
Abstract
Transcription factor Myb is overexpressed in most colorectal cancers (CRC). Patients with CRC expressing the highest Myb are more likely to relapse. We previously showed that mono-allelic loss of Myb in an Adenomatous polyposis coli (APC)-driven CRC mouse model (ApcMin/+) significantly improves survival. Here we directly investigated the association of Myb with poor prognosis and how Myb co-operates with tumor suppressor genes (TSGs) (Apc) and cell cycle regulator, p27. Here we generated the first intestinal-specific, inducible transgenic model; a MybER transgene encoding a tamoxifen-inducible fusion protein between Myb and the estrogen receptor-α ligand-binding domain driven by the intestinal-specific promoter, Gpa33. This was to mimic human CRC with constitutive Myb activity in a highly tractable mouse model. We confirmed that the transgene was faithfully expressed and inducible in intestinal stem cells (ISCs) before embarking on carcinogenesis studies. Activation of the MybER did not change colon homeostasis unless one p27 allele was lost. We then established that MybER activation during CRC initiation using a pro-carcinogen treatment, azoxymethane (AOM), augmented most measured aspects of ISC gene expression and function and accelerated tumorigenesis in mice. CRC-associated symptoms of patients including intestinal bleeding and anaemia were faithfully mimicked in AOM-treated MybER transgenic mice and implicated hypoxia and vessel leakage identifying an additional pathogenic role for Myb. Collectively, the results suggest that Myb expands the ISC pool within which CRC is initiated while co-operating with TSG loss. Myb further exacerbates CRC pathology partly explaining why high MYB is a predictor of worse patient outcome.
Similar content being viewed by others


Introduction
Aberrant activation of the WNT pathway (Adenomatous polyposis coli (APC) or β-catenin mutation) is the pre-eminent early event in colorectal cancers (CRC). Nevertheless, parallel activation of other oncogenes including MYB occurs commonly and is reasonably thought to participate in CRC.1 Furthermore, Myb and β-catenin cooperate to regulate the CRC genes MYC and PTGS2 (COX2)2 along with the intestinal stem cell (ISC) gene, LGR5.3 Elevated MYB transcripts are a common feature of CRC2, 4, 5 and high Myb protein is a poor-prognosis indicator.6 Others have identified MYB and WNT-target gene, AXIN2 as among nine essential and differentially expressed genes in colon cancer.7 However, although MYB is on occasion amplified in CRC it is rarely (if ever) found to have mutations in coding regions but rather in transcriptional regulatory regions.1 Thus, it remains unclear how MYB specifically participates in colorectal carcinogenesis.
The intestine is a rapidly self-renewing tissue maintained by ISCs. The colonic epithelium is composed of the absorptive and the secretory lineages continuously renewed by rapidly cycling intestinal progenitor cells (IPC) generated from ISC.8 ISC are targets for transformation.9 Two kinds of ISCs have been proposed; crypt basal cells (CBC)10 and another, radiosensitive +4 ISCs.11, 12 These share the expression of Musashi-1, Pten and Sox-9 but can be discriminated by specific markers13, 14, 15 such as Lgr5 and Olfm4 for CBCs or Hopx, Bmi1 and mTert for +4 ISCs. These two populations of ISCs are phylogenetically related and can give rise to each other.15 IPCs also exhibit a high degree of phenotypic plasticity in emergency situations and can revert to a state of 'stem-ness'.16
Deregulation of ISC signaling pathways are evident in intestinal cancer exemplified in the Wnt-driven ApcMin/+ mouse model,17 which recapitulates both genetic (Apc mutations) and environmental aspects (inflammation) of human CRC.18 Importantly, activation of the Wnt pathway in either Lgr5+ or Bmi1+ ISCs but mostly not IPCs has been shown to initiate intestinal cancer.9 Here we sought to understand why patients with high MYB have worse outcomes and we demonstrate the direct involvement of Myb in activating ISC gene expression, CRC initiation and progression. Accordingly, we generated a transgenic mouse with latent Myb function that can be activated to confer constitutive Myb activity throughout the intestinal epithelium including in the context of a Wnt-driven tumorigenesis model and loss of the cell cycle regulator p27. Importantly, this work revealed the effect of Myb in the ISC compartment, co-operation with tumor suppressor gene (TSG) loss and a new role for Myb in influencing hypoxia and Vegfa expression.
Results
MybER transgenic mice recapitulate constitutive Myb expression in CRC
We have shown that Myb is frequently elevated in CRC1 and that those patients with tumors that have the higher level of Myb had a commensurate poorer prognosis.6 This remains the case for even early stage CRC (pT3 or pT4N0M0) tumors (Millen et al., submitted). Loss of Myb function leads to reduced IPC proliferation in the colon of adult mice.19 To test whether sustained Myb expression is pathogenic we enforced Myb activity, rather than its level of expression, in the gastrointestinal tract by taking advantage of the gastrointestinal tract-epithelial-specific expression pattern conferred by the Gpa33 promoter.20 A Gpa33-driven expression construct that fuses mouse Myb to a mutant ERα ligand-binding domain (Figures 1a and b) was engineered to generate transgenic mice. Transgene DNA sequence integrity was confirmed as well as its tight negative control and sensitive induction by 4-hydroxy-tamoxifen (4OHT) (Supplementary Figure 1b and c). Of six founders, two lines were used in experiments (Supplementary Figure 2). Insertion of the several transgene copies was demonstrated by Southern blotting (Figure 1c), whereas MybER fusion protein expression was confirmed by western blotting with anti-ERα antibody (Figure 1d) as well as anti-Myb antibodies in colonic crypts to levels comparable to endogenous Myb found in colon cancer cell line, CT2621 but is, as expected absent in WT crypts (Figure 1e).

Generation and characterization of MybER transgenic mice. (a) A fusion of mouse Myb to a mutant ERT2 estrogen receptor ligand-binding domain was generated under the control of the intestinal Gpa33-specific antigen promoter. (b) Model of tamoxifen (4OHT)-activation of the MybER protein to reveal the DNA-binding domain and transactivation domain (T/A) of Myb. (c) Insertion of the transgene was confirmed by Southern blotting using a Myb probe. (d) MybER protein expression (black arrow) was confirmed by western blotting with an anti-ERα antibody in the colonic crypt lysates from a WT and a MybER mouse, respectively, exposed to tamoxifen for 2 weeks. Pan actin was used a protein-loading control (gray arrow). (e) Colonic crypt lysates were also probed for Myb expression in parallel with CRC cell line CT26 with a cocktail to anti-Myb antibodies that binds amino-terminal to the fusion junction point of Myb in the MybER protein to detect a protein of the predicted size. (f) Organoid cultures derived from colonic crypts seeded in Matrigel in the presence of 0.1 μM 4OHT show increased colony size in MybER cultures. (g) Quantification of colonies formed/plated crypt nests and overall increased growth as assessed by the MTT assay. Cultures were grown for 7–10 days in the presence of 0.1 μM 4OHT before harvesting. Data presented for individual samples are shown; n>5; mean ± s.e.m.; *P<0.05, two sided t-test. Primary organoids formed in presence of 4OHT were then dissociated and secondary organoids generated ± 4OHT were scored after 5 days. (h) ChIP assay showing the occupancy of Lgr5 promoter by the fusion protein MybER following activation by 4OHT. ChIP was quantified by qRT–PCR and data were normalized for Gapdh promoter (n=4 independent experiments in triplicate). Data presented for individual samples (n>4) with mean ± s.e.m.; *P<0.05, ordinary one-way ANOVA.
In vitro activation of MybER increases IPC function
To understand the cell intrinsic effect of MybER-activation in primary colon cells, IPC were studied in vitro using the organoid assay22 (Figure 1f). MybER-activation led to an increase in organoids formed per plated crypt nest and greater growth as measured by MTT assays (Figure 1g). Then to test whether activated Myb is important for organoid maintenance of superior colon organoid propagation MybER organoids were treated with 4OHT for the entire period of the primary culture before dissociation into single cells. Dissociated organoids were then challenged to grow in the presence or absence of 4OHT to test whether 4OHT-withdrawal affects secondary organoid formation. Figure 1g shows that organoids require sustained MybER-activation to achieve superior growth and that the effect of MybER-induction is reversible.
We then tested the promoter occupancy of a well-characterized Myb target gene Lgr5 by MybER fusion protein in colonic crypts isolated from MybER mice exposed to tamoxifen or in untreated mice. Single-cell suspensions were cross-linked and chromatin immunoprecipitation assays were then performed using anti-ERα antibody whereby the binding of MybER to Lgr5 promoter was confirmed and this was further found to be tamoxifen-dependent (Figure 1h).
To dissect the effect of MybER-activation on different crypt cell compartments we conducted gene expression analysis in various cell population based on the activity of aldehyde dehydrogenase. Colonic stem cells have been shown to be characterized by high-aldehyde dehydrogenase activity. We analyzed the expression of cell cycle gene (CyclinE1; Ccne1) and ISC gene Lgr5, the TSG Pten and the pro-angiogenic gene Vegfa (Figure 2a) in populations with high (AldeHi), low (AldeLo) and no (AldeNEG) aldehyde dehydrogenase activity. Predicted Myb target genes, Lgr5, Ccne119 and Pten (unpublished data) were elevated in various populations of cells. Notably Lgr5 was elevated in both AldeHi and AldeLo population in contrast to Vegfa and Ccne1, which were elevated in AldeHi population and Pten, which was significantly induced only in the AldeLo population.

Molecular analysis of WT and MybER AldeHi, AldeLo and AldeNEG colonic crypt cell populations shows differential intestinal stem cell gene expression increased following MybER-activation and is reversible in adenomas. (a) Gene expression analyses were performed using qRT–PCR on RNA extracted from WT and MybER FACS sorted colonic crypt cells based on ALDH activity. Myb target gene Lgr5 mRNA expression was elevated in both AldeHi and AldeLo population. By contrast, mRNA of predicted target genes Vegfa and Ccne1, which were only significantly elevated in AldeHi population while Pten mRNA, was significantly elevated in the AldeLo population. (b) ON/OFF control of Myb-target Lgr5 by MybER following addition and withdrawal of 4OHT in MybER:Apc±-derived adenoma organoids cultured for 6 days were treated with 4OHT for 24 h before harvesting or were washed twice in DMEM/F12 and re-incubated in 4OHT-free adenoma media for 48 h before harvesting. Data presented for individual samples (n>4) are mean ± s.e.m.; *P<0.05 two-sided t-test.
To evaluate the role of Myb-activation during tumor initiation and progression we established organoid cultures from early stage adenomas in non-symptomatic MybER:Apcmin/+ mice (Supplementary Figure 2). Our results demonstrate that activated MybER is required at least during the first 48 h of initiation of culture to significantly increase organoid numbers (Supplementary Figure 2a–c). Sustained MybER-activation was also required to achieve optimal growth, whereas 4OHT-withdrawal abrogated the enhanced growth of MybER adenoma organoids (Supplementary Figure 2a and c). These data suggest that Myb is required to support both maximum adenoma initiation and progression and that the effects of MybER-activation are reversible.
By using adenomas from MybER:Apcmin/+ mice we were also able to establish cultures of relatively uniform cells that typically express the Wnt and Myb target gene, Lgr5 and ask whether modulating MybER activity with 4-OHT affects Lgr5 expression. Data in Figure 2b confirm that this strategy shows Lgr5 expression is subject to the ON/OFF control by MybER activity even in the context of activated Wnt signaling.
Activated Myb in vivo does not change IPC homeostasis unless p27 is lost
In vivo effects of sustained Myb activity on crypt morphology was investigated along with proliferation. We found that the crypt morphology was largely unaltered following MybER-activation (Supplementary Figure 3a). Furthermore, IPC proliferation was similar to that prior to tamoxifen-treatment (Supplementary Figure 3b). Two weeks of Tamoxifen produced modest but not significant effects on proliferation (Supplementary Figure 3a, d and e) when PCNA+ cells were scored according to crypt location or total number.
In view of such modest effects of activated MybER alone we postulated that homeostatic mechanisms might be restricting the in vivo effect of elevated Myb activity particularly in view of the increased Ccne1 expression in the colons of all MybER transgenic lines (Supplementary Figure 3c). As Pten mRNA expression was elevated in the AldeLo progenitor/precursor population (Figure 2a) we decided to explore Pten expression at the protein level in vivo within crypts finding significantly higher expression in MybER mice treated with tamoxifen compared with similarly treated controls (Supplementary Figure 4). These observations are consistent with reduced Pten expression observation in Myb-hypomorphic mutant colons (unpublished observations). As Pten is a negative regulator of the cell cycle and is molecularly considered upstream of p2723, 24 and that p27 in turn is a negative-regulator of Cdk/CyclinE1 activity25 we hypothesized that WT levels of p27 might mask effects of MybER in vivo. Accordingly, we established compound MybER:p27+/− mice. As expected we found p27 was reduced in both p27+/− and p27+/−:MybER colons. We also found that its expression was strongly induced in discrete population of cells located at the bottom of the crypts in MybER colon (Supplementary Figure 5a).
Although crypts of p27−/− mice show increased proliferation, this was not observed in p27+/− mice19 where IPC proliferation was comparable to WT mice (Supplementary Figure 6b, d and e); however, analysis of proliferation in p27+/−:MybER colons using immunohistochemistry (IHC) showed a significant increase in the total number of PCNA-positive cells, more remained in cycle up to crypt positions 14 compared with tamoxifen-treated WT mice (Supplementary Figure 6b, d and e). Thus, we hypothesized that p27 loss may increase the number of CyclinE1-positive cells. To confirm reduced p27 expression in p27+/−:MybER mouse we performed p27 IHC. We found that p27 is prominently expressed in the IPC zone (Supplementary Figure 5a). Quantitation of CyclinE1 IHC (Supplementary Figure 5b) showed that the number of CyclinE1-positive cells was not increased by MybER-activation alone. By contrast, when MybER was activated in p27+/− mice their number was significantly increased.
Activated Myb accelerates AOM-driven tumorigenesis
In view of this association between elevated Myb and CRC patient outcome patients we treated MybER mice with tamoxifen for an extended period of time (7 months) and at this point started to detect sick mice that once examined revealed elevated aberrant crypt foci (ACF) and a modest increase in colon tumors (Figures 3a and b). The cooperation we had previously found between Myb and activated β-catenin in driving Myc, Cox-2 and Lgr5 expression in the GI prompted us to generate compound MybER:Apcmin/+ mice. Cohorts of these mice were exposed to tamoxifen but their disease-free survival was not different to Apcmin/+ mice alone on tamoxifen (Figure 3c).

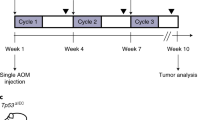
MybER-activation accelerates CRC initiation and progression of AOM-driven CRC. (a) ACF in tamoxifen-treated MybER mice compared with WT mice on Tamoxifen at 7 months (n=3). (b) Tumor number is slightly increased in MybER mice (n=8). (c) Activation of MybER in mice on an Apcmin/+ background does not influence survival. (d) Diagram depicting the pre-treatment and continual access to tamoxifen in chow (8 weeks) and 2 weeks later the initiation of weekly AOM injections used to test the effect of Myb-induction in the initiation phase of CRC. (e) Pre-treatment by tamoxifen increases the percentage of cells with high aldehyde dehydrogenase activity identified by FACS (n>5). (f) Survival experiments were conducted to determine whether MybER-activation on a WT or Apcmin/+ (n>10) background translates into poorer survival outcome owing to AOM-induced CRC. Individual mice were harvested when they reached ethical end points defined by either bleeding from the anus, anaemia (pale feet), hunching, severe diarrhea, prolapsed anus or body weight loss >20%. Survival analysis revealed that MybER-activation on a WT and Apcmin/+ backgrounds significantly accelerated the initiation and progression of disease and reduced the life expectancy of mice after treatment. (g) Tumors generated on an Apcmin/+ background in presence of MybER were significantly larger compared with tumors arising in Apcmin/+ mice and MybER-activation did not alter the number of tumors. (h) Tumors derived from all groups were sectioned and analyzed by IHC for PCNA to assess cell proliferation and for goblet cell differentiation (i–j) using periodic acid staining (PAS). Data presented for individual samples with means ±s.e.m.; *P<0.05, **P<0.01. Survival curves were analyzed using log-rank (Mantel–Cox) Test.
As we have found that inflammation appears to be responsible for an increase Myb expression during early events of CRC, for example, ACF and adenoma formation (Pereira et al., in press) prior to detectable mutated Myb regulatory sequences we decided to activate Myb in the initiation phase of CRC. MybER mice were treated with tamoxifen over a period of 8 weeks, six of which also employed weekly colon carcinogen azoxymethane (AOM) injections26 (Figure 3d). Hence, MybER was activated for 2 weeks prior to AOM-treatment to allow the expansion of the ISC pool. To demonstrate this we have employed the Aldefluor assay following tamoxifen-treatment for 2 weeks, to identify colonic ISC. Such cells have been shown to act as tumor-initiating precursor cells in the colon and to be responsible for the transition from colitis to cancer.27 As predicted, we found that this population was expanded (Figure 3e).
Subsequent exposure of tamoxifen-treated mice to AOM markedly accelerated colon tumorigenesis in MybER mice on either WT or Apcmin/+ backgrounds (Figure 3f) and led to more frequent, as well as larger tumors, on an Apcmin/+ background (Figure 3g). As Myb overexpression or Apc mutation both drive proliferation and block cyto-differentiation in colon cells28 we evaluated PCNA staining and indirectly explored the state of goblet cell differentiation by measuring the extent of mucin staining in tumors with Period Acid Schiffs (PAS). Mucin production was significantly reduced in MybER tumors compared with AOM-induced tumors from WT mice, whereas only a trend toward increased proliferation was evident in the presence of MybER activation (Figures 3h–j).
To characterize the molecular profile of the tumors arising in the context of the MybER in vivo following AOM-treatment, we performed quantitative RT–PCR analyses. Following confirmation of MybER mRNA expression we investigated the cell cycle genes Ccne1 and Ccnd1, the Wnt and Myb target genes Myc2 and Lgr53 and the ISC genes29 Olfm4, Bmi1, Aldh1 and Pten (Figure 4). We found that expression of both Ccne1 and Ccnd1 was increased in MybER transgenic mice on either an ApcMin/+ or WT background. Shared Wnt/Myb target genes, Myc and Lgr5, increased in transgenic compared with non-transgenic mice on an ApcMin/+ background. Olfm4 mRNA was increased in MybER tumors on both an ApcMin/+ and WT background. When the expression of the Myb target gene Bmi130 was assessed we found that it was increased in MybER tumors but not in ApcMin/+ tumors, suggesting adenocarcinomas may arise from, or be sustained by, different ISC populations when MybER is activated. Aldh1 mRNA was not changed, whereas Pten mRNA was significantly increased in MybER:ApcMin/+ compared with ApcMin/+ tumors further suggesting that this TSG may restrict tumorigenesis in this context.

MybER-activation alters the molecular expression profile of tumors. qRT–PCR was used to analyse expression of MybER, CcneE1, Ccnd1, Myc, Lgr5, Olfm4, Bmi1, Aldh1 and Pten in tumors. Data presented for individual samples (n>6) with means ±s.e.m.; *P<0.05, **P<0.01; ANOVA.
To understand the effect of TSGs, Apc and p27 in terms of MybER-associated adenoma initiation mice were evaluated in groups according to genotype and when the first mouse in the paired groups became sick. Mice on an ApcMin/+ background were first to get sick. MybER:ApcMin/+ and ApcMin/+ mice were harvested 4 weeks after the last AOM injection; the p27±:MybER and p27± cohorts were harvested at 10 weeks, whereas MybER and WT mice were culled later again at 18 weeks. No significant differences in either number nor average size of ACF was detected between p27±:MybER and p27±, or MybER:ApcMin/+ and ApcMin/+ groups (Supplementary Figure 7a and b). Nevertheless, the MybER-activation on an ApcMin/+ background resulted in a larger class of ACF compared with ApcMin/+ alone (Supplementary Figure 7c), confirming cooperation between Myb and the Wnt pathway in CRC initiation. The number and size of tumors were also assessed in these animals (Supplementary Figure 7d and e). Although changes were not significant the size of tumors generated on an ApcMin/+ background following MybER-activation trended to being larger.
Activated Myb in vivo reproduces human CRC clinical symptoms
In view of the increased morbidity of the MybER:ApcMin/+ and MybER mice compared with their ApcMin/+ and WT counterparts we decided to take a CRC-clinical perspective in evaluating symptoms that collectively precipitate the culling of mice following AOM-induced carcinogenesis. In both comparisons, the presence of MybER was associated with a more severe morbidity (Figures 5a and b) except when MybER was activated on an ApcMin/+ background where the symptom of anus bleeding shifted toward another symptom, hunching. Hunching relates to intestinal occlusion by tumors and this is globally increased on the ApcMin/+ background given that tumors also developed in the small intestines (SI) and are more numerous. Hunching was exacerbated by MybER-activation in ApcMin/+:MybER mice probably owing to the fact that there is a significant increase in tumor size in the colon in these mice (Figure 3g). There was no change of distribution between colon and SI in ApcMin/+:MybER compared with ApcMin/+ mice. Mice on an ApcMin/+ background also showed increased anaemia. As mice suffering anemia commonly initiate splenomegaly we measured spleen weights of all mice at the time of cull and found they were significantly larger in MybER mice on an ApcMin/+ background (Figure 5c).

MybER-activation promotes CRC-associated clinical symptoms. (a) AOM-treated mice on a WT or (b) Apcmin/+ background were euthanized owing to a spectrum of symptoms. (c) Splenomegaly was significantly higher in Apcmin/+:MybER mice (grey zone indicates normal spleen weights in age-matched WT mice). (d) The distribution of tumors between small intestines and colon was similar in AOM treated Apcmin/+:MybER and Apcmin/+ mice. (e) Tumors arising on a MybER background were consistently redder indicative of an increase in red blood cells (RBC) (red arrows show red tumors). (f) The tumors were also significantly more hypoxic as shown by HIF1α IHC and (g) Vegfa mRNA expression was significantly higher in mice on a MybER background. (h–i) Although vascularization was not found to increase in MybER mice (see Supplementary Figure 8) the amount of RBCs in tumors on a WT background was most apparently increased (red arrows). Data presented for individual samples (n>5) with means ±s.e.m.; *P<0.05, two-sided t-test.
Because of the more severe anemia and anal bleeding in MybER mice we examined colon lesions for evidence of increased vascularization. We consistently observed adenomas with excessive hemorrhage in MybER mice and hypoxia (as shown by hypoxia inducible factor-1α IHC in Figure 5f and Supplementary Figure 8a and elevated expression of Myb and hypoxia inducible factor-1α target gene Vegfa 31, 32 (Figure 5g). The amount of CD31-positive endothelium and PCNA staining were not increased (Supplementary Figure 8). When the abundance of red blood cells was evaluated in adenomas/carcinomas from MybER mice they were significantly increased compared with WT mice (Figures 5h and i). These data suggest that Myb-activation influences blood vessel leakage, leading to the clinical symptoms that precipitate culling of mice in AOM studies. By contrast, hypoxia-inducible factor-1α and red blood cell leakage in ApcMin/+and ApcMin/+:MybER tumors was not significantly different.
Discussion
High Myb expression is a robust predictor of CRC patient outcome and by employing a novel gastrointestinal tract transgenic mouse model, we have been able to reveal a parallel malignant role of elevated Myb observed in human CRC2, 4, 5 not only intrinsic to cancer cells but also in the tumor microenvironment promoting hypoxia and leakiness of the tumor vasculature.
The malignant potential of MYB in CRC has been ascribed previously1 but has not been formally demonstrated. With the very recent revelation that MYB-NFIB fusion genes are pathogenic in adenoid cystic carcinoma33, 34, 35, 36 often in the context of few other promalignant genomic changes, expands the spectrum of solid tumors where MYB shows oncogenic association. Our transgenic mice use a fusion construct that essentially truncates the Myb protein. This was necessary to allow the ER ligand-binding domain to be tightly modulated by Tamoxifen. With specific reference to CRC recent insights into how this oncogenic potential might be driven, and in what cells, come from studies such as those that show that loss of Myb function decreased ISC activity and proliferation plus improved survival of ApcMin/+ mice.2, 3 Although loss of Myb function was effective in instilling a loss of ISC activity by itself this is the first occasion where the effect of Myb hyperactivation has been evaluated. First, the full effects of hyperactivated-Myb on ISC function under homeostasis was only achieved, when at the same time, one allele of CyclinE1-Cdk inhibiting p27 gene was lost.25 This was consistent with our previous work suggesting that p27 complete loss resulted in increased proliferation in colonic crypt IPCs.19 We also suggest that the effect of p27 haploinsufficiency on intestinal homeostasis is achieved by allowing CyclinE1 to exert its pro-proliferative action on IPCs. Although MybER-induced tumorigenicity in mice by itself required extended periods of time (~6–7 months), it is markedly increased with AOM-treatment and on a Apcmin± background. These collective observations clearly demonstrate that Myb needs to act in concert with other factors in keeping with the generally accepted view that CRC requires multiple oncogenic 'hits'.37
Apcmin/+:MybER compound mice were not without complexities as the Min mutation does progressively compromise the hematological and immunological competency, which is very likely to accelerate the progressive decline of mice carrying tumors evident in 80-day-old mice and to modulate the aetiology of clinical symptoms such as blood loss and anaemia.38, 39, 40 Accordingly, the use of the intestinal-specific activation of Myb alone in the tumor induction phase (first ~60 days) in the presence of AOM-treatment and with the knowledge that induced-MybER expands the ISC compartment may explain the shortened time to cull of MybER mice compared with WT littermates (160 vs 273 days, P<0.01). By contrast in mice on an Apcmin/+ background, mice when treated with AOM require culling on average at 95 days and only slightly (but significantly) earlier, when MybER is activated (average at 90 days).
Interestingly, some mice required euthanizing much earlier in the Apcmin/+:MybER cohort than the Apcmin/+alone cohort. The co-morbidities that arise because of systemic effects of the Min mutation highlight the need to investigate ACF and tumor burden at specific times following AOM-treatment particularly when the presence of SI adenomas inherent to this mouse genotype compromise the health of Apcmin/+ mice.
Using combinations of mice with TSG defects we observed that the Pten/p27 axis might be responsible for keeping Myb activity in-check presumably via CyclinE1/Cdk2 inhibition. However, although p27 monoallelic-loss increases IPC proliferation as seen in extended PCNA staining in colonic crypts, this did not influence MybER oncogenic potential. Thus, it is rather unlikely that Pten/p27 influences the ability of Myb to promote ISC properties in adenomas. On the other hand, Myb clearly acts in concert with ApcMin to enhance ISC activity and gene expression in normal and tumor tissue, a property ultimately responsible for adenoma formation.41
In this study, we found that Myb-activation co-operates with mutational events induced by AOM to faithfully reproduce human clinical symptoms of CRC better than found in ApcMin mice. Myb-hyperactivation also leads to an increase in Vegfa expression associated with an increase in hemorrhaging tumors as well as hypoxia and red blood cell leakage into surrounding stroma. This leakage we speculate is due to VegfA-driven vessel permeablization.3, 27, 42, 43 In this context Vegfa was positively correlated with MybER-induction and this observation was consistent with RNAseq data obtained from human CRC (data not shown).
More generally, these observations are potentially important as the presence of hemoglobin (that is, lysed red blood cells) is the basis for population-based fecal occult blood testing as a screening modality for deducing CRC burden,44, 45 and targeting VEGFA in CRC metastasis augments clinical management.46 A working model of MybER in inducing genes and initiating ACFs plus subsequent progression to AOM-induced adenoma is mapped-out in Figure 6.

Model describing the effects of MybER-activation on crypt biology and adenoma gene expression as initiating events for CRC. Pink cells identify differentiated goblet cells that are reduced during aberrant crypt focus (ACF) formation following MybER-activation. Further gene expression changes and function consequences are established following progression from ACFs to adenomas.
In conclusion, we report the generation of the first gastrointestinal tract-specific, inducible transgenic Myb mouse that exacerbates CRC initiation and reproduces aspects of human CRC pathology. Myb co-operates with the loss of TSG Apc where both alleles need to be affected, whereas our studies demonstrate another instance where p27 haploinsufficiency unleashes aspects of the pro-proliferative potential of Myb. This is consistent with the manner in which p27 seems to act as a tumor progression rheostat47 but this alone is not sufficient for accelerating CRC. Here we demonstrate that Myb confers a malignant phenotype to cancer cells and we provide understanding why high MYB equates to poor prognosis and discriminates patients who are likely to do poorly and relapse from patients who fare better. These data are in accordance with our previous reporting that elevated Myb is a feature of metastatic CRC.6 Several MYB target genes serve as therapeutic targets, for example, BCL-2, VEGFA, COX-2 and GRP-78, placing MYB at the hub for the regulation of a range of pro-malignant genes. Thus, MYB represents an 'Achilles heel' in CRC and we have shown that Myb can be targeted immunologically in the setting of mouse CRC,48, 49 raising the prospect of considering this strategy in the human disease.
Materials and methods
Generation of MybER transgenic mice
Complementary DNA encoding the MybERT2 fusion protein was inserted in to a described expression construct encompassing a 4.1 kb promoter sequence of the murine Gpa33 gene.20 The Gpa33-MybERT2 fragment was excised and microinjected into the male pronuclei of fertilized mouse oocytes (CBA/CAH/C57BL/6). Injected embryos were transferred to pseudo-pregnant mice. The transgenic lines obtained were intercrossed with C57BL/6 females.
Southern blot analysis and PCR genotyping
Ten μg of genomic DNA was extracted from MybER or WT liver using DNA easy Kit (Qiagen, Chadstone, Victoria, Australia) and was digested using EcoRI. Following precipitation the DNA was suspended in 10 μl resolved on a 0.8% agarose gel and capillary transferred onto Gene screen+ hybridization transfer paper. Membranes were prehybridized for 30 min at 42 oC in ULTRAhyb buffer (AMBION, Scorsesby, Victoria, Australia) and hybridized overnight at 42 °C with [32P] labeled probes. Probes were generated from restriction enzyme digestion of the MybERT2 plasmid; gel purified and labeled using the DECAprime II kit (AMBION). For routine genotyping 50 ng of genomic tail DNA was obtained and the MybER transgene was amplified using HotStar-Taq DNA polymerase (Qiagen). MybER primers for genotyping were:- forward 5′ -ggagctggagttgctcctgatgtc -3′ and reverse 5′catcgaagcttcactgaagggtctgg-3′.
Western blotting
Soluble protein was extracted from mouse colon and colon adenomas using radioimmunoprecipitation assay buffer (25 mM HEPES pH8, 300 mM KCl, 1 mM EDTA, 0.15% sodium dodecyl sulphate, 0.15% sodium deoxycholate, 1% Triton X, Complete Protease Inhibitor cocktail). Lysates were incubated on ice for 30 min and then mechanically lysed using a dounce homogenizer. Lysates were subsequently centrifuged at 14 krpm, 4 °C for 30 min and the soluble protein extracts were resolved on 4–12% NuPAGE MOPs gels (Invitrogen, Scorsesby, Victoria, Australia) and transferred to polyvinylidene difluoride membrane. Membranes were probed with anti-estrogen receptor antibody (TE111.5D11; Thermo Scientific, Scorsesby, Victoria, Australia) and anti-Myb 5.1 plus Mab 6.2.50 Membranes were probed with mouse horseradish peroxidase secondary antibodies (Bio-Rad, Gladesville, New South Wales, Australia) and developed by enhanced chemiluminescence or with alkaline phosphatase secondary antibody plus nitro-blue tetrazolium chloride and 5-bromo-4-chloro-3′-indolyphosphate p-toluidine salt substrate (Roche, Castle Hill, New South Wales, Australia).
Chromatin immunoprecipitation assays
Mice were treated with Tamoxifen chow for a week prior to crypt extraction and cross linking. Chromatin immunoprecipitation assays were performed on the Lgr5 promoter as described previously.3 Anti-estrogen receptor antibody (TE111.5D11; Thermo Scientific) was used to chromatin immunoprecipitation the MybER fusion protein.
Oligonucleotide sequences for quantitative real-time PCR
Quantitative real-time PCR reactions were conducted using an ABI Prism 7000 Sequence Detection System (Applied Biosystems, Scorseby, Victoria, Australia). For one reaction, 8 μl complementary DNA (1:10 dilution) was combined with 10 μl SYBR Green PCR Master Mix (Applied Biosystems) and 200 nM primers (Geneworks, Thebarton, South Australia, Australia) and amplified using temperatures of 50 oC for 2 min and 95 oC for 15 min. These initial steps were followed by 45 cycles of 95 oC for 15 s and 60 oC for 1 min. RNA was extracted using Trizol (Invitrogen) and treated with RNAase-free DNAseI (1 U μl−1; Promega, Madison, WI, USA). Superscript III Reverse Transcriptase (Invitrogen) was used for first-strand complementary DNA synthesis. Gene expression was normalized to β2-microglobulin. Primers sequences were designed against the complementary DNA sequence of target genes and spanned exon–exon junctions.

IHCs
Mice were culled and the colons were cut open in cold phosphate-buffered saline (PBS) and fixed overnight in neutral buffered formalin at ambient temperature. For CD31 (Pharmingen, North Ryde, New South Wales, Australia) staining samples were fixed in 4% paraformaldehyde at 4 oC overnight. Colons were processed for paraffin embedding and sectioning at 4 μm. Periodic Acid Schiff’s reagent stain and IHC for PCNA (BD Transduction Laboratories, North Ryde, New South Wales, Australia), p27 (Santa-Cruz, Dallas, TX, USA), Pten (Cell signalling, Danver, MA, USA) and CyclinE1 (AbCam, Melbourne, Victoria, Australia) as previously published.19
Aldefluor assay
Colon crypts were extracted as previously published and were dissociated for 30 min in accumax before gentle trituration to obtain a single-cell suspension. Colon cells were stained as per manufacturer's instruction (Aldefluor assay STEMCELLS Technologies, Tullamarine, Victoria, Australia). Cells were analyzed using FACS Diva (BD Biosciences) or RNA was extracted using RNAeasy kit (Qiagen) as per manufacturer’s instructions to perform target gene analysis.
Organoid assays
Organoid cultures were established using a modified version of published protocols.22 Cultures were assessed for growth using the MTT assay51 and the number of organoids formed was assessed after 10 days. In brief, colons were dissected in cold PBS and cut open. Following 30 min incubation in PBS containing Nystatin and Gentamycin, colons were incubated in PBS containing 2 mM EDTA and 0.5 mM dithiothreitol for 60 min under gentle agitation in a cold room. Samples were washed in PBS and pipetted up and down 10 times in 10 ml of cold PBS prior to detaching colon crypts by vigorous shaking. Crypts were centrifuged at 1000 rpm for 5 min and 500 crypts were resuspended in 50 μl of Matrigel. Matrigel was set for 5–10 min prior to the addition of 500 μl of 1:1 phenol red-free DMEM/F12 and Wnt-3a phenol red-free conditioned DMEM/F12 (obtained from Wnt3a producing L cells52) and containing growth factors (B27 supplement from Life Technology (Rocky Hill, NJ, USA), R-spondin 500 ng ml−1 from Sino Biological (Beijing, China), 20 ng ml−1 Noggin from Peprotech, EGF 100 ng ml−1 from Roche). For mouse adenoma cultures the media used was phenol red-free DMEM/F12 and contained growth factors (B27 supplement from Life Technology, and EGF 100 ng ml−1 from Roche).
Mice, experimental treatments and tumorigenesis studies
MybER transgenic mice were backcrossed onto a C57/BL6 background and experiments were conducted between generations 2–8 and used to generate compound p27±:MybER53 and Apcmin/+:MybER54 mice approved by the PMCC Animal Ethic Committee. Mice were fed Tamoxifen-chow (1 g Kg−1) for 2 weeks prior to the start of AOM intraperitoeal injections (10 mg Kg−1 body weight) once weekly over 6 consecutive weeks26 during which mice were provided with tamoxifen.
Quantitation and statistical analysis
The number and location of PCNA+ cells were recorded in 10–20 crypts under high power magnification. PCNA, Periodic Acid Schiff’s reagent stain and CD31 staining on tumor samples were quantified using Metamorph software (Universal Imaging, Sunnydale, CA, USA).19 Statistical analyses were carried out using GraphPad Prism (GraphPad Software, San Diego, CA, USA).
References
Ramsay RG, Gonda TJ . MYB function in normal and cancer cells. Nat Rev Cancer 2008; 8: 523–534.
Ciznadija D, Tothill R, Waterman ML, Zhao L, Huynh D, Yu RM et al. Intestinal adenoma formation and MYC activation are regulated by cooperation between MYB and Wnt signaling. Cell Death Differ 2009; 16: 1530–1538.
Cheasley D, Pereira L, Lightowler S, Vincan E, Malaterre J, Ramsay RG . Myb controls intestinal stem cell genes and self-renewal. Stem Cells 2011; 29: 2042–2050.
Hugo H, Cures A, Suraweera N, Drabsch Y, Purcell D, Mantamadiotis T et al. Mutations in the MYB intron I regulatory sequence increase transcription in colon cancers. Genes Chromosomes Cancer 2006; 45: 1143–1154.
Thompson MA, Flegg R, Westin EH, Ramsay RG . Microsatellite deletions in the c-myb transcriptional attenuator region associated with over-expression in colon tumour cell lines. Oncogene 1997; 14: 1715–1723.
Biroccio A, Benassi B, D'Agnano I, D'Angelo C, Buglioni S, Mottolese M et al. c-Myb and Bcl-x overexpression predicts poor prognosis in colorectal cancer: clinical and experimental findings. Am J Pathol 2001; 158: 1289–1299.
Cheung HW, Cowley GS, Weir BA, Boehm JS, Rusin S, Scott JA et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc Natl Acad Sci USA 2011; 108: 12372–12377.
Rothenberg ME, Nusse Y, Kalisky T, Lee JJ, Dalerba P, Scheeren F et al. Identification of a cKit(+) colonic crypt base secretory cell that supports Lgr5(+) stem cells in mice. Gastroenterology 2012; 142: 1195–1205 e1196.
Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009; 457: 608–611.
Cheng H, Leblond CP . Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. III. Entero-endocrine cells. Am J Anat 1974; 141: 503–519.
Potten CS, Kovacs L, Hamilton E . Continuous labelling studies on mouse skin and intestine. Cell Tissue Kinet 1974; 7: 271–283.
Potten CS . Extreme sensitivity of some intestinal crypt cells to X and gamma irradiation. Nature 1977; 269: 518–521.
Umar S . Intestinal stem cells. Current Gastroenterol Rep 2010; 12: 340–348.
Lin SA, Barker N . Gastrointestinal stem cells in self-renewal and cancer. J Gastroenterol 2011; 46: 1039–1055.
Takeda N, Jain R, LeBoeuf MR, Wang Q, Lu MM, Epstein JA . Interconversion between intestinal stem cell populations in distinct niches. Science 2011; 334: 1420–1424.
van Es JH, Sato T, van de Wetering M, Lyubimova A, Yee Nee AN, Gregorieff A et al. Dll1(+) secretory progenitor cells revert to stem cells upon crypt damage. Nat Cell Biol 2012; 14: 1099–1104.
Ramsay RG, Barton AL, Gonda TJ . Targeting c-Myb expression in human disease. Expert Opin Ther Targets 2003; 7: 235–248.
Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012; 491: 254–258.
Malaterre J, Carpinelli M, Ernst M, Alexander W, Cooke M, Sutton S et al. c-Myb is required for progenitor cell homeostasis in colonic crypts. Proc Natl Acad Sci USA 2007; 104: 3829–3834.
Flentjar N, Chu PY, Ng AY, Johnstone CN, Heath JK, Ernst M et al. TGF-betaRII rescues development of small intestinal epithelial cells in Elf3-deficient mice. Gastroenterology 2007; 132: 1410–1419.
Corbett TH, Griswold DP Jr, Roberts BJ, Peckham JC, Schabel FM Jr . Tumor induction relationships in development of transplantable cancers of the colon in mice for chemotherapy assays, with a note on carcinogen structure. Cancer Res 1975; 35: 2434–2439.
Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology 2011; 141: 1762–1772.
Gottschalk AR, Basila D, Wong M, Dean NM, Brandts CH, Stokoe D et al. p27Kip1 is required for PTEN-induced G1 growth arrest. Cancer Res 2001; 61: 2105–2111.
Tsutsui S, Inoue H, Yasuda K, Suzuki K, Tahara K, Higashi H et al. Inactivation of PTEN is associated with a low p27Kip1 protein expression in breast carcinoma. Cancer 2005; 104: 2048–2053.
Tsihlias J, Kapusta L, Slingerland J . The prognostic significance of altered cyclin-dependent kinase inhibitors in human cancer. Annu Rev Med 1999; 50: 401–423.
Altemani A, Costa AF, Montalli VA, Mosqueda-Taylor A, Paes de Almeida O, Leon JE et al. Signet-ring cell change in adenoid cystic carcinoma: a clinicopathological and immunohistochemical study of four cases. Histopathology 2013; 62: 531–542.
Roeb E, Dreyer T, Steiner D, Brauninger A, Gattenlohner S . A 75-year-old female patient with pleural effusion and gastric metastases of a poorly differentiated carcinoma. Internist (Berl) 2013; 54: 244–248.
Ramsay RG, Ciznadija D, Sicurella C, Reyes N, Mitchelhill K, Darcy PK et al. Colon epithelial cell differentiation is inhibited by constitutive c-myb expression or mutant APC plus activated RAS. DNA Cell Biol 2005; 24: 21–29.
Ziskin JL, Dunlap D, Yaylaoglu M, Fodor IK, Forrest WF, Patel R et al. In situ validation of an intestinal stem cell signature in colorectal cancer. Gut 2012; 62: 1012–1013.
Waldron T, De Dominici M, Soliera AR, Audia A, Iacobucci I, Lonetti A et al. c-Myb and its target Bmi1 are required for p190BCR/ABL leukemogenesis in mouse and human cells. Leukemia 2012; 26: 644–653.
Kim SY, Yang YS, Hong KH, Jang KY, Chung MJ, Lee DY et al. Adenovirus-mediated expression of dominant negative c-myb induces apoptosis in head and neck cancer cells and inhibits tumor growth in animal model. Oral Oncol 2008; 44: 383–392.
Lutwyche JK, Keough RA, Hunter J, Coles LS, Gonda TJ . DNA binding-independent transcriptional activation of the vascular endothelial growth factor gene (VEGF) by the Myb oncoprotein. Biochem Biophys Res Commun 2006; 344: 1300–1307.
Stephens PJ, Davies HR, Mitani Y, Van Loo P, Shlien A, Tarpey PS et al. Whole exome sequencing of adenoid cystic carcinoma. J Clin Invest 2013; 123: 2965–2968.
Ho AS, Kannan K, Roy DM, Morris LG, Ganly I, Katabi N et al. The mutational landscape of adenoid cystic carcinoma. Nat Genet 2013; 45: 791–798.
Mitani Y, Li J, Rao PH, Zhao YJ, Bell D, Lippman SM et al. Comprehensive analysis of the MYB-NFIB gene fusion in salivary adenoid cystic carcinoma: incidence, variability, and clinicopathologic significance. Clin Cancer Res 2010; 16: 4722–4731.
Persson M, Andren Y, Mark J, Horlings HM, Persson F, Stenman G . Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc Natl Acad Sci USA 2009; 106: 18740–18744.
Stenman G . Fusion oncogenes in salivary gland tumors: molecular and clinical consequences. Head Neck Pathol 2013; 7: 12–19.
Bell D, Roberts D, Karpowicz M, Hanna EY, Weber RS, El-Naggar AK . Clinical significance of Myb protein and downstream target genes in salivary adenoid cystic carcinoma. Cancer Biol Ther 2011; 12: 569–573.
You S, Ohmori M, Pena MM, Nassri B, Quiton J, Al-Assad ZA et al. Developmental abnormalities in multiple proliferative tissues of Apc(Min/+) mice. Int J Exp Pathol 2006; 87: 227–236.
Coletta PL, Muller AM, Jones EA, Muhl B, Holwell S, Clarke D et al. Lymphodepletion in the ApcMin/+ mouse model of intestinal tumorigenesis. Blood 2004; 103: 1050–1058.
von Holstein SL, Fehr A, Persson M, Therkildsen MH, Prause JU, Heegaard S et al. Adenoid cystic carcinoma of the lacrimal gland: MYB gene activation, genomic imbalances, and clinical characteristics. Ophthalmology 2013.
Bates DO, Harper SJ . Regulation of vascular permeability by vascular endothelial growth factors. Vascul Pharmacol 2002; 39: 225–237.
Nagy JA, Benjamin L, Zeng H, Dvorak AM, Dvorak HF . Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis 2008; 11: 109–119.
Ouyang DL, Chen JJ, Getzenberg RH, Schoen RE . Noninvasive testing for colorectal cancer: a review. Am J Gastroenterol 2005; 100: 1393–1403.
Vernon SW . Participation in colorectal cancer screening: a review. J Natl Cancer Inst 1997; 89: 1406–1422.
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004; 350: 2335–2342.
Philipp-Staheli J, Payne SR, Kemp CJ . p27(Kip1): regulation and function of a haploinsufficient tumor suppressor and its misregulation in cancer. Exp Cell Res 2001; 264: 148–168.
Williams BB, Wall M, Miao RY, Williams B, Bertoncello I, Kershaw MH et al. Induction of T cell-mediated immunity using a c-Myb DNA vaccine in a mouse model of colon cancer. Cancer Immunol Immunother 2008; 57: 1635–1645.
Cross RS, Malaterre J, Davenport AJ, Carpinteri S, Anderson RL, Darcy PK et al. Therapeutic DNA vaccination against colorectal cancer by targeting the MYB oncoprotein. Clin Transl Immunol 2015; 4: e30.
Ramsay RG, Ishii S, Nishina Y, Soe G, Gonda TJ . Characterization of alternate and truncated forms of murine c-myb proteins. Oncogene Res 1989; 4: 259–269.
Xu H, Balakrishnan K, Malaterre J, Beasley M, Yan Y, Essers J et al. Rad21-cohesin haploinsufficiency impedes DNA repair and enhances gastrointestinal radiosensitivity in mice. PLoS One 2010; 5: e12112.
Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T et al. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 2003; 423: 448–452.
Moser AR, Luongo C, Gould KA, McNeley MK, Shoemaker AR, Dove WF . ApcMin: a mouse model for intestinal and mammary tumorigenesis. Eur J Cancer 1995; 31 A: 1061–1064.
Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E et al. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell 1996; 85: 733–744.
Acknowledgements
The authors thank the Peter MacCallum Histology and Microscopy departments and animal facility as well as the NHMRC Senior Research Fellowship Scheme (RR, ME) and Program Grant scheme (RGR, JM and ME).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Malaterre, J., Pereira, L., Putoczki, T. et al. Intestinal-specific activatable Myb initiates colon tumorigenesis in mice. Oncogene 35, 2475–2484 (2016). https://doi.org/10.1038/onc.2015.305
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2015.305
This article is cited by
-
Genome-wide mapping of DNA-binding sites identifies stemness-related genes as directly repressed targets of SNAIL1 in colorectal cancer cells
Oncogene (2019)
-
CircHIPK3 promotes colorectal cancer growth and metastasis by sponging miR-7
Cell Death & Disease (2018)
-
Overexpression of c-Myb is associated with suppression of distant metastases in colorectal carcinoma
Tumor Biology (2016)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}