
Abstract
Background:
Type 2 diabetes stems from obesity-associated insulin resistance, and in the genetically susceptible, concomitant pancreatic β-cell failure can occur, which further exacerbates hyperglycemia. Recent work by our group and others has shown that the natural polyphenol curcumin attenuates the development of insulin resistance and hyperglycemia in mouse models of hyperinsulinemic or compensated type 2 diabetes. Although several potential downstream molecular targets of curcumin exist, it is now recognized to be a direct inhibitor of proteasome activity. We now show that curcumin also prevents β-cell failure in a mouse model of uncompensated obesity-related insulin resistance (Leprdb/db on the Kaliss background).
Results:
In this instance, dietary supplementation with curcumin prevented hyperglycemia, increased insulin production and lean body mass, and prolonged lifespan. In addition, we show that short-term in vivo treatment with low dosages of two molecularly distinct proteasome inhibitors celastrol and epoxomicin reverse hyperglycemia in mice with β-cell failure by increasing insulin production and insulin sensitivity.
Conclusions:
These studies suggest that proteasome inhibitors may prove useful for patients with diabetes by improving both β-cell function and relieving insulin resistance.
Similar content being viewed by others

Introduction
Diabetes is a disease in which the body does not sufficiently produce and/or respond to insulin, a pancreatic endocrine hormone crucial for maintaining glucose homeostasis. Insulin deficiency leads to hyperglycemia, which, if uncontrolled, will acutely cause life-threatening ketoacidosis and in the long-term foster vascular disease leading to potentially devastating end-organ failure. There are nearly 21 million Americans, or 7% of the population, who have diabetes, an epidemic mainly attributable to the marked increase in the incidence of obesity in this country.1, 2, 3, 4
Obesity is associated with enhanced adipocyte death and infiltration of macrophages into white adipose tissue.5, 6 Many inflammation-provoking 'adipokines', such as PAI-1 (plasminogen activator-1) and MCP-1 (monocyte chemoattractant protein-1), and macrophage-specific genes, including tumor necrosis factor-α and interleukin-6, are significantly upregulated in the white adipose tissue of obese subjects.7, 8 Within liver tissue, obesity and inflammation are linked by activation of the nuclear factor-κB (NF-κB) transcription factor. Genetic studies in mice have shown that interruption of NF-κB activity in hepatocytes and macrophages attenuates the development of insulin resistance in the setting of obesity.9 Thus, the development and progression of diabetes in obese individuals stems, in part, from these proinflammatory alterations that limit the response to insulin.
In the early stages of insulin resistance, hyperglycemia is usually averted due to pancreatic islet cell hyperplasia and hyperinsulinemia.10 The duration of this hyperinsulinemic, normoglycemic phase depends on both the severity of the insulin resistance and individual genetic factors. In patients with prolonged or severe insulin resistance, however, the metabolic stress imposed upon their β-cells often initiates their apoptotic death, necessitating exogenous insulin administration.
For most patients with diabetes, exercise and dietary management will prove insufficient to maintain euglycemia, and roughly 80% at some point will require either oral hypoglycemic agents and/or insulin injections for satisfactory blood glucose control. These drugs can be expensive and can induce unpleasant side effects or harmful toxicities.11, 12 Given the staggering prevalence and expense of treating diabetes and its associated complications, it is imperative to investigate alternative and/or complementary treatments that would ideally be safe, effective, inexpensive and readily available.
The dried ground rhizome of the perennial herb turmeric (Curcuma longa) is both a popular dietary spice as well as an integral part of the ancient Hindu medicinal system called Ayurveda.13, 14 Curcumin (diferuloylmethane) comprises 2–8% of most turmeric preparations and is generally regarded as its most active component. Commercial grade curcumin comes as a 95% standardized extract and contains the curcuminoids curcumin (~80%), desmethoxycurcumin (~10–20%) and bisdesmethoxycurcumin (<5%). Studies in vitro as well as in preclinical animal models of disease have shown that curcumin inhibits carcinogenesis15, 16, 17, 18, 19, 20, 21, 22, 23, 24 and inflammation.13, 25, 26, 27, 28, 29 In addition, we have recently demonstrated that dietary curcumin treatment attenuates insulin resistance and hyperglycemia in high-fat diet fed and genetically obese leptin-deficient C57BL/6 Lepob/ob mice by decreasing inflammation in adipose tissue and liver.30
Multiple different mechanisms, including NF-κB inhibition,17, 31 peroxisome proliferator-activated receptor-γ agonism,32, 33 cyclooxygenase-2 pathway inhibition34, 35, 36, 37, 38 and inhibition of the c-Jun N-terminal kinase signaling pathway,39, 40 have been implicated in curcumin’s pleiotropic beneficial effects. Most recently, however, it has been established that curcumin is a direct inhibitor of the proteolytic core of the mammalian 26 S proteasome complex,41, 42, 43, 44 and evidence suggests that it is this ability that may, in fact, underlie its multiple downstream effects.31, 44, 45, 46, 47, 48, 49, 50, 51, 52
The proteasome works in concert with a tagging protein, ubiquitin, to create the ubiquitin-proteasome pathway, the major proteolytic pathway of eukaryotes. It controls the intracellular levels of a wide range of proteins, including NF-κB and others involved in the control of the cell cycle, transcriptional activation,53, 54, 55, 56, 57 apoptosis43, 58, 59 and cell signaling.59, 60, 61, 62 We postulated that proteasome inhibition may be contributing to the antidiabetogenic effects that we have shown curcumin to exert.
In this series of studies, we demonstrate that curcumin treatment enhances β-cell function and proliferation both in vitro and in vivo, and we implicate its inhibitory actions on the proteasome in its therapeutic activities. To model β-cell failure in the setting of insulin-resistant obesity, we used obese leptin receptor-deficient mice Leprdb/db on the Kaliss (Ks) background. Unlike obese mice on the C57BL/6 background, the Ks Leprdb/db mice are highly susceptible to β-cell failure in the setting of insulin resistance, and they develop early insulin depletion, severe hyperglycemia, ketoacidosis and early death. We found that feeding these animals a diet supplemented with curcumin markedly attenuated the development of hyperglycemia, increased circulating insulin levels, prevented β-cell loss and lengthened the lifespan.
In addition, short-term treatment of Ks Leprdb/db mice with two structurally distinct proteasome inhibitors, epoxomicin and celastrol, significantly reduced hyperglycemia by concomitantly increasing insulin sensitivity and increasing insulin production. Taken together, these data suggest that one mechanism by which dietary supplementation with curcumin treats diabetes is by inhibiting the proteasome, a mechanism heretofore not described to possess antidiabetogenic properties.
Materials and methods
Experimental animals
Dietary-induced obese C57BL/6J mice: After 2–3 months, inbred male C57BL/6J wild-type mice will become obese and moderately diabetic if fed a high-fat diet (35% fat by weight). C57BL/6J ob/ob mice possess a spontaneous knockout mutation of the leptin gene that produces hyperphagia, decreased metabolic rate, severe obesity and moderate diabetes, which is eventually compensated for by pancreatic β-cell hyperplasia and hyperinsulinemia. C57BL/Ks db/db mice possess a spontaneous knockout mutation of the leptin receptor, which generates a phenotype initially very similar to that of the ob/ob mice. However, the loss of leptin effect on the C57BL/Ks background is not compensated for β-cell hyperplasia and hyperinsulinemia. At a very young age these mice become severely obese, hyperglycemic, hyperphagic and polydipsic. As they mature, they start to lose weight, develop nephropathy and ultimately die around age 40 weeks from diabetic complications.
All mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). Mice were housed five mice per cage and maintained on a 12:12 light–dark cycle with ad libitum access to food and water. In the dietary curcumin experiments, the male Lepob/ob and Leprdb/db mice were received at age 8–10 weeks and then randomized to receive a standardized 4% fat by weight meal diet (D12450B-I; Research Diets, New Brunswick, NJ, USA) containing either a 3% by weight admixture of curcumin or no additive. Assuming that each mouse weighed on average 40 g and consumed 2 g of food per day, their daily curcumin intake would be equivalent to 60 mg per day or 1.5 g kg−1 per day. The wild-type mice were obtained at age 3–5 weeks and randomized to receive either a standardized 4% fat by weight diet or, in an attempt to induce obesity, a high-fat diet containing 35% fat by weight (D12492-I; Research Diets). At the age of 20 weeks, these wild-type mice were further randomized with regard to the addition of a 3% by weight admixture of curcumin or no additive to their predesignated diet. Curcumin admixture dosage was based on our previous work with diabetic mice.30 Owing to obvious differences in food being consumed, blind analysis of mice experimental variables was not feasible.
The curcumin used was Curcumin C3 Complex (Sabinsa Corporation, Newark, NJ, USA), which is a 95% standardized curcumin extract. All mice remained on their assigned diet until being killed by CO2 asphyxiation at the age of ~28 weeks. All protocols were approved by the Columbia University Institutional Animal Care and Use Committee.
Body composition analysis
The fat and lean tissue masses were calculated from living, non-anesthetized mice using a Bruker nuclear magnetic resonance machine (Bruker BioSpin Corp., Billerica, MA, USA). Pelleted food was weighed daily to assess total grams of food consumed per cage per day and then divided by 5 to estimate average daily food consumed per mouse.
Glucose/HbA1c testing
Tail tip blood was collected from live mice to ascertain glucose and hemoglobin A1c (HbA1c) levels. Approximately 5 μl was used to assess blood glucose levels using a Freestyle Flash glucometer (Abbott, Abbott Park, IL, USA) and 25 μl was used to assess HbA1c using a Bayer DCA 2000+ analyzer (Siemens, Tarrytown, NY, USA).
Insulin tolerance test
After fasting mice for 6 h to stabilize dietary glucose and insulin levels, ~25 μl tail tip blood was collected to ascertain fasting glucose and insulin levels. Mice then received 1.5 U kg−1 of regular insulin (Lilly, Indianapolis, IN, USA) by intraperitoneal injection. Sample tail tip blood glucose levels were assessed by glucometer at 15, 30, 45, 60 and 120 min.
Histology
Pancreata were removed from mice at the end of treatment period and were fixed overnight at room temperature in zinc-formalin fixative ('Z-Fix'; Anatech Ltd, Battle Creek, MI, USA) and then embedded in paraffin. Tissue was sliced into 5-μm sections cut at 50-μm intervals and then mounted on charged glass slides, deparaffinized in xylene and then stained. Slides were stained with polyclonal antibodies specific to insulin (sc-9168; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and the proliferative marker Ki-67 (sc-7846; Santa Cruz Biotechnology).
Short-term effect of proteasome inhibitors on murine islet cell transcription
Approximately 12-week-old male C57BL/Ks Leprdb/db mice were given a single 100 μl intraperitoneal injection of vehicle (10% dimethyl sulfoxide, 70% 3:1 cremophor/ethanol and 20% phosphate-buffered saline), or vehicle with added curcumin (3 mg kg−1), celastrol (3 mg kg−1) (Calbiochem, Darmstadt, Germany) or epoxomicin (0.1 mg kg−1) (Calbiochem). Dosages of celastrol and epoxomicin were based on previous work in mice using these compounds.63, 64 After 24 h, they were killed by cervical dislocation, and collagenase (1.0 mg ml−1 in Hanks' balanced salt solution) was then injected into the pancreatic duct (5.0 ml per mouse). The distended pancreata were then incubated in a shaking water bath at 37 °C in 1.0 mg ml−1 collagenase in Hanks' balanced salt solution for 10 min, and the islets were recovered using Histopaque gradient solutions (3 ml of 1.119 g l−1, 3 ml of 1.083 g l−1 and 3 ml of 1.077 g l−1). After centrifugation for 20 min at 2500 g, islets were removed from the top layer and washed two times with RPMI-1640. N=5 per group.
Quantitative real-time PCR
Total RNA was extracted from freshly removed pancreatic islet cells, 100 mg of flash-frozen mouse adipose or liver tissue or from freshly scraped cultured Ins-1 cells, using a commercially available acid-phenol reagent (TRIzol; Invitrogen, Carlsbad, CA, USA). First-strand cDNA was synthesized using SuperScript III reverse transcriptase and random hexamer primers as described in the manufacturer’s protocol (Invitrogen). Samples of cDNA were diluted 1:25 in nuclease-free water (Qiagen Inc., Hilden, Germany). Samples from each cDNA pool were diluted 1:10, 1:30, 1:90 and 1:270 to create a standard curve for calculation of relative gene expression levels. PCR amplification mixtures (20 μl) contained 10 μl of 2x PCR SYBR Green I QuantiTect Master Mix (Qiagen Inc.), 0.4 μl of a mixture of 25 μm reverse and forward primers and 11.6 μl diluted cDNA template. Real-time quantitative PCR was carried out using the DNA Engine Opticon 2 Instrument (MJ Research Inc., Waltham, MA, USA) with the following cycling parameters: polymerase activation for 15 min at 95 °C and amplification for 40 cycles of 15 s at 94 °C, 10 s at 58 °C and 10 s at 72 °C. After amplification, melting-curve analysis confirmed the presence of a single amplicon in each instance.
For expression analysis, we used prevalidated real-time PCR primers (QuantiTect; Qiagen Inc.) for the genes analyzed including the 'housekeeping' control Rps3.
For each cDNA and standard curve sample, quantitative PCR reactions were performed to assay the expression of each internal control gene. To calculate the normalized relative expression levels of each gene assayed in each sample, we divided the relative gene expression value for that sample by the geometric mean of the relative expression values of the control genes. Separate analyses in which relative expression values were normalized with the relative expression values of each control gene yielded similar results.
Cell viability assays
After treating Ins-1 cells with vehicle, curcumin, celastrol or epoxomicin, as described, 20 μl of a resazurin-containing reagent (CellTiter-Blue; Promega, Madison, WI, USA) was then added directly to each well. After a further incubation at 37 °C for 4 h, the fluorescence per well (560Ex/590Em) produced by the conversion of resazurin to resorufin, which is directly proportional to viable cell number, was quantified using a fluorescent plate reader.
In vitro studies
INS-1 cell studies: INS-1 (832/13) β-cells (20) were plated at a density of ~2000 cells per well in 96-well trays in 100 μl of RPMI-1640 containing 11 mmol l−1 glucose, 10% (vol vol−1) fetal calf serum, 4 mmol l−1 l-glutamine, 100 IU ml−1 penicillin, 100 μg ml−1 streptomycin, 10 mmol l−1 HEPES, 1 mmol l−1 sodium pyruvate and 0.05 mmol l−1 β-mercaptoethanol. During the experiments, when achieving ~70% confluency, the Ins-1 cells were washed and given fresh media containing vehicle or the indicated concentrations of curcumin, celastrol or epoxomicin for 12 or 24 h. N=24 wells per group.
3T3-L1 cell studies: 3T3-L1 preadipocytes at 75% confluency were harvested from 25 mm2 flasks by trypsinization and then seeded in complete media (DMEM-high glucose, 10% fetal bovine serum, 1% penicillin) on 6-well plates at ~8000 cells per well. The cells were grown to confluency and then kept in this state for another 2 days to arrest cell division. They were then treated with adipocyte differentiation media (complete media containing dexamethasone 1 μm, 3-isobutyl-1-methylxanthine 0.5 mm and insulin 10 μg ml−1). After 4 days in this media, the media were carefully aspirated and the cells were then treated with adipocyte maturation media (complete media and insulin 10 μg ml−1 insulin), which was changed thereafter every 2 days for 10 days. At this point, the differentiated 3T3-L1 cells were subjected to treatment with adipocyte maturation media containing vehicle or varying concentrations of curcumin, celastrol or epoxomicin for 24 h. The cells were then harvested for RNA analyses.
All cell lines were kindly provided by the laboratory of Rudolph Leibel, MD, Columbia University Medical Center, New York, NY, USA.
Hormone assays
After fasting for 6 h, mice were weighed and then had their blood glucose measured by glucometer (Glucometer Elite, Elkhart, IN, USA) using ~5 μl of tail tip blood. They were then killed by cervical dislocation. Their blood was then obtained by cardiac puncture, allowed to clot on ice for 3 h and then centrifuged at 10 000 g for 10 min. The sera were then transferred to clean vials for storage at −80 °C until the day of assay. Mouse serum insulin, leptin, MCP-1, adiponectin, tPAI-1 (tissue PAI-1) were quantified by ELISA (Linco Research Inc., St Charles, MO, USA). All inter- and intra-assay coefficients of variation were <10%.
Statistics and definitions
Two-tailed Student’s t-tests were used to compare serum analytes between C57BL/6J lean and obese experimental groups. Sample sizes of five to six mice were used in the treatment groups to generate means that were compared by two-tailed t-tests. To acquire at least 75% power, α was set at 0.05. A treatment effect of 50% difference in population means and a standard deviation of 30% was presumed. P<0.05 was considered statistically significant. All data were examined using the Sigmastat 2.0 software (Jandel Scientific, San Rafael, CA, USA).
Results
Effects of curcumin on glucose homeostasis in obese diabetic mice
To investigate the effects of curcumin in a mouse model of severe β-cell failure stemming from obesity and insulin resistance, we randomized 10-week-old Leprdb/db male Ks mice to a 4% fat by weight diet (D12451; Research Diets, New Brunswick, NJ, USA) that was or was not supplemented with a 3% by weight 95% pure curcumin admixture (Sabinsa Corporation, Piscataway, NJ, USA). As a frame of reference, to assess the effect of dietary curcumin on normoglycemic mice with varying degrees of insulin sensitivity, we also subjected several other inbred mouse strains to dietary curcumin or not, namely wild-type male C57BL/6 J, wild-type male DBA/2 J, male Ks Leprdb/+ heterozygotes and male C57BL/6 J Lepob/ob homozygotes (Figure 1).

Most mice groups, lean and obese, consumed more food when curcumin (3%) was added to their diet. A notable exception was the C57BL/Ks Leprdb/db mice, which actually consumed less food, likely due to an improvement in their glucose utilization. N=5 per group; *P<0.05 by two-tailed t-test.
Similar to Lepob/ob mice on a C57BL/6 J (B6) background, the Leprdb/db male Ks mice display hyperphagia secondary to a lack of leptin signaling and develop early-onset obesity.65 The addition of curcumin to the diet was associated with an increase in the grams of food consumed in the wild-type B6 mice as well as in the Ks Leprdb/+ heterozygotes (Figure 1). Interestingly, the total amount of food consumed by the Ks Leprdb/db homozygotes was actually decreased by the addition of curcumin to their diet, an effect presumably due to their improved glucose utilization rather than any curcumin flavor aversion given the fact that the normoglycemic Ks Leprdb/+ heterozygotes behaved in an opposite manner (Figure 1). Mice with absolute leptin deficiency on the Ks background display early-onset insulin resistance with progressive β-cell failure that leads to severe insulin deficiency, wasting and death from ketoacidosis.65 Owing to genetic background differences, the male Lepob/ob mice on the C57BL6 background are able to rapidly compensate for their early diabetes through hyperinsulinemia66 stemming from pancreatic β-cell hyperplasia. It is not surprising therefore to see that the C57BL/6J Lepob/ob homozygotes showed no significant alteration in their food consumption (Figure 1).
As early as 2 weeks following initiation of dietary curcumin, we noted a striking decline in the random glucose values of Leprdb/db Ks mice in the curcumin-treated group compared with the controls, and this difference persisted over the 2.5-month treatment period (Figure 2). Accordingly, we also observed markedly decreased HbA1c percentages among all obese mice in the curcumin-treated group compared with controls (Figure 3). The most marked improvement, however, occurred in the Leprdb/db Ks mice in which the curcumin treatment was associated with a >50% reduction in the HbA1c level (Figure 3). Curcumin had no effect on glucose levels or HbA1c percentages in lean mice and therefore does not appear to cause hypoglycemia. Unlike the Lepob/ob and dietary-induced obese mice, which actually lost weight and fat mass, as determined by nuclear magnetic resonance on curcumin treatment, the improvement in glucose metabolism mediated by curcumin in Leprdb/db Ks mice allowed them to avoid the late-onset cachexia associated with diabetic ketosis and they weighed significantly more at the end of the treatment (Figure 4) compared with their control group, despite displaying decreased food intake (Figure 1). The increased body weight of the curcumin-fed Leprdb/db Ks mice was attributable to an increase in their lean body mass as determined by nuclear magnetic resonance analysis (Figure 4). The salutary effects of curcumin on their glucose metabolism were sufficient to significantly extend the lifespan of curcumin-fed mice Leprdb/db Ks compared with their controls (Figure 5).

Dietary curcumin (3%) confers significant protection against hyperglycemia in male C57BL/Ks Leprdb/db mice. N=5 per group; *P<0.05 by two-tailed t-test.

Dietary curcumin decreases HbA1c percentage in all mouse models of diabetes tested. Non-diabetic mice were not affected. N=6 per group. *P<0.05 by two-tailed t-test.

Effect of dietary curcumin on body composition. Nuclear magnetic resonance (NMR) spectroscopy revealed that dietary curcumin was associated with significantly diminished body weight and body fat percentage in dietary-induced obese (DIO) and C57BL/6 J Lepob/ob mouse groups, while it was associated with an increase in body weight and lean mass in the C57BL/Ks Leprdb/db mice, likely by allowing them avoid the cachectic effects of poor glucose utilization and terminal ketoacidosis.

Dietary curcumin significantly improves survival in severely diabetic male C57BL/Ks Leprdb/db mice. Beginning at 30 weeks of age, 40 C57BL/Ks Lepdb/db were randomized to either continue on their current diet or receive one supplemented by 3% curcumin. After 1 month of treatment, there was a statistically significant difference in the lifespans of the two groups demonstrating a beneficial effect of curcumin.
Effects of curcumin on β-cell function in obese diabetic mice
To investigate the mechanism by which curcumin prevents hyperglycemia in Leprdb/db Ks mice, we measured fasting circulating insulin levels in lean Ks and obese Leprdb/db Ks mice on control and curcumin-supplemented diets. We also examined β-cell proliferation and β-cell mass using immunohistochemical staining for insulin and the proliferation marker Ki-67 in pancreatic sections from these mice.
Obese Leprdb/db Ks mice on the control diet had similar circulating insulin levels as lean animals despite severe obesity and hyperglycemia (Figure 6)—a manifestation of severe β-cell failure. Accordingly, pancreatic sections of obese Ks Leprdb/db mice on a regular diet showed no evidence of islet hypertrophy or β-cell proliferation compared with those of lean mice (Supplementary Figures 1d–f). In contrast, Leprdb/db Ks mice on a curcumin-supplemented diet were able to generate a hyperinsulinemic response similar to that of the Lepob/ob B6 mice and had significantly increased circulating insulin levels compared with lean animals and their control-fed Leprdb/db Ks mice (Figure 6). As expected, the obese Ks Leprdb/db mice had the highest serum leptin and PAI-1 concentrations of all mice scrutinized, although curcumin did not significantly affect these levels (Figure 6).

Dietary curcumin significantly increased serum insulin levels in male C57BL/Ks Leprdb/db mice, while it was associated with a decrease in serum leptin levels in male C57BL/6J Lepob/ob mice. N=5 per group. *P<0.05 by two-tailed t-test.
Consistent with their elevated serum insulin levels, the pancreatic sections of Ks Leprdb/db mice fed a curcumin-supplemented diet contained markedly enlarged islets with numerous Ki-67-positive β-cells (Supplementary Figures 1g–i) similar to those observed in obese Lepob/ob B6 animals with compensatory hyperinsulinemia (Supplementary Figures 1a–c). This suggests that curcumin may foster islet hyperplasia by increasing β-cell proliferation.
Effects of proteasome inhibitors on glucose homeostasis and β-cell function in obese diabetic mice
Several molecular targets of curcumin have been described including NF-κB, as well as key regulators of the cell cycle and apoptosis. However, direct interactions between curcumin and these molecules have not been described. Recent work has demonstrated that curcumin binds to and directly inhibits the chymotrypsin-like activity of the 20S proteasome, the proteolytic core of the mammalian proteasome complex. We reasoned that if the therapeutic actions of curcumin were mediated to some degree by proteasome inhibition, then structurally distinct proteasome inhibitors would have similar therapeutic activities as curcumin. We selected two compounds for evaluation, celastrol and epoxomicin, both of which are potent and selective direct proteasome inhibitors with activities, similar to curcumin, that have been demonstrated both in vivo and in cell-free systems.67, 68 Owing to limitations imposed by the stability and cost of these compounds, they could not be administered over long periods of time as food admixtures.
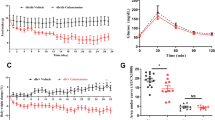
We treated obese and hyperglycemic Ks Leprdb/db mice with one dose of intraperitoneal celastrol (3 mg kg−1), epoxomicin (0.1 mg kg−1) or vehicle, and followed blood glucose values for 72 h postinjection. During this period, vehicle-treated mice were food entrained to the celastrol- and epoxomicin-treated mice to ensure that any changes in food intake induced by drug treatment would not influence blood glucose values. Between 12 and 40 postinjection, blood glucose levels in epoxomicin- (Supplementary Figure 2) and celastrol- (Supplementary Figure 3) treated mice remained significantly lower compared with those in vehicle-treated controls. Increased serum insulin levels were observed in celastrol- and epoxomicin-treated mice at time points corresponding to the peak hypoglycemic effects of these compounds (Supplementary Figure 4). In contrast, serum insulin values did not change significantly during the treatment period in vehicle-injected mice (Supplementary Figure 4).
Effects of proteasome inhibitors on systemic insulin sensitivity
We have previously shown that long-term treatment with curcumin improves insulin sensitivity in obese mice, in part, by increasing adiponectin expression in adipose tissue and reducing expression of inflammatory mediators in adipose tissue. To determine if treatment with other proteasome inhibitors also increases insulin sensitivity in obese mice, we performed insulin tolerance tests on Ks Leprdb/db mice 24 h after a single dose of celastrol (3 mg kg−1, intraperitoneally) or vehicle. Following an intraperitoneal bolus of insulin, celastrol-treated mice exhibited a significantly greater hypoglycemic response to insulin compared with vehicle-treated mice. Three days of daily celastrol intraperitoneal injections resulted in significantly decreased adipose tissue expression of Ccl2 and significantly increased expression of adiponectin in the white adipose tissue of male Ks Leprdb/db mice (Supplementary Figure 5).
Effects of proteasome inhibitors on proliferative gene transcription
We used quantitative real-time PCR to identify transcriptional changes in islets that may contribute to the improved β-cell function induced by proteasome inhibition. RNA was extracted from islets purified from whole pancreas by collagenase digestion followed by gradient density centrifugation. Expression of Islet neogenesis-associated protein, which has been shown to directly increase β-cell mass, was significantly upregulated in islets from celastrol- and epoxomicin-treated mice compared with controls. Expression of the phosphatase and tensin homolog, a negative regulator of phosphoinositide kinase signaling, β-cell proliferation and survival (reference needed), and FOXO3a, a protein known to mitigate that compensatory adaptation of β-cell mass,69 was significantly downregulated in islets from celastrol- and epoxomicin-treated mice compared with controls (Supplementary Figure 6). Taken together, these gene expression changes may contribute to the beneficial effects of proteasome inhibitors on β-cell function.70, 71
Effect of proteasome inhibitors on proliferation and insulin secretion in the β-cell line Ins-1
To investigate the ability of proteasome inhibition to directly improve β-cell function, we performed experiments using the rat insulinoma β-cell line Ins-1. We determined that the number of viable Ins-1 cells (CellTiter-Blue Cell Viability Assay; Promega) after 24 h in culture with varying concentrations of proteasome inhibitors was increased, except at the highest concentrations of celastrol and epoxomicin, which proved cytotoxic (Supplementary Figure 7a). When Ins-1 cells were cultured overnight in serum-free RPMI media containing varying concentrations of proteasome inhibitors, insulin secretion was significantly increased by proteasome inhibition, except at the highest concentrations of epoxomicin, which again proved cytotoxic (Supplementary Figure 7b).
Effect of proteasome inhibitors on transcription of inflammatory and endoplasmic reticulum stress response proteins the adipocyte cell line 3T3-L1
Preadipocyte cells from the cell line 3T3-L1 were differentiated in culture until they achieved a state phenotypically compatible with mature adipocytes. They were then incubated overnight in varying concentrations of epoxomicin, curcumin or celastrol. Significant increases in the expression mRNA encoding the heat-shock proteins 70 and 90 were noted (Supplementary Figures 8a and b). Curcumin and epoxomicin were also noted to increase expression of the histone deacetylase activators sirt1 and sirt2.
Discussion
The proteasome is thought to mediate the turnover of ~80% of cellular proteins. However, drugs that inhibit its core proteolytic activity have proven to be of clinical benefit and to have acceptable safety profiles. For example, the tripeptide proteasome inhibitor bortezomib is now routinely used in humans for relapsed and refractory multiple myeloma. Preclinical studies have shown promising antitumor activity and toxicity profiles of proteasome inhibitors in other animal models of human cancer.
Several mechanisms are postulated to underlie bortezomib’s efficacy, but perhaps the most important is its ability to block NF-κB activation in a dose- and time-dependent manner by inhibiting the degradation of its binding partner and negative regulator, IκBα.72 Enhanced levels of IκBα lead to increased sequestration of the proinflammatory transcription factor NF-κB outside the nucleus. As a result, several NF-κB-dependent genes that foster carcinogenesis, angiogenesis, metastasis and severe inflammation are turned off, and NF-κB-dependent genes that foster inflammation are downregulated.
Obesity is associated with overproduction of multiple proinflammatory mediators that contribute to insulin resistance by activation of NF-κB.73, 74 Genetic or pharmacologic inhibition of this pathway with aspirin,75 adiponectin,76 thiazolidinediones77 or statins78 has been shown to ameliorate obesity-induced insulin resistance.9, 79, 80, 81 The hyperglycemia of diabetes, if unchecked, fosters further inflammation via oxidative damage. This phenomenon, referred to as 'glucose toxicity',82 is believed to be responsible for the progressive β-cell failure noted in poorly controlled type 2 diabetes.
In a previous report, we demonstrated that curcumin, a non-toxic compound purified from turmeric, prevented the development of obesity-induced insulin resistance and glucose intolerance primarily by attenuating hepatic and adipose tissue inflammation. The anti-inflammatory effect of curcumin in other systems and disease models is well established; however, its direct molecular target had not been identified until a recent report provided compelling evidence using both in vivo and cell-free systems that curcumin directly inhibits proteasome activity.
Here we have tested the hypothesis that curcumin’s antidiabetes activity stems from proteasome inhibition by comparing its efficacy at treating type 2 diabetes in mice with that of two other structurally distinct proteasome inhibitors. We found that all three proteasome inhibitors ameliorate insulin resistance and β-cell failure.
Previously, we found that prolonged treatment with oral curcumin prevented insulin resistance and inflammation in obese mice that were not genetically susceptible to β-cell failure (C57BL/6 background). Here we tested the effects of curcumin therapy on obese mice with a strong genetic susceptibility to β-cell failure (Ks background). Our studies reveal that administration of proteasome inhibitors to these mice corrects their hyperglycemia, and appears to do so by increasing insulin sensitivity and by preserving β-cell function. In this system, the effects of curcumin on β-cell function were quite pronounced. After long-term dietary curcumin administration, the curcumin-treated male Leprdb/db Ks mice had similar levels of islet hyperplasia and circulating insulin as male Lepob/ob C57BL/6 mice, indicating that curcumin almost completely overcame their genetic susceptibility to β-cell failure.
We also evaluated the effects of two structurally distinct proteasome inhibitors, celastrol and epoxomicin, on glucose levels, insulin sensitivity and β-cell function in obese diabetic mice. A single injection of these compounds raised circulating insulin levels within 24 h and substantially reduced hyperglycemia in these mice for nearly 48 h. Expression studies performed on the insulin-producing pancreatic islets of these mice showed that proteasome inhibition upregulated the expression of islet neogenesis-associated protein—a factor known to increase β-cell mass—and downregulated the expression of Pten—a negative regulator of β-cell growth and insulin secretion. In addition, short-term celastrol treatment improved systemic insulin responsiveness and increased the expression of adiponectin in adipose tissue.
We infer from these data that the common therapeutic activity of the structurally distinct compounds celastrol, curcumin and epoxomicin in obese and diabetic mice is related to their shared molecular target—the proteasome. It is also possible, however, that the antidiabetes activity of these three compounds is related to common off-target effects on molecules other than the proteasome. Further studies to evaluate the antidiabetes activity of a larger spectrum of specific proteasome inhibitors are needed to address this question.
Clinical trials in human patients using curcumin and celastrol to treat diseases such as arthritis and amyotrophic lateral sclerosis are currently underway. Although toxicities associated with celastrol are less well defined, curcumin has been shown to be non-toxic at very high dosages in humans.83 The data presented here suggest that curcumin, celastrol or other proteasome inhibitors warrant further evaluation for the treatment of type 2 diabetes in humans.
References
Engelgau MM, Geiss LS, Saaddine JB, Boyle JP, Benjamin SM, Gregg EW et al. The evolving diabetes burden in the United States. Ann Intern Med 2004; 140: 945–950.
Pi-Sunyer FX . The obesity epidemic: pathophysiology and consequences of obesity. Obes Res 2002; 10 (Suppl 2): 97S–104S.
Wyatt SB, Winters KP, Dubbert PM . Overweight and obesity: prevalence, consequences, and causes of a growing public health problem. Am J Med Sci 2006; 331: 166–174.
DeStefano F, Ford ES, Newman J, Stevenson JM, Wetterhall SF, Anda RF et al. Risk factors for coronary heart disease mortality among persons with diabetes. Ann Epidemiol 1993; 3: 27–34.
Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res 2005; 46: 2347–2355.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr . Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003; 112: 1796–1808.
Matsuzawa Y . The metabolic syndrome and adipocytokines. FEBS Lett 2006; 580: 2917–2921.
Guzik TJ, Mangalat D, Korbut R . Adipocytokines—novel link between inflammation and vascular function? J Physiol Pharmacol 2006; 57: 505–528.
Eldor R, Yeffet A, Baum K, Doviner V, Amar D, Ben-Neriah Y et al. Conditional and specific NF-kappaB blockade protects pancreatic beta cells from diabetogenic agents. Proc Natl Acad Sci USA 2006; 103: 5072–5077.
Ahren B . Type 2 diabetes, insulin secretion and beta-cell mass. Curr Mol Med 2005; 5: 275–286.
Chitturi S, George J . Hepatotoxicity of commonly used drugs: nonsteroidal anti-inflammatory drugs, antihypertensives, antidiabetic agents, anticonvulsants, lipid-lowering agents, psychotropic drugs. Semin Liver Dis 2002; 22: 169–183.
Spiller HA, Sawyer TS . Toxicology of oral antidiabetic medications. Am J Health Syst Pharm 2006; 63: 929–938.
Chainani-Wu N . Safety and anti-inflammatory activity of curcumin: a component of tumeric (Curcuma longa. J Altern Complement Med 2003; 9: 161–168.
Lodha R, Bagga A . Traditional Indian systems of medicine. Ann Acad Med Singapore 2000; 29: 37–41.
Aggarwal S, Ichikawa H, Takada Y, Sandur SK, Shishodia S, Aggarwal BB . Curcumin (diferuloylmethane) down-regulates expression of cell proliferation and antiapoptotic and metastatic gene products through suppression of IkappaBalpha kinase and Akt activation. Mol Pharmacol 2006; 69: 195–206.
Banerji A, Chakrabarti J, Mitra A, Chatterjee A . Effect of curcumin on gelatinase A (MMP-2) activity in B16F10 melanoma cells. Cancer Lett 2004; 211: 235–242.
Belakavadi M, Salimath BP . Mechanism of inhibition of ascites tumor growth in mice by curcumin is mediated by NF-kB and caspase activated DNase. Mol Cell Biochem 2005; 273: 57–67.
Busquets S, Carbo N, Almendro V, Quiles MT, Lopez-Soriano FJ, Argiles JM . Curcumin, a natural product present in turmeric, decreases tumor growth but does not behave as an anticachectic compound in a rat model. Cancer Lett 2001; 167: 33–38.
Campbell FC, Collett GP . Chemopreventive properties of curcumin. Fut Oncol 2005; 1: 405–414.
Chakraborty S, Ghosh U, Bhattacharyya NP, Bhattacharya RK, Roy M . Inhibition of telomerase activity and induction of apoptosis by curcumin in K-562 cells. Mutat Res 2006; 569: 81–90.
Chen A, Xu J, Johnson AC . Curcumin inhibits human colon cancer cell growth by suppressing gene expression of epidermal growth factor receptor through reducing the activity of the transcription factor Egr-1. Oncogene 2006; 25: 278–287.
Cheng AL, Hsu CH, Lin JK, Hsu MM, Ho YF, Shen TS et al. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res 2001; 21: 2895–2900.
Choudhuri T, Pal S, Agwarwal ML, Das T, Sa G . Curcumin induces apoptosis in human breast cancer cells through p53-dependent Bax induction. FEBS Lett 2002; 512: 334–340.
Chuang SE, Kuo ML, Hsu CH, Chen CR, Lin JK, Lai GM et al. Curcumin-containing diet inhibits diethylnitrosamine-induced murine hepatocarcinogenesis. Carcinogenesis 2000; 21: 331–335.
Banerjee M, Tripathi LM, Srivastava VM, Puri A, Shukla R . Modulation of inflammatory mediators by ibuprofen and curcumin treatment during chronic inflammation in rat. Immunopharmacol Immunotoxicol 2003; 25: 213–224.
Brouet I, Ohshima H . Curcumin, an anti-tumour promoter and anti-inflammatory agent, inhibits induction of nitric oxide synthase in activated macrophages. Biochem Biophys Res Commun 1995; 206: 533–540.
Chan MM, Ho CT, Huang HI . Effects of three dietary phytochemicals from tea, rosemary and turmeric on inflammation-induced nitrite production. Cancer Lett 1995; 96: 23–29.
Cole GM, Morihara T, Lim GP, Yang F, Begum A, Frautschy SA . NSAID and antioxidant prevention of Alzheimer's disease: lessons from in vitro and animal models. Ann NY Acad Sci 2004; 1035: 68–84.
Giri RK, Rajagopal V, Kalra VK . Curcumin, the active constituent of turmeric, inhibits amyloid peptide-induced cytochemokine gene expression and CCR5-mediated chemotaxis of THP-1 monocytes by modulating early growth response-1 transcription factor. J Neurochem 2004; 91: 1199–1210.
Weisberg SP, Leibel R, Tortoriello DV . Dietary curcumin significantly improves obesity-associated inflammation and diabetes in mouse models of diabesity. Endocrinology 2008; 149: 3549–3558.
Brennan P, O'Neill LA . Inhibition of nuclear factor kappaB by direct modification in whole cells—mechanism of action of nordihydroguaiaritic acid, curcumin and thiol modifiers. Biochem Pharmacol 1998; 55: 965–973.
Chen A, Xu J . Activation of PPAR{gamma} by curcumin inhibits Moser cell growth and mediates suppression of gene expression of cyclin D1 and EGFR. Am J Physiol Gastrointest Liver Physiol 2005; 288: G447–G456.
Zheng S, Chen A . Activation of PPARgamma is required for curcumin to induce apoptosis and to inhibit the expression of extracellular matrix genes in hepatic stellate cells in vitro. Biochem J 2004; 384: 149–157.
Zhang F, Altorki NK, Mestre JR, Subbaramaiah K, Dannenberg AJ . Curcumin inhibits cyclooxygenase-2 transcription in bile acid- and phorbol ester-treated human gastrointestinal epithelial cells. Carcinogenesis 1999; 20: 445–451.
Goel A, Boland CR, Chauhan DP . Specific inhibition of cyclooxygenase-2 (COX-2) expression by dietary curcumin in HT-29 human colon cancer cells. Cancer Lett 2001; 172: 111–118.
Reddy BS, Rao CV . Novel approaches for colon cancer prevention by cyclooxygenase-2 inhibitors. J Environ Pathol Toxicol Oncol 2002; 21: 155–164.
Kang G, Kong PJ, Yuh YJ, Lim SY, Yim SV, Chun W et al. Curcumin suppresses lipopolysaccharide-induced cyclooxygenase-2 expression by inhibiting activator protein 1 and nuclear factor kappab bindings in BV2 microglial cells. J Pharmacol Sci 2004; 94: 325–328.
Wallace JM . Nutritional and botanical modulation of the inflammatory cascade—eicosanoids, cyclooxygenases, and lipoxygenases—as an adjunct in cancer therapy. Integr Cancer Ther 2002; 1: 7–37; discussion.
Chen YR, Tan TH . Inhibition of the c-Jun N-terminal kinase (JNK) signaling pathway by curcumin. Oncogene 1998; 17: 173–178.
Parra M, Lluis F, Miralles F, Caelles C, Munoz-Canoves P . The cJun N-terminal kinase (JNK) signaling pathway mediates induction of urokinase-type plasminogen activator (uPA) by the alkylating agent MNNG. Blood 2000; 96: 1415–1424.
Dikshit P, Goswami A, Mishra A, Chatterjee M, Jana NR . Curcumin induces stress response, neurite outgrowth and prevent NF-kappaB activation by inhibiting the proteasome function. Neurotox Res 2006; 9: 29–37.
Dikshit P, Goswami A, Mishra A, Nukina N, Jana NR . Curcumin enhances the polyglutamine-expanded truncated N-terminal huntingtin-induced cell death by promoting proteasomal malfunction. Biochem Biophys Res Commun 2006; 342: 1323–1328.
Jana NR, Dikshit P, Goswami A, Nukina N . Inhibition of proteasomal function by curcumin induces apoptosis through mitochondrial pathway. J Biol Chem 2004; 279: 11680–11685.
Wyke SM, Russell ST, Tisdale MJ . Induction of proteasome expression in skeletal muscle is attenuated by inhibitors of NF-kappaB activation. Br J Cancer 2004; 91: 1742–1750.
Singh S, Aggarwal BB . Activation of transcription factor NF-kappa B is suppressed by curcumin (diferuloylmethane) [corrected]. J Biol Chem 1995; 270: 24995–25000.
Kumar A, Dhawan S, Hardegen NJ, Aggarwal BB . Curcumin (diferuloylmethane) inhibition of tumor necrosis factor (TNF)-mediated adhesion of monocytes to endothelial cells by suppression of cell surface expression of adhesion molecules and of nuclear factor-kappaB activation. Biochem Pharmacol 1998; 55: 775–783.
Han SS, Chung ST, Robertson DA, Ranjan D, Bondada S . Curcumin causes the growth arrest and apoptosis of B cell lymphoma by downregulation of egr-1, c-myc, bcl-XL, NF-kappa B, and p53. Clin Immunol 1999; 93: 152–161.
Jobin C, Bradham CA, Russo MP, Juma B, Narula AS, Brenner DA et al. Curcumin blocks cytokine-mediated NF-kappa B activation and proinflammatory gene expression by inhibiting inhibitory factor I-kappa B kinase activity. J Immunol 1999; 163: 3474–3483.
Thaloor D, Miller KJ, Gephart J, Mitchell PO, Pavlath GK . Systemic administration of the NF-kappaB inhibitor curcumin stimulates muscle regeneration after traumatic injury. Am J Physiol 1999; 277: C320–C329.
Deeb D, Jiang H, Gao X, Hafner MS, Wong H, Divine G et al. Curcumin sensitizes prostate cancer cells to tumor necrosis factor-related apoptosis-inducing ligand/Apo2L by inhibiting nuclear factor-kappaB through suppression of IkappaBalpha phosphorylation. Mol Cancer Ther 2004; 3: 803–812.
Zheng M, Ekmekcioglu S, Walch ET, Tang CH, Grimm EA . Inhibition of nuclear factor-kappaB and nitric oxide by curcumin induces G2/M cell cycle arrest and apoptosis in human melanoma cells. Melanoma Res 2004; 14: 165–171.
Al-Sadi RM, Kreydiyyeh SI . Mediators of interleukin-1 beta action Na(+)-K(+)ATPase in Caco-2 cells. Eur Cytokine Netw 2003; 14: 83–90.
Callige M, Kieffer I, Richard-Foy H . CSN5/Jab1 is involved in ligand-dependent degradation of estrogen receptor {alpha} by the proteasome. Mol Cell Biol 2005; 25: 4349–4358.
Chen JJ, Huang WC, Chen CC . Transcriptional regulation of cyclooxygenase-2 in response to proteasome inhibitors involves reactive oxygen species-mediated signaling pathway and recruitment of CCAAT/enhancer-binding protein delta and CREB-binding protein. Mol Biol Cell 2005; 16: 5579–5591.
Guo W, Shang F, Liu Q, Urim L, Zhang M, Taylor A . Ubiquitin-proteasome pathway function is required for lens cell proliferation and differentiation. Invest Ophthalmol Vis Sci 2006; 47: 2569–2575.
Liacini A, Sylvester J, Li WQ, Huang W, Dehnade F, Ahmad M et al. Induction of matrix metalloproteinase-13 gene expression by TNF-alpha is mediated by MAP kinases, AP-1, and NF-kappaB transcription factors in articular chondrocytes. Exp Cell Res 2003; 288: 208–217.
Marinovic AC, Zheng B, Mitch WE, Price SR . Tissue-specific regulation of ubiquitin (UbC) transcription by glucocorticoids: in vivo and in vitro analyses. Am J Physiol Renal Physiol 2006; 292: F660–F666.
Ward C, Chilvers ER, Lawson MF, Pryde JG, Fujihara S, Farrow SN et al. NF-kappaB activation is a critical regulator of human granulocyte apoptosis in vitro. J Biol Chem 1999; 274: 4309–4318.
Yew EH, Cheung NS, Choy MS, Qi RZ, Lee AY, Peng ZF et al. Proteasome inhibition by lactacystin in primary neuronal cells induces both potentially neuroprotective and pro-apoptotic transcriptional responses: a microarray analysis. J Neurochem 2005; 94: 943–956.
Butts BD, Hudson HR, Linseman DA, Le SS, Ryan KR, Bouchard RJ et al. Proteasome inhibition elicits a biphasic effect on neuronal apoptosis via differential regulation of pro-survival and pro-apoptotic transcription factors. Mol Cell Neurosci 2005; 30: 279–289.
Kitiphongspattana K, Mathews CE, Leiter EH, Gaskins HR . Proteasome inhibition alters glucose-stimulated (pro)insulin secretion and turnover in pancreatic {beta}-cells. J Biol Chem 2005; 280: 15727–15734.
Yang W, Monroe J, Zhang Y, George D, Bremer E, Li H . Proteasome inhibition induces both pro- and anti-cell death pathways in prostate cancer cells. Cancer Lett 2006; 243: 217–227.
Yang H, Chen D, Cui QC, Yuan X, Dou QP . Celastrol, a triterpene extracted from the Chinese 'Thunder of God Vine,' is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res 2006; 66: 4758–4765.
Garrett IR, Chen D, Gutierrez G, Zhao M, Escobedo A, Rossini G et al. Selective inhibitors of the osteoblast proteasome stimulate bone formation in vivo and in vitro. J Clin Invest 2003; 111: 1771–1782.
Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ et al. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell 1996; 84: 491–495.
Bock T, Pakkenberg B, Buschard K . Increased islet volume but unchanged islet number in ob/ob mice. Diabetes 2003; 52: 1716–1722.
Dai Y, Desano J, Tang W, Meng X, Meng Y, Burstein E et al. Natural proteasome inhibitor celastrol suppresses androgen-independent prostate cancer progression by modulating apoptotic proteins and NF-kappaB. PLoS One 2010; 5: e14153.
Fricker LD, Gelman JS, Castro LM, Gozzo FC, Ferro ES . Peptidomic analysis of HEK293T cells: effect of the proteasome inhibitor epoxomicin on intracellular peptides. J Proteome Res 2012; 11: 1981–1990.
Abdelli S, Bonny C . JNK3 maintains expression of the insulin receptor substrate 2 (IRS2) in insulin-secreting cells: functional consequences for insulin signaling. PLoS One 2012; 7: e35997.
Stiles BL, Kuralwalla-Martinez C, Guo W, Gregorian C, Wang Y, Tian J et al. Selective deletion of Pten in pancreatic beta cells leads to increased islet mass and resistance to STZ-induced diabetes. Mol Cell Biol 2006; 26: 2772–2781.
Barbosa H, Bordin S, Stoppiglia L, Silva K, Borelli M, Del Zotto H et al. Islet neogenesis associated protein (INGAP) modulates gene expression in cultured neonatal rat islets. Regul Pept 2006; 136: 78–84.
Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T et al. NF-kappa B as a therapeutic target in multiple myeloma. J Biol Chem 2002; 277: 16639–16647.
Deans KA, Sattar N . 'Anti-inflammatory' drugs and their effects on type 2 diabetes. Diabetes Technol Ther 2006; 8: 18–27.
Sjoholm A, Nystrom T . Inflammation and the etiology of type 2 diabetes. Diabetes Metab Res Rev 2006; 22: 4–10.
Hundal RS, Petersen KF, Mayerson AB, Randhawa PS, Inzucchi S, Shoelson SE et al. Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J Clin Invest 2002; 109: 1321–1326.
Whitehead JP, Richards AA, Hickman IJ, Macdonald GA, Prins JB . Adiponectin—a key adipokine in the metabolic syndrome. Diabetes Obes Metab 2006; 8: 264–280.
Buckingham RE . Thiazolidinediones: pleiotropic drugs with potent anti-inflammatory properties for tissue protection. Hepatol Res 2005; 33: 167–170.
Takebayashi K, Matsumoto S, Wakabayashi S, Inukai Y, Matsutomo R, Aso Y et al. The effect of low-dose atorvastatin on circulating monocyte chemoattractant protein-1 in patients with type 2 diabetes complicated by hyperlipidemia. Metabolism 2005; 54: 1225–1229.
Norlin S, Ahlgren U, Edlund H . Nuclear factor-{kappa}B activity in {beta}-cells is required for glucose-stimulated insulin secretion. Diabetes 2005; 54: 125–132.
Sen P, Bhattacharyya S, Wallet M, Wong CP, Poligone B, Sen M et al. NF-kappa B hyperactivation has differential effects on the APC function of nonobese diabetic mouse macrophages. J Immunol 2003; 170: 1770–1780.
Jaeschke A, Rincon M, Doran B, Reilly J, Neuberg D, Greiner DL et al. Disruption of the Jnk2 (Mapk9) gene reduces destructive insulitis and diabetes in a mouse model of type I diabetes. Proc Natl Acad Sci USA 2005; 102: 6931–6935.
Wu L, Nicholson W, Knobel SM, Steffner RJ, May JM, Piston DW et al. Oxidative stress is a mediator of glucose toxicity in insulin-secreting pancreatic islet cell lines. J Biol Chem 2004; 279: 12126–12134.
Gupta SC, Patchra S, Aggarwal BB . Therapeutic roles of curcumin: lessons learned from clinical trials. AAPS 2013; 15: 195–218.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Nutrition & Diabetes website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Weisberg, S., Leibel, R. & Tortoriello, D. Proteasome inhibitors, including curcumin, improve pancreatic β-cell function and insulin sensitivity in diabetic mice. Nutr & Diabetes 6, e205 (2016). https://doi.org/10.1038/nutd.2016.13
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nutd.2016.13
This article is cited by
-
Botanical Interventions to Improve Glucose Control and Options for Diabetes Therapy
SN Comprehensive Clinical Medicine (2021)
-
Curcumin reverses the depressive-like behavior and insulin resistance induced by chronic mild stress
Metabolic Brain Disease (2017)
