TEAD, which was proved by both in vitro and in vivo experiments.42 TEAD1 was reported to be involved in a feedback loop of miR-222/Vgll4/TEAD1, in which miR-222 repressed the activity of Vgll4.36 Hence, TEAD proteins activate Hippo–YAP1 signaling pathway either through direct binding to YAP or indirectly by suppression of Vgll4. All these findings revealed a critical role of TEAD transcription factors and their co-activators in cancer development, and here we demonstrated their impact on GC development. Consistent with our data, a recent study uncovered a tripartite signaling complex involving RUNX3, which acted as a potent inhibitor of YAP1–TEAD-driven GC. They justified that RUNX3 physically interacted with the N-terminal region of TEAD through its Runt domain, which markedly reduced the DNA-binding ability of TEAD and attenuated signals conveyed by TEAD–YAP1 complex to the downstream.43 Besides, recently a group of Korean scientists proposed TEAD4 as a therapeutic target for clinical intervention during GC treatment. More importantly, based on their 108 Korean cohorts, they attributed TEAD4 upregulation to hypomethylation.37 The CpG spot proposed in their report, cg21637033, was also included in our study. Our results suggested that hypomethylation is one of the multiple mechanisms for the upregulation of TEAD1 and TEAD4 in gastric carcinogenesis.
Even though both genomic alteration and epigenetic change of TEAD1/4 were found significantly correlated in TCGA cohort, we still sought to improve the integrity of regulatory mechanisms of TEADs. Therefore, from a different angle, we investigated the role of miRNA dysregulation during the activation of TEAD1/4. Three novel tumor-suppressive miRNAs, miR-377-3p, miR-1343-3p and miR-4269, were confirmed to directly regulate the expression of TEAD1 or TEAD4. In particular, miR-4269 targeted both TEAD1 and TEAD4, and regulated the expression of downstream, including CTGF, CYR61 and c-Myc. By rescue experiments, we confirmed that TEAD1 and TEAD4 are functional targets for miR-4269 in GC. All these findings enriched our horizon of TEAD1/4 regulation by miRNAs, and ulteriorly revealed novel mechanisms underlying the aberrant activation Hippo–YAP1 pathway in GC tumorigenesis.
miR-377-3p, miR-1343-3p and miR-4269 are novel tumor-suppressive miRNAs whose functions are largely unknown. There were no reports about the functional role of miR-1343-3p and miR-4269 in tumorigenesis, while only several groups reported the tumor-suppressive role of miR-377-3p in various cancer types. In clear cell renal cell carcinoma, miR-377-3p exerted its tumor-suppressive function by targeting E26 transformation specific-1.44 In hepatocellular carcinoma, miR-377-3p targeted T-lymphoma invasion and metastasis 1 and impaired T-lymphoma invasion and metastasis 1-promoted cell proliferation and invasion.45 Our findings revealed that TEAD1 is a functional target for miR-377-3p and enriched the target pool for this tumor-suppressive miRNA. On the other hand, a salient anti-GC marker, miR-4269, was attested in our study for the first time. Owing to prominent P-values in both univariate and multivariate analysis, as well as high sensitivity and specificity, this miRNA served as a promising prognostic marker among GC patients.
In conclusion, the increasing knowledge about TEAD function and its regulation by dysregulated miRNAs will not only enhance our understanding of the underlying mechanisms of gastric tumorigenesis but also facilitate identification of novel prognostic biomarkers in the Hippo–YAP1 cascade and develop intervention therapies for GC.
Materials and methods
GC cell lines and clinical samples
Eleven human GC cell lines (AGS, KatoIII, MGC-803, MKN1, MKN7, MKN28, MKN45, NCI-N87, SGC-7901, SNU1 and SNU16) and GES-1, an immortalized gastric epithelial cell line, were cultured as reported.46 A total of 129 patients diagnosed as GC between 1999 and 2006 at the Prince of Wales Hospital were recruited, whose formalin-fixed paraffin-embedded tissues were applied for this project. Another 76 paired frozen samples were also obtained from the same hospital (diagnosed between 1999 and 2010). The CUHK Clinical Research Ethics Committee approved the usage of human samples and Reference No. is CREC 2016.050.
Treatment of cell lines with 5-Aza and TSA
Cell lines, including AGS, NCI-N87, MGC-803 and MKN1, were treated with a demethylating agent (5-Aza) and histone deacetylases inhibitor.47 For 5-Aza (Sigma, St Louis, MO, USA) treatment group, the cells were treated with 10 μm 5-Aza for 3 days. For TSA (Sigma) treatment group, 100 nm TSA was added to cells for 24 h. For combination, we treated the cells with 5-Aza for 4 days. In the following 24 h, TSA was added at 100 nm concentration. Control cultures were treated with an equal amount of vehicle dimethylsulfoxide (Sigma).
RNA extraction and quantitative reverse transcription–PCR
The procedures of RNA extraction was reported previously.48 TRIzol reagent was purchased from Invitrogen, Carlsbad, CA, USA, and High-Capacity cDNA Reverse Transcription Kit was also from Applied Biosystems, Carlsbad, CA, USA. The primers of quantitative reverse transcription–PCR used were listed in Supplementary Table S5. Quantitative reverse transcription–PCR was performed as before.49 Kits and reagents used were as follows: miR-377-3p (assay ID: #000566, Life Technologies, Carlsbad, CA, USA); miR-1343-3p (#463957); miR-4269 (#242701); and RNU6B (#001093). We repeated the experiment three times to get s.d.’s. Averages were defined as center values and s.d.’s were used for error bars.
Western blot analysis
Western blot analysis was performed in our previous study.5 Some of the western blot results were quantified by densitometric scans using ImageJ (version 1.48p; W Rasband, National Institutes of Health; available at http://rsb.info.nih.gov/ij/) for the following Pearson’s correlation analysis. The primary antibodies of TEAD1 (sc-376113), TEAD4 (sc-134071), CTGF (L-20) (sc-14939) and Cyr-61 (sc-374129) were commercially available from Santa Cruz (Dallas, TX, USA). YAP1 (ab52771) antibody was achieved from Abcam (Cambridge, MA, USA). TEAD2 (SAB2102402) and TEAD3 (AV38278) antibodies were obtained from Sigma-Aldrich (St Louis, MO, USA). Other primary antibodies were from Cell Signaling (Danvers, MA, USA), including p21 (#2946), p27 (#2552), p-Rb (Ser807/811) (#9308), cleaved-caspase 3 (Asp175) (#9661), cleaved-caspase 7 (Asp198) (#9491), cleaved-PARP (Asp214) (#9541), cyclin D1 (#2978), cyclin D3 (#2936), CDK4 (#12790), CDK6 (#3136), c-Myc (#9402) and GAPDH (#2118). Anti-Mouse IgG-HRP (Dako, Glostrup, Denmark, 00049039, 1:30 000) and anti-Rabbit IgG-HRP (Dako, 00028856, 1:10 000) were used for secondary antibodies.
Immunohistochemistry
Immunohistochemistry was also performed as in our earlier paper.48 TEAD4 primary antibody (1:25, HPA056896) was from Protein Atlas Antibodies (Voltavägen, Bromma, Sweden). The nuclear accumulation of TEAD4 was assessed according to the ratio of GC cells with positive nuclear staining (low expression, ⩽5%; high expression, >5%).
miRNA and siRNA transfection for functional assays
miRNA precursors, including miR-377-3p (PM10524, Life Technologies), miR-1343-3p (PM20896), miR-4269 (PM16968), miR-873-5p (PM12405), miR-340 (PM12670), miR-124 (PM10691), miR-375 (PM10327) and scramble control (AM17110), were from Life Technologies. siRNAs, such as siTEAD1-1 (SI04181261), siTEAD1-2 (SI04237205), siTEAD4-1 (SI04131127) and siTEAD4-2 (SI04136069), were purchased from Qiagen (Valencia, CA, USA). We used Lipofectamine 2000 (Invitrogen) to perform all transfection assays. Cell functional tests were performed as before.5 Flow cytometry was applied to perform cell cycle and apoptosis assays.50
In senescence experiments, MKN28, MGC-803 and SGC-7901 cells were transfected with siTEAD1, siTEAD4 or siScramble for 3 days. Cells were then stained with β-galactosidase (Kit, #9860, Cell Signaling) for 8 h and the senescence-positive cell populations were shown in pale green.6 For rescue experiments, we transfected miR-4269 precursors or negative control in GC cells. Twenty-four hours after transfection, TEAD1- or TEAD4-expressing plasmid (#33109 and #24638, Addgene, Cambridge, MA, USA) or empty vector negative control (pcDNA3.1, Life Technologies) were then transfected with FuGENE HD (Roche, Nutley, NJ, USA). We collected cells after 24 h for western blot analysis and MTT assay.
Luciferase activity assays
The putative miR-377-3p- and miR-4269-binding sites in 3′-UTR of TEAD1, as well as the miR-1343-3p- and miR-4269-binding sites in TEAD4 3′-UTR were separately sub-cloned into pMIR-REPORT vector (Ambion, Austin, TX, USA). The sense and anti-sense of the oligonucleotides were listed in Supplementary Table S6. All the experimental procedures were reported previously.48
In vivo tumorigenicity model
The protocol of in vivo tumorigenicity model was described before.48 Tumor weights were measured on day 25. All animal experimental procedures were approved by Department of Health, Hong Kong and CUHK Animal Ethics Committee. The Reference No. is 15-745 in DH/HA&P/8/2/1 Pt.53.
Statistical analysis
Some of the values are adjusted (log transformation) to be approximately normally distributed to meet the requirement of parametric tests. Corresponding statistical methods for each comparison and correlation was as previous.48 We performed all the statistical analysis via SPSS software (version 22.0; SPSS Inc., Chicago, IL, USA; two-tailed, P<0.05, statistically significant; two-tailed, P<0.001, highly statistically significant).
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A . Global cancer statistics, 2012. CA Cancer J Clin 2015; 65: 87–108.
Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med 2001; 345: 784–789.
Lauren P . The two histological main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma. an attempt at a histo-clinical classification. Acta Pathol Microbiol Scand 1965; 64: 31–49.
Wadhwa R, Song S, Lee JS, Yao Y, Wei Q, Ajani JA . Gastric cancer-molecular and clinical dimensions. Nat Rev Clin Oncol 2013; 10: 643–655.
Kang W, Tong JH, Chan AW, Lee TL, Lung RW, Leung PP et al. Yes-associated protein 1 exhibits oncogenic property in gastric cancer and its nuclear accumulation associates with poor prognosis. Clin Cancer Res 2011; 17: 2130–2139.
Kang W, Tong JH, Lung RW, Dong Y, Zhao J, Liang Q et al. Targeting of YAP1 by microRNA-15a and microRNA-16-1 exerts tumor suppressor function in gastric adenocarcinoma. Mol Cancer 2015; 14: 52.
Bai N, Zhang C, Liang N, Zhang Z, Chang A, Yin J et al. Yes-associated protein (YAP) increases chemosensitivity of hepatocellular carcinoma cells by modulation of p53. Cancer Biol Ther 2013; 14: 511–520.
Guo C, Wang X, Liang L . LATS2-mediated YAP1 phosphorylation is involved in HCC tumorigenesis. Int J Clin Exp Pathol 2015; 8: 1690–1697.
Mao B, Hu F, Cheng J, Wang P, Xu M, Yuan F et al. SIRT1 regulates YAP2-mediated cell proliferation and chemoresistance in hepatocellular carcinoma. Oncogene 2014; 33: 1468–1474.
Wanjin H, Xia Y, Zhang Y-L, Yu C, Chang T, Fan H-Y . YAP/TEAD co-activator regulated pluripotency and chemoresistance in ovarian cancer initiated cells. PLoS One 2014; 9: e109575.
Wang C, Nie Z, Zhou Z, Zhang H, Liu R, Wu J et al. The interplay between TEAD4 and KLF5 promotes breast cancer partially through inhibiting the transcription of p27 Kip1. Oncotarget 2015; 6: 17685–17697.
Knight JF, Shepherd CJ, Rizzo S, Brewer D, Jhavar S, Dodson AR et al. TEAD1 and c-Cbl are novel prostate basal cell markers that correlate with poor clinical outcome in prostate cancer. Br J Cancer 2008; 99: 1849–1858.
Pobbati AV, Hong W . Emerging roles of TEAD transcription factors and its coactivators in cancers. Cancer Biol Ther 2013; 14: 390–398.
Shin G, Kang T-W, Yang S, Baek S-J, Jeong Y-S, Kim S-Y . GENT: Gene Expression Database of Normal and Tumor Tissues. Cancer Informatics 2011; 10: 149–157.
Zhang X, Ni Z, Duan Z, Xin Z, Wang H, Tan J et al. Overexpression of E2F mRNAs associated with gastric cancer progression identified by the transcription factor and miRNA co-regulatory network analysis. PLoS One 2015; 10: e0116979.
Győrffy B, Surowiak P, Budczies J, Lánczky A . Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One 2013; 8: e82241.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6: pl1–pl1.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA et al. The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2: 401–404.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005; 102: 15545–15550.
Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003; 34: 267–273.
Qian Z, Zhu G, Tang L, Wang M, Zhang L, Fu J et al. Whole genome gene copy number profiling of gastric cancer identifies PAK1 and KRAS gene amplification as therapy targets. Genes Chromosomes Cancer 2014; 53: 883–894.
Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B et al. Focal gains of vascular endothelial growth factor A and molecular classification of hepatocellular carcinoma. Cancer Res 2008; 68: 6779–6788.
Lee J-S, Chu I-S, Heo J, Calvisi DF, Sun Z, Roskams T et al. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology 2004; 40: 667–676.
Tsukamoto Y, Nakada C, Noguchi T, Tanigawa M, Nguyen LT, Uchida T et al. MicroRNA-375 is downregulated in gastric carcinomas and regulates cell survival by targeting PDK1 and 14-3-3zeta. Cancer Res 2010; 70: 2339–2349.
Ding L, Xu Y, Zhang W, Deng Y, Si M, Du Y et al. MiR-375 frequently downregulated in gastric cancer inhibits cell proliferation by targeting JAK2. Cell Res 2010; 20: 784–793.
Xia J, Wu Z, Yu C, He W, Zheng H, He Y et al. miR-124 inhibits cell proliferation in gastric cancer through down-regulation of SPHK1. J Pathol 2012; 227: 470–480.
Murray-Stewart T, Sierra JC, Piazuelo MB, Mera RM, Chaturvedi R, Bravo LE et al. Epigenetic silencing of miR-124 prevents spermine oxidase regulation: implications for Helicobacter pylori-induced gastric cancer. Oncogene 2016; 35: 5480–5488.
Zhou Y, Huang T, Cheng AS, Yu J, Kang W, To KF . The TEAD family and its oncogenic role in promoting tumorigenesis. Int J Mol Sci 2016; 17: 138.
Lamar JM, Stern P, Liu H, Schindler JW, Jiang ZG, Hynes RO . The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc Natl Acad Sci USA 2012; 109: E2441–E2450.
Wang C, Nie Z, Zhou Z, Zhang H, Liu R, Wu J et al. The interplay between TEAD4 and KLF5 promotes breast cancer partially through inhibiting the transcription of p27Kip1. Oncotarget 2015; 6: 17685–17697.
Nowee ME, Snijders AM, Rockx DA, de Wit RM, Kosma VM, Hamalainen K et al. DNA profiling of primary serous ovarian and fallopian tube carcinomas with array comparative genomic hybridization and multiplex ligation-dependent probe amplification. J Pathol 2007; 213: 46–55.
Skotheim RI, Autio R, Lind GE, Kraggerud SM, Andrews PW, Monni O et al. Novel genomic aberrations in testicular germ cell tumors by array-CGH, and associated gene expression changes. Cell Oncol 2006; 28: 315–326.
Schutte U, Bisht S, Heukamp LC, Kebschull M, Florin A, Haarmann J et al. Hippo signaling mediates proliferation, invasiveness, and metastatic potential of clear cell renal cell carcinoma. Transl Oncol 2014; 7: 309–321.
Fernandez LA, Northcott PA, Dalton J, Fraga C, Ellison D, Angers S et al. YAP1 is amplified and up-regulated in hedgehog-associated medulloblastomas and mediates Sonic hedgehog-driven neural precursor proliferation. Genes Dev 2009; 23: 2729–2741.
Perra A, Kowalik MA, Ghiso E, Ledda-Columbano GM, Di Tommaso L, Angioni MM et al. YAP activation is an early event and a potential therapeutic target in liver cancer development. J Hepatol 2014; 61: 1088–1096.
Li N, Yu N, Wang J, Xi H, Lu W, Xu H et al. miR-222/VGLL4/YAP-TEAD1 regulatory loop promotes proliferation and invasion of gastric cancer cells. Am J Cancer Res 2015; 5: 1158–1168.
Lim B, Park JL, Kim HJ, Park YK, Kim JH, Sohn HA et al. Integrative genomics analysis reveals the multilevel dysregulation and oncogenic characteristics of TEAD4 in gastric cancer. Carcinogenesis 2014; 35: 1020–1027.
Chu CY, Chang CC, Prakash E, Kuo ML . Connective tissue growth factor (CTGF) and cancer progression. J Biomed Sci 2008; 15: 675–685.
Sala-Torra O, Gundacker HM, Stirewalt DL, Ladne PA, Pogosova-Agadjanyan EL, Slovak ML et al. Connective tissue growth factor (CTGF) expression and outcome in adult patients with acute lymphoblastic leukemia. Blood 2007; 109: 3080–3083.
de la Cova C, Johnston LA . Myc in model organisms: a view from the flyroom. Semin Cancer Biol 2006; 16: 303–312.
Vita M, Henriksson M . The Myc oncoprotein as a therapeutic target for human cancer. Semi Cancer Biol 2006; 16: 318–330.
Jiao S, Wang H, Shi Z, Dong A, Zhang W, Song X et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 25: 166–180.
Qiao Y, Lin SJ, Chen Y, Voon DCC, Zhu F, Chuang LSH et al. RUNX3 is a novel negative regulator of oncogenic TEAD-YAP complex in gastric cancer. Oncogene 2015; 35: 2664–2674.
Wang R, Ma Y, Yu D, Zhao J, Ma P . miR-377 functions as a tumor suppressor in human clear cell renal cell carcinoma by targeting ETS1. Biomed Pharmacother 2015; 70: 64–71.
Chen G, Lu L, Liu C, Shan L, Yuan D . MicroRNA-377 suppresses cell proliferation and invasion by inhibiting TIAM1 expression in hepatocellular carcinoma. PLoS One 2015; 10: e0117714.
Kang W, Tong JH, Chan AW, Lung RW, Chau SL, Wong QW et al. Stathmin1 plays oncogenic role and is a target of microRNA-223 in gastric cancer. PLoS One 2012; 7: e33919.
Zehentmayr F, Hauser-Kronberger C, Zellinger B, Hlubek F, Schuster C, Bodenhofer U et al. Hsa-miR-375 is a predictor of local control in early stage breast cancer. Clin Epigenetics 2016; 8: 28.
Huang T, Kang W, Zhang B, Wu F, Dong Y, Tong JH et al. miR-508-3p concordantly silences NFKB1 and RELA to inactivate canonical NF-kappaB signaling in gastric carcinogenesis. Mol Cancer 2016; 15: 9.
Kang W, Tong JH, Lung RW, Dong Y, Yang W, Pan Y et al. let-7b/g silencing activates AKT signaling to promote gastric carcinogenesis. J Transl Med 2014; 12: 281.
Kang W, Tong J, Chan A, Zhao J, Dong Y, Wang S et al. Yin Yang 1 contributes to gastric carcinogenesis and its nuclear expression correlates with shorter survival in patients with early stage gastric adenocarcinoma. J Transl Med 2014; 12: 1–11.
Acknowledgements
We acknowledge the TCGA research Network (http://cancergenome.nih.gov/), The UCSC Cancer Genomics Browser (https://genome-cancer.ucsc.edu/) and NCI Center for Cancer Genomics Office (http://gdc.nci.nih.gov/) for providing the gastric cancer data set and analysis. This study is supported by General Research Fund (RGC Reference No. CUHK14114414 and CUHK14110016) from The Research Grants Council of Hong Kong.
Author contributions
KFT and WK designed the experiments, offered direction and help on the whole project. YZ, TH, JZ, CCW, YD, FW and BZ conducted the experiments, analyzed the results and performed bioinformatics analysis. YZ, TH and WK drafted the manuscript. WKKW, ASLC and JY reviewed the manuscript and made significant revisions on the drafts. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Zhou, Y., Huang, T., Zhang, J. et al. TEAD1/4 exerts oncogenic role and is negatively regulated by miR-4269 in gastric tumorigenesis. Oncogene 36, 6518–6530 (2017). https://doi.org/10.1038/onc.2017.257
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2017.257
This article is cited by
-
CircGLIS3 promotes gastric cancer progression by regulating the miR-1343-3p/PGK1 pathway and inhibiting vimentin phosphorylation
Journal of Translational Medicine (2024)
-
Abnormal activation of RFC3, A YAP1/TEAD downstream target, promotes gastric cancer progression
International Journal of Clinical Oncology (2024)
-
Hippo pathway dysregulation in gastric cancer: from Helicobacter pylori infection to tumor promotion and progression
Cell Death & Disease (2023)
-
TEAD4 antagonizes cellular senescence by remodeling chromatin accessibility at enhancer regions
Cellular and Molecular Life Sciences (2023)
-
Long noncoding RNA LINC00941 promotes pancreatic cancer progression by competitively binding miR-335-5p to regulate ROCK1-mediated LIMK1/Cofilin-1 signaling
Cell Death & Disease (2021)

{kind=link}
{kind=link}
{kind=link}
{kind=link}