
Abstract
The ERK1/2 (extracellular signal-regulated kinase 1 and 2) pathway, comprising the protein kinases RAF (v-raf-1 murine leukemia viral oncogene homolog 1), MEK1/2 (mitogen-activated protein kinase or ERK kinase 1 and 2) and ERK1/2 is frequently de-regulated in human cancers, due to mutations in RAS or BRAF (v-raf-1 murine leukemia viral oncogene homolog B1). New, highly selective inhibitors of BRAF and MEK1/2 have shown promise in clinical trials, including in previously intractable diseases such as melanoma. However, drug-resistant tumour cells invariably emerge leading to disease progression. It is important to understand the mechanisms underlying such acquired resistance since this may lead to the development of rational strategies either to delay its onset or to overcome it once established. It also offers unique insights into the plasticity of signalling pathways, which may in turn inform our understanding of the basic biology of these pathways and lead to the validation of new drug targets. Several recent reports have identified diverse mechanisms of acquired resistance to MEK1/2 or BRAF inhibitors. In this article, we review these studies, discuss the different mechanisms, identify common themes and consider their therapeutic implications.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 50 print issues and online access
$259.00 per year
only $5.18 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others
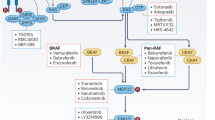


References
Yoon S, Seger R . The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 2006; 24: 21–44.
Karnoub AE, Weinberg RA . Ras oncogenes: split personalities. Nat Rev Mol Cell Biol 2008; 9: 517–531.
Meloche S, Pouysségur J . The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 2007; 26: 3227–3239.
Balmanno K, Cook SJ . Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ 2009; 16: 368–377.
Gillings AS, Balmanno K, Wiggins CM, Johnson M, Cook SJ . Apoptosis and autophagy: BIM as a mediator of tumour cell death in response to oncogene-targeted therapeutics. FEBS J 2009; 276: 6050–6062.
Schulze A, Lehmann K, Jefferies HB, McMahon M, Downward J . Analysis of the transcriptional program induced by Raf in epithelial cells. Genes Dev 2001; 15: 981–994.
Dougherty MK, Müller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD et al. Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell 2005; 17: 215–224.
Ritt DA, Monson DM, Specht SI, Morrison DK . Impact of feedback phosphorylation and Raf heterodimerization on normal and mutant B-Raf signaling. Mol Cell Biol 2010; 30: 806–819.
Buday L, Warne PH, Downward J . Downregulation of the Ras activation pathway by MAP kinase phosphorylation of Sos. Oncogene 1995; 11: 1327–1331.
Porfiri E, McCormick F . Regulation of epidermal growth factor receptor signaling by phosphorylation of the ras exchange factor hSOS1. J Biol Chem 1996; 271: 5871–5877.
Heisermann GJ, Wiley HS, Walsh BJ, Ingraham HA, Fiol CJ, Gill GN . Mutational removal of the Thr669 and Ser671 phosphorylation sites alters substrate specificity and ligand-induced internalization of the epidermal growth factor receptor. J Biol Chem 1990; 265: 12820–12827.
Li X, Huang Y, Jiang J, Frank SJ . ERK-dependent threonine phosphorylation of EGF receptor modulates receptor downregulation and signaling. Cell Signal 2008; 20: 2145–2155.
Owens DM, Keyse SM . Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 2007; 26: 3203–3213.
Sturm OE, Orton R, Grindlay J, Birtwistle M, Vyshemirsky V, Gilbert D et al. The mammalian MAPK/ERK pathway exhibits properties of a negative feedback amplifier. Sci Signal 2010; 3: ra90.
Birtwistle MR, Kolch W . Biology using engineering tools: the negative feedback amplifier. Cell Cycle 2011; 10: 2069–2076.
Hanahan D, Weinberg RA . The hallmarks of cancer. Cell 2000; 100: 57–70.
Hanahan D, Weinberg RA . Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674.
Weinstein IB, Joe A . Oncogene addiction. Cancer Res 2008; 68: 3077–3080.
Ji Z, Flaherty KT, Tsao H . Targeting the RAS pathway in melanoma. Trends Mol Med 2012; 18: 27–35.
Joseph EW, Pratilas CA, Poulikakos PI, Tadi M, Wang W, Taylor BS et al. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc Natl Acad Sci USA 2010; 107: 14903–14908.
Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R et al. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther 2007; 6: 2209–2219.
Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol 2011; 29: 3085–9306.
Engelman JA, Settleman J . Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Curr Opin Genet Dev 2008; 18: 73–79.
Hammerman PS, Janne PA, Johnson BE . Resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res 2009; 15: 7502–7509.
Milojkovic D, Apperley J . Mechanisms of resistance to imatinib and second-generation tyrosine inhibitors in chronic myeloid leukemia. Clin Cancer Res 2009; 15: 7519–7527.
Ercan D, Zejnullahu K, Yonesaka K, Xiao Y, Capelletti M, Rogers A et al. Amplification of EGFR T790M causes resistance to an irreversible EGFR inhibitor. Oncogene 2010; 29: 2346–2356.
Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316: 1039–1043.
Scaltriti M, Eichhorn PJ, Cortés J, Prudkin L, Aura C, Jiménez J et al. Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. Proc Natl Acad Sci USA 2011; 108: 3761–3766.
Fedorenko IV, Paraiso KH, Smalley KS . Acquired and intrinsic BRAF inhibitor resistance in BRAF V600E mutant melanoma. Biochem Pharmacol 2011; 82: 201–209.
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417: 949–954.
Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP et al. BRAF mutations in hairy-cell leukemia. N Engl J Med 2011; 364: 2305–2315.
Xi L, Arons E, Navarro W, Calvo KR, Stetler-Stevenson M, Raffeld M et al. Both variant and IGHV4-34-expressing hairy cell leukemia lack the BRAF V600E mutation. Blood 2012; 119: 3330–3332.
Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010; 363: 809–819.
Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010; 467: 596–599.
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364: 2507–2516.
Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 2010; 140: 209–221.
Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N . RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010; 464: 427–430.
Röring M, Herr R, Fiala GJ, Heilmann K, Braun S, Eisenhardt AE et al. Distinct requirement for an intact dimer interface in wild type, V600E and kinase dead BRAF signaling. EMBO J 2012 31: 2629–2647.
Montagut C, Sharma SV, Shioda T, McDermott U, Ulman M, Ulkus LE et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res 2008; 68: 4853–4861.
Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010; 18: 683–695.
Balmanno K, Chell SD, Gillings AS, Hayat S, Cook SJ . Intrinsic resistance to the MEK1/2 inhibitor AZD6244 (ARRY-142886) is associated with weak ERK1/2 signalling and/or strong PI3K signalling in colorectal cancer cell lines. Int J Cancer 2009; 125: 2332–2341.
Meng J, Peng H, Dai B, Guo W, Wang L, Ji L et al. High level of AKT activity is associated with resistance to MEK inhibitor AZD6244 (ARRY-142886). Cancer Biol Ther 2009; 8: 2073–2080.
Halilovic E, She QB, Ye Q, Pagliarini R, Sellers WR, Solit DB et al. PIK3CA mutation uncouples tumor growth and cyclin D1 regulation from MEK/ERK and mutant KRAS signaling. Cancer Res 2010; 70: 6804–6814.
Xing F, Persaud Y, Pratilas CA, Taylor BS, Janakiraman M, She QB et al. Concurrent loss of the PTEN and RB1 tumor suppressors attenuates RAF dependence in melanomas harboring (V600E)BRAF. Oncogene 2012; 31: 446–457.
Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010; 468: 968–972.
Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010; 468: 973–977.
Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011; 480: 387–390.
Shi H, Moriceau G, Kong X, Lee M-K, Lee H, Koya RC et al. Melanoma whole-exome sequencing identifies V600EB-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun 2012; 3: 724.
Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci USA 2009; 106: 20411–20416.
Whittaker S, Kirk R, Hayward R, Zambon A, Viros A, Cantarino N et al. Gatekeeper mutations mediate resistance to BRAF-targeted therapies. Sci Transl Med 2010; 2: 35ra41.
Delaney AM, Printen JA, Chen H, Fauman EB, Dudley DT . Identification of a novel mitogen-activated protein kinase kinase activation domain recognized by the inhibitor PD 184352. Mol Cell Biol 2002; 22: 7593–7602.
Ohren JF, Chen H, Pavlovsky A, Whitehead C, Zhang E, Kuffa P et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Mol Biol 2004; 11: 1192–1197.
Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature 2006; 439: 358–362.
Carragher LA, Snell KR, Giblett SM, Aldridge VS, Patel B, Cook SJ et al. V600EBraf induces gastrointestinal crypt senescence and promotes tumour progression through enhanced CpG methylation of p16INK4a. EMBO Mol Med 2010; 2: 458–471.
Little AS, Balmanno K, Sale MJ, Smith PD, Cook SJ . Tumour cell responses to MEK1/2 inhibitors: acquired resistance and pathway remodelling. Biochem Soc Trans 2012; 40: 73–78.
Wang H, Daouti S, Li WH, Wen Y, Rizzo C, Higgins B et al. Identification of the MEK1(F129L) activating mutation as a potential mechanism of acquired resistance to MEK inhibition in human cancers carrying the B-RafV600E mutation. Cancer Res 2011; 71: 5535–5545.
Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL . Conger BA, Cruz MS et al. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science 2006; 311: 1287–1290.
Nava C, Hanna N, Michot C, Pereira S, Pouvreau N, Niihori T et al. Cardio-facio-cutaneous and Noonan syndromes due to mutations in the RAS/MAPK signalling pathway: genotype-phenotype relationships and overlap with Costello syndrome. J Med Genet 2007; 44: 763–771.
Rodriguez-Viciana P, Rauen KA . Biochemical characterization of novel germline BRAF and MEK mutations in cardio-facio-cutaneous syndrome. Methods Enzymol 2008; 438: 277–289.
Hatzivassiliou G, Liu B, O'Brien C, Spoerke JM, Hoeflich KP, Peter M et al. ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol Cancer Ther 2012, ( doi:10.1158/1535-7163).
Corcoran RB, Dias-Santagata D, Bergethon K, Iafrate AJ, Settleman J, Engelman JA . BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Sci Signal 2010; 3: ra84.
Little AS, Balmanno K, Sale MJ, Newman S, Dry JR, Hampson M et al. Amplification of the driving oncogene, KRAS or BRAF, underpins acquired resistance to MEK1/2 inhibitors in colorectal cancer cells. Sci Signal 2011; 4: ra17.
Soh J, Okumura N, Lockwood WW, Yamamoto H, Shigematsu H, Zhang W et al. Oncogene mutations, copy number gains and mutant allele specific imbalance (MASI) frequently occur together in tumor cells. PLoS One 2009; 4: e7464.
Modrek B, Ge L, Pandita A, Lin E, Mohan S, Yue P et al. Oncogenic activating mutations are associated with local copy gain. Mol Cancer Res 2009; 7: 1244–1252.
Witzel II, Koh LF, Perkins ND . Regulation of cyclin D1 gene expression. Biochem Soc Trans 2010; 38: 217–222.
Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB et al. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci USA 2009; 106: 4519–4524.
Woods D, Parry D, Cherwinski H, Bosch E, Lees E, McMahon M . Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol Cell Biol 1997; 17: 5598–5611.
Zhu J, Woods D, McMahon M, Bishop JM . Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev 1998; 12: 2997–3007.
Elgendy M, Sheridan C, Brumatti G, Martin SJ . Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol Cell 2011; 42: 23–35.
Shi H, Kong X, Ribas A, Lo RS . Combinatorial treatments that overcome PDGFRβ-driven resistance of melanoma cells to V600EB-RAF inhibition. Cancer Res 2011; 71: 5067–5074.
Smalley KS, Lioni M, Dalla Palma M, Xiao M, Desai B, Egyhazi S et al. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E-mutated melanomas. Mol Cancer Ther 2008; 7: 2876–2883.
Tolcher AW, Baird RD, Patnaik A, Moreno Garcia V, Papadopoulos KP, Garrett CR et al. A phase I dose-escalation study of oral MK-2206 (allosteric AKT inhibitor) with oral selumetinib (AZD6244; MEK inhibitor) in patients with advanced or metastatic solid tumors. J Clin Oncol 2011; 29 (Suppl)): abstr 3004.
Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010; 141: 69–80.
Paraiso KH, Haarberg HE, Wood E, Vito W, Rebecca VW, Chen YA et al. The heat shock protein-90 inhibitor XL888 overcomes BRAF inhibitor resistance mediated through diverse mechanisms. Clin Cancer Res 2012, ( doi:10.1158/1078-0432).
Prahallad A, Sun C, Huang S, Nicolantonio FD, Salazar R, Zecchin D et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012; 483: 100–103.
Acknowledgements
We apologise to colleagues whose work we have not been able to cite due to space limitations. Dr Simon Cook's laboratory was supported by the Babraham Institute, which receives strategic support from the Biotechnology and Biological Sciences Research Council. Work in Dr Cook's laboratory on mechanisms of resistance to ERK1/2 pathway inhibitors was funded by a collaborative research grant from AstraZeneca, which provided Dr Annette Little's salary and research consumables. Neither Dr Cook or Dr Little received any personal renumeration from AstraZeneca. This article is dedicated to the memory of Maureen Cook, a devoted, loving and much-loved mother whose life was blighted by dementia; she is sorely missed.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Dr Paul Smith is a paid employee of AstraZeneca. The remaining authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Little, A., Smith, P. & Cook, S. Mechanisms of acquired resistance to ERK1/2 pathway inhibitors. Oncogene 32, 1207–1215 (2013). https://doi.org/10.1038/onc.2012.160
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2012.160
Keywords
This article is cited by
-
The Agonist of Adenosine A1 Receptor Induced Desensitization of delta Opioid receptor-mediated Raf-1/MEK/ERK Signaling by Feedback Phosphorylation of Raf-1-Ser289/296/301
Neurochemical Research (2023)
-
RAF kinase dimerization: implications for drug discovery and clinical outcomes
Oncogene (2020)
-
Mitotic checkpoint kinase Mps1/TTK predicts prognosis of colon cancer patients and regulates tumor proliferation and differentiation via PKCα/ERK1/2 and PI3K/Akt pathway
Medical Oncology (2020)
-
The effects of mutant Ras proteins on the cell signalome
Cancer and Metastasis Reviews (2020)
-
Selective ERK1/2 agonists isolated from Melia azedarach with potent anti-leukemic activity
BMC Cancer (2019)
