
Abstract
Background and Aims:
Ingestion of food stimulates the secretion of incretin peptides glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide 1 to ensure the proper absorption and storage of nutrients. Menin is the 67 kDa protein product of the MEN1 gene recently reported to have a role in metabolism. In this study, we will determine the regulation of menin in the proximal duodenum by food intake and diet in correlation with GIP levels in the proximal duodenum of mice after an 18 h fast followed by 4 and 7 h refeeding and 3 months of high-fat diet.
Methods:
A dual luciferase assay was used to determine GIP promoter activity and ELISA was used to measure the levels of GIP after inhibition of menin through small interfering RNA (siRNA) and exposure to MAPK and AKT inhibitors. Colocalization of menin and GIP were determined by immunofluorescence.
Results:
Menin and GIP expression are regulated by fasting, refeeding and diet in the proximal duodenum. Overexpression of menin in STC-1 cells significantly inhibited GIP mRNA and promoter activity, whereas menin siRNA upregulated GIP levels. Inhibition of GIP expression by the PI3/AKT inhibitor, LY294002, was abrogated in STC-1 cells with reduced menin levels, whereas the MAPK inhibitor, UO126, inhibited the expression of GIP independent of menin. Exposure of STC-1 cells to GIP reduced menin expression in a dose-dependent manner via PI3K-AKT signaling.
Conclusion:
Feeding and diet regulates the expression of menin, which inversely correlates with GIP levels in the proximal duodenum. In vitro assays indicate that menin is a negative regulator of GIP via inhibition of PI3K-AKT signaling. We show menin colocalizing with GIP in K cells of the proximal gut and hypothesize that downregulation of menin may serve as a mechanism by which GIP is regulated in response to food intake and diet.
Similar content being viewed by others

Introduction
Feeding and diet initiate a complex network of hormonal and neural signals for the absorption and storage of nutrients from the gastrointestinal tract. Factors produced by K and L cells located within the intestinal mucosa in response to nutrient ingestion are capable of stimulating the release of hormones from the pancreas for regulation of blood glucose. K cells are located predominately in the proximal duodenum of the intestinal tract, whereas L cells are present in the lower jejenum and terminal ileum.1, 2, 3
Incretin peptides are glucose-lowering intestinal derived factors that consist of glucagon-like peptide 1 (GLP-1) secreted by L cells and glucose-dependent insulinotropic polypeptide (GIP) produced by K cells. Both incretin peptides share common actions on the islet beta cells of the pancreas. GIP is a 42-amino acid incretin hormone secreted into the bloodstream in response to the ingestion of carbohydrates, proteins and fat.1, 45 GIP binds to its receptor (GIP receptor, GIPR) in the pancreas and potentiates glucose-stimulated insulin secretion.
The enteroinsular axis is an essential component of carbohydrate metabolism and consists of the pancreas, GLP-1 and GIP. It has been reported that hypersecretion of endogenous GIP is responsible for the hyperfunction of enteroinsular signaling in patients with duodenal ulcers and total gastrectomy,6 supporting GIP’s role as a principal mediator of the enteroinsular axis.7, 8 Both GIP and GLP-1 are responsible for the incretin effect in healthy subjects,9 with GIP considered to have a predominant role10
The GIP repertoire of functions include potentiating glucose-dependent insulin release upon activation of the GIPR.8, 11 This leads to nutrient deposition via glucose uptake, fatty acid synthesis and fatty acid incorporation in adipocytes.12 Genetic ablation of GIPR demonstrates a critical role of this receptor in the enteroinsular axis13, 14 and genetic variation of GIPR in humans is associated with a reduction in early phase insulin reaction and elevation of blood glucose.15, 16 This suggests that defective GIPR signaling might have a critical role in the early pathophysiology of impaired glucose tolerance and type 2 diabetes.
Previous reports have shown that the tumor suppressor, menin, is expressed highest in the proximal duodenum of the gastrointestinal tract and during fasting17 compared with feeding. Menin is a tumor suppressor protein implicated in the development of tumors that overexpress the AKT and MAPK signaling pathways.18, 19 Menin inhibits AKT kinase activation and uncouples Elk-1 from MAP kinase activation sites.20, 21 Menin is expressed in the pancreas and when down-regulated is associated with increased beta cell proliferation.19, 22, 23 MEN1 syndrome is associated with the development of gastrinomas in the duodenum and tumors in endocrine organs such as the pancreas and pituitary, but not the liver.24 The reason for organ-specific preferences still remains unknown.24
The serine/threonine protein kinase PI3/AKT has an important role in cell growth, survival, transcription, differentiation and modulation of insulin signaling for glucose metabolism.25, 26 Since menin directly inhibits AKT activation and expression,20 we hypothesized that inhibition of GIP expression by menin may be a consequence of inactivation of the PI3K-AKT kinase signal transduction cascade.
The development of type 2 diabetes and obesity has been linked to an ‘anti-incretin’ theory with the authors postulating the existence of a negative regulatory mechanism to oppose the actions of incretins, such as GIP.12, 17 An imbalance in the equilibrium between ‘anti-incretin’ factors and incretins could lead to delayed insulin response and impaired insulin action.27
In this study, we investigated the presence of menin in K cells of the proximal duodenum and determined if it regulated the expression of GIP via PI3/AKT signaling in response to fasting and refeeding, and high-fat (HF) diet.
Materials and methods
Animal maintenance
C57BL/6 wild-type male mice, 5 fasted for 18 h, 4 fasted for 18 h and refed at 4 h, and 5 fasted for 18 h and refed at 7 h, were used for the proposed studies. An additional 3 animals per group, fed regular diet (RD) or HF diet, were utilized for the diet studies. All animals were kept in a 12 h dark/light cycle and control animals were fed RD (ad libitum). All procedures were approved by the University of Toledo Animal Care and Utilization Committee. For the feeding experiments, 3-month-old wild-type mice were fed a RDand a high-fat (HF) diet for 3 months (Research Diets Inc., New Brunswick, NJ, USA; Cat #D12451 and D12079B respectively). The RD contains 69% carbohydrates and 11% fat and the HF diet contains 35% carbohydrates and 45% fat.
Cell culture
The enteroendocrine intestinal cell line, STC-1, was obtained from ATCC with permission from Douglas Hanahan (UCSF-CA) and maintained at 37 °C and 5% CO2 in DMEM medium containing 1% penicillin and streptomycin. All experiments were performed on cells passaged five times at 80% confluency. LY294002, an inhibitor of the PI3K-AKT kinase signaling pathway, and UO126, a MAP kinase inhibitor, were both obtained from Cell Signaling Technology (Beverly, MA, USA) and were used at concentrations of 10 nM from a 10 mM stock reconstituted in DMSO. Aliquots were stored at −20 °C and vehicle was used for control experiment.
Transfections
The 2.9 and 0.210 kb fragments of the human GIP promoter were cloned into the luciferase-containing pGL4 reporter plasmid, as previously described.28 STC-1 cells were plated in 6-well plates (2 × 105 cells/well) and allowed to adhere for 24 h. Thereafter, cells were transfected with a mixture of 4 μg of plasmid DNA (pBABE) containing menin (Addgene Inc., Cambridge, MA, USA), 0.5 μg of the control plasmid (PGL4) or GIPLuc reporter plasmid28 using Lipofectamine 2000 reagent according to the manufacturer's protocol (Invitrogen, Carlsbad, CA, USA). After 48 h of incubation with complete media, luciferase and renilla activities were assayed according to manufacturer’s instruction (Dual Luciferase Reporter Assay; Promega-Madison, WI, USA). Firefly luciferase activity was normalized to renilla luciferase expression and is presented as percent activity over samples transfected with pGL4. All experiments were analyzed in duplicate in at least three separate experiments. Menin small interfering RNA (siRNA) oligos and the non-targeting control siRNA and reagents were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and the assay was performed according to the manufacturer’s protocol.
Western blot
STC-1 cells were plated in 6-wells in triplicate, serum starved overnight and exposed to 5 or 20 nM GIP for 24 h. The concentrations of proteins from total cell lysates were quantified prior to analysis on 10 or 4–12% gradient SDS-PAGE and immunoprobed with specific antibodies. Rabbit anti-menin (Bethyl Labs/Abcam, Cambridge, MA, USA),29 phospho-p38 (Cell Signaling, Beverly, MA, USA),30 goat anti-GIP (Santa Cruz Biotechnology, Santa Cruz, CA, USA)31 and GAPDH (Sigma-Aldrich, St, Louis, MO, USA) antibodies were used. Proteins were detected using an Odyssey Infrared Imaging System (Lincoln, NE, USA), using corresponding secondary antibodies conjugated to fluorescent dyes of different wavelengths.
GIP ELISA assay
STC-1 cells were plated in 6-well plates in triplicate, serum starved overnight and transfected with siRNA oligos for menin or scrambled controls according to manufacturer’s protocol (Santa Cruz Biotechnology). The transfected cells were exposed to LY294002, an inhibitor of PI3K-AKT kinase signaling, or the MAP kinase inhibitor, UO126, overnight prior to harvest and determination of protein concentration. 20 μg of protein in a volume of 10 μl total lysate and 10 μl media were used for a rat/mouse GIP ELISA (Cat. #EZRMGIP-55K, Millipore, Billerica, MA, USA) according to manufacturer’s protocol.
Real-time PCR
RNA was extracted using TRIzol (Life Technologies, Grand Island, NY, USA). Following DNAse digestion (DNAfree, Applied biosystems/Ambion, Austin, TX, USA), 1 μg RNA was transcribed into cDNA in a 20 μl reaction using High-Capacity cDNA achieve kit (Applied Biosystems/Ambion) and amplified (ABI 7900 HT system). PCR was performed in a 15 μl reaction, containing 5 μl cDNA (1/10 diluted), 1 × SYBR Green PCR Master Mix (Applied Biosystems/Ambion) and 300 nM of each primer. Primers are listed in 5'–3' orientation.
Menin forward primer: TCATTGCTGCCCTCTATGCC
Menin reverse primer: TCCAGTTTGGTGCCTGTGATG
GIP forward primer: GTGGCTTTGAAGACCTGCTC
GIP reverse primer: AAGTCCCCTCTGCGTACCTT
GAPDH forward primer: CCACCAGCCCCAGCAAGAGC
GAPDH reverse primer: GGCAGGGACTCCCCAGCAGT
18S forward primer: ATACATGCCGACGGGCGCTG
18S reverse primer: GGGAGGGAGCTCACCGGGTT
Ct values (cycle threshold) were used to calculate the amount of amplified PCR product relative to GAPDH and 18S. The relative amount of mRNA was calculated as 2–DCT. Results are expressed as transcript of interest mean normalized to GAPDH and 18S with s.e.m.
Immunofluorescence
One-cm tissue starting from the pyloric sphincter including the brunners gland and into the proximal duodenum was taken from the upper section of the gut from 5 C57BL/6 wild-type male mice fasted for 18 h and 5 C57BL/6 wild-type male mice fed ad libitum. Additional 2 sets of mice for each time point were also used for all described studies and consisted of a group of mice fasted for 18 h, refed and sacrificed after 4 h of feeding, and a second set of mice fasted for 18 h, refed and sacrificed after 7 h.
Tissues were harvested and fixed in 4% paraformaldehyde/phosphate-buffered saline for 18–20 h at room temperature followed by embedding in paraffin. Tissue blocks were obtained and 5 μM thick sections were cut and mounted on poly-L-lysine coated glass slides, blocked with 20% normal donkey serum/phosphate-buffered saline and 0.1% Triton X-100 for 30 min after citrate antigen retrieval. The slides were incubated for 1 h with a 1:50 dilution of primary antibodies (Bethyl labs, Montgomery, TX, USA) and a 1:200 dilution of fluorescein isothiocynate-conjugated anti-rabbit or goat (Jackson Laboratories, Bar Harbor, ME, USA) used as secondary antibodies for 1 h, and DAPI for blue staining of nuclei. Negative controls were performed on similar slides using secondary antibodies alone without incubation of primary antibodies.
All colocalization studies were performed on the same sections with specific antibodies raised in different species. Incubations were performed with anti-rabbit menin overnight followed by 1 h incubation with fluorescein isothiocynate-conjugated donkey anti rabbit-green and anti-goat GIP overnight followed by streptavidin-Texas Red-conjugated donkey anti-goat for 1 h. Control staining included (a) replacement of the first layer of antibody by non-immune serum and by the diluent alone, and (b) secondary antibodies tested in relation to the specificity of the species in which the primary antibodies were raised, with the secondary antibody in question being replaced by secondary antibodies from different animal species.
Sections were examined with an Olympus IX70 inverted fluorescence microscope (Olympus; Tokyo, Japan) equipped with filters (Olympus) giving excitation at wavelengths of 475–555 nm for Texas Red and 453–488 nm for fluorescein isothiocynate, with a digital camera. Merged images were viewed by superimposing both photographs at 10 × and 40 × magnification.
Statistical analysis
Data were analyzed with SPSS software (Armonk, NY, USA) using one-factor analysis of variance analysis or Student’s t-test as appropriate. P<0.05 were considered statistically significant.
Results
Menin colocalizes with GIP in the villi of proximal gut
Previous studies have shown that menin is expressed in the duodenum22 and that K cells express and secrete GIP.11 To examine whether menin in the gut is expressed in GIP-expressing K cells, we performed double stain fluorescent co-immunohistochemistry on the proximal duodenum, 1 cm from the pylorus. We determined that menin and GIP colocalize in cells on the villi as shown in Figure 1.

Menin colocalizes with GIP in the villi of proximal gut. (a) Representative immunoflourescence picture from a 1 cm section of the proximal duodenum analyzed for GIP (a), menin (b) and menin/GIP coexpression on villi (c). All images are captured at 40 × magnification as described in Materials and Methods. Colored arrows indicate expression of respective staining. White double arrowheads represent a 100 × magnification shown as a boxed insert. (40 × ), (d) represents a merged picture of menin and GIP (orange) as a cytoplasmic expression around the nuclei (in blue) at 40 × magnification. (e) represents a merged picture of menin and GIP (orange) as a cytoplasmic expression around the nuclei (in blue) at 100 × magnification.
Menin and GIP are inversely regulated by fasting and refeeding in the proximal duodenum
We have previously shown that 4 and 7 h of refeeding are the times that induce the highest spikes of serum insulin with metabolic events implicated during these times.32 We analyzed menin and GIP levels in the proximal duodenum of mice after an 18 h fast followed by 4 and 7 h of refeeding using western blot analysis and a GIP specific ELISA kit, respectively. We found that menin expression was inversely correlated with GIP levels (Figure 2) such that significantly higher menin levels are observed at fasting compared with refeeding, as opposed to the level of GIP expression at fasting and refeeding in the same tissue samples. Specifically, after 4 and 7 h of refeeding, menin expression declined significantly as the duration of feeding time increased, with the lowest menin levels observed at 7 h of refeeding (Figures 2a and b). However, GIP expression was lowest at fasting and highest at 7 h of refeeding (Figure 2c). Although our data confirms a previous report that menin expression is highest in the gastrointestinal tract during fasting17 and at feeding, serum GIP is increased,33 the effect of refeeding on menin levels and the inverse correlation with GIP expression is entirely novel.

Menin and GIP are inversely correlated during fasting and refeeding. Menin and GIP expression were analyzed using western blot and an ELISA respectively. Menin expression is higher during fasting and significantly decreases when refed at 4 and 7 h opposite of GIP expression, which is lowest during fasting and increases with 4 and 7 h refeeding. N=5 for fasted mice; N=4 for fasted and refed at 4 h; N=5 for fasted and refed at 7 h. (a) A representative western blot showing menin protein expression with each lane representing an individual mouse. (b) Graph showing relative expression of menin protein of mice per each group × 3 experiments. **P<0.005 (c) Represents the mean values of GIP expression analyzed from the proximal gut and analyzed by a GIP specific ELISA kit. **P<0.005.
Feeding inversely modulates menin and GIP expression at the mRNA level
We investigated if the effect of feeding on menin and GIP expression is at the level of mRNA by analyzing the proximal duodenum after an 18 h fast followed by 4 and 7 h of refeeding by reverse transcripion-PCR. In Figure 3a, we show that menin mRNA levels steadily decline the with length of refeeding after the 18 h fast, consistent with the levels of protein shown in Figures 2a and b. This steady decline in menin mRNA was associated with an increase in GIP mRNA expression peaking at 7 h of refeeding (Figure 3b), and consistent with the ELISA data in Figure 2c.

Menin and GIP mRNA are inversely correlated during fasting and refeeding. Menin mRNA expression decreases with refeeding whereas GIP mRNA expression increases. (a) Menin expression was determined and expressed relative to GAPDH. **P<0.0004, ***P<0.0001. (b) GIP expression was determined and expressed relative to GAPDH. N⩾8 from each group of mice, values are mean±s.e. expressed as ratio relative to GAPDH. *P=0.0218.
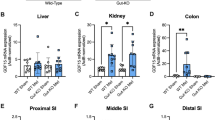
High-fat diet downregulates menin expression in the duodenum and is associated with increase in GIP expression
To determine the effect of diet on duodenal menin expression in relation to GIP, we subjected C57BL/6 mice to 3 months of HF diet and analyzed menin and GIP expression in the proximal duodenum by western blot analysis and reverse transcripion-PCR. In Figure 4a, we show that HF diet downregulates menin protein levels in the proximal gut of these mice. Furthermore, we observed a significant decrease in menin mRNA expression, (Figure 4b) consistent with reduction in protein levels as shown in Figure 4a. The reduction of menin expression inversely correlated with the upregulation of GIP caused by the HF diet (Figure 4c) relative to expression from mice on RD and consistent with reports that GIP is regulated by dietary fat and glucose.34, 35, 36

Inverse relationship of menin and GIP in proximal duodenum of mice fed a HF diet. Menin expression is downregulated after 3 months on a HF diet opposite of GIP, which is significantly increased after 3 months on a HF diet. (a) is a representative western blot analysis showing downregulation of menin protein expression after 3 months on HF diet. Reverse transcripion-PCR was used to determine the relative expression of menin and GIP after normalizing with 18S. N=6 from each feeding group of mice. Values are mean±s.e. (b) shows menin expression in RD; HF diet (HF) **P<0.004; (c) is GIP expression in RD and HF diet (HF) *<0.005.
Menin overexpression decreases endogenous GIP mRNA
To study the mechanism underlying the inverse correlation observed between the expression of menin and GIP, we tested the hypothesis that menin is a negative regulator of GIP in vitro. We overexpressed menin in STC-1 cells by transient transfection using Lipofectamine 2000 (Figure 5a), and determined the effect on endogenous GIP levels. We show in Figure 5b that overexpression of menin in STC-1 cells correlates with significant downregulation of GIP mRNA.

Menin overexpression decreases endogenous GIP mRNA. Overexpression of menin in STC cells shows a resultant decrease in GIP levels. (a) Western blot analysis showing level of menin overexpression using a menin expression vector (meninO/E). (b) Reverse transcripion-PCR data showing downregulation of GIP mRNA following menin overexpression (meninO/E).
Menin regulates GIP promoter activity and expression via AKT signaling
Previously, when a 2.9 kB human GIP promoter luciferase reporter gene construct was transfected into STC-1 cells, there was a 40-fold elevated GIP promoter activity over the response in IEC cells (a cell line derived from the ileum).28 However, the greatest promoter activity was detected with the highly conserved 0.210 kB promoter construct, suggesting that more distal regions may contain repressor elements.28 We therefore compared the effect of menin on both the 0.210 and 2.9 kB GIP promoters to determine if menin is part of a repressor component that regulates GIP expression in STC-1 cells. Using whole cell lysates derived from STC-1 cells overexpressing menin (Figure 5a), we determined the effect of menin on the GIP promoter by luciferase reporter assay. In Figure 6a, we show that the activity of the short 0.210 kB promoter for GIP was not affected by menin overexpression; however, overexpression of menin significantly inhibited the 2.9 kB GIP promoter (Figure 6b), supporting our hypothesis that menin may be part of a repressor component that negatively regulates GIP. These results suggest that menin may negatively regulate the transcription of GIP in cells and explain the in vivo inverse correlation observed with previous results shown.

Menin regulates GIP promoter activity and expression and abrogates PI3K-AKT regulation in STC-1 cells. Overexpression of menin in the 0.210 kb GIP did not change GIP activity levels, (a), however overexpression in the 2.9 kb promoter significantly inhibited relative GIP activity (b), supporting our hypothesis that menin may be part of a repressor component that negatively regulates GIP. In (c and d), using AKT and MAPK inhibitors, we concluded that menin regulates the expression of GIP through the AKT pathway. (a) represents the % activity of the 0.210 kB construct and (b) is a representation of % activity of the 2.9 kb construct, ***P=0.0001. (c) represents expression of GIP in whole cell lysates. GIP expression in the media of cells described in (c) was also determined by ELISA and is shown in (d). All ELISA results were calculated as means of two separate experiments. *P<0.005; ** and ##P<0.0024. (e) Western blot analysis showing level of menin downregulation using menin specific siRNA oligos (simenin). Protein lysates were prepared and GIP expression in whole cell lysates determined by ELISA with LYsimenin and UOsimenin representing treatment of LY294002 and UO126 on cells transfected with menin siRNA.
In line with our hypothesis and consistent with menin’s possible role as a GIP repressor, we found that menin siRNA (simenin) significantly increased GIP expression as determined by ELISA performed on STC-1 whole cell lysates (Figure 6c). The PI3K-AKT inhibitor LY294002 (LY) caused downregulation of GIP expression; however, the effect was inhibited with menin siRNA (LYsimenin) (Figure 6c). Furthermore, the MAP kinase inhibitor UO126 (UO) inhibited GIP expression independent of menin, since menin siRNA did not reverse the effect of UO as observed with LY294002. Expression of secreted GIP in the media followed a similar trend as observed in the lysates (Figure 6d). In Figure 6e, we demonstrate that the addition of menin-specific siRNA to STC-1 cells indeed leads to a significant decrease in menin expression and that the changes observed with simenin and LYsimenin can be attributed to the significant downregulation of menin expression in the transiently transfected STC-1 cells. With this data, we conclude that the PI3K-AKT-mediated regulation of GIP is dependent on menin whereas MAP kinase-mediated GIP expression is via a menin-independent pathway.
GIP regulates menin expression via PI3K-AKT signaling
GIP is known to suppress p38 MAPK activation via AKT to induce pro-survival responses and inhibit apoptosis in pancreatic beta cells.37 Menin induces apoptosis and when down-regulated has also been associated with proliferation of beta cells.22 In contrast, GIP directly stimulates proliferation by binding to its receptor on target beta cells.38 Since it has been reported that GIP exerts its effects on cell survival via an AKT-dependent pathway,37 we postulated that GIP inhibits menin expression via the PI3K-AKT signaling cascade. In Figure 7a, we show that STC-1 cells exposed to GIP resulted in significant upregulation of pAKT associated with the downregulation of menin protein. This activation was abrogated with LY294002. Since GIP is known to inhibit MAPK signaling via downregulation of phospho-p38, we demonstrated that 20 nM GIP not only downregulates menin expression but also reduces phospho-p38 levels in the STC-1 cells (Figure 7b). Furthermore, we confirm that the ability of PI3K/AKT inhibitor LY294002 (LYg20) to abrogate GIP (g) downregulation of menin protein expression is via inhibition of pAKT (Figure 7a), as phospho-p38 levels did not change with LY treatment as shown in Figure 7b. Indeed the AKT-mediated GIP suppression of p38 is confirmed with the activation of pAKT (Figure 7a) and the significant reduction in phospho-p38 (Figure 7b). This was followed by LY294002’s ability to abrogate the inactivation of p38 by GIP, as demonstrated by the high expression of phospho-p38 in the LYg20 lane compared to g20 in Figure 7b.

GIP is a negative regulator of menin protein expression. STC cells treated with (a) and (b) 5 or 20 nM GIP (g5 or g20) resulted in decreased menin levels. STC cells treated with 10 μM LY294002 (LY) alone or LY294002 with 5 and 20 nM GIP, respectively (LYg5; LYg20 ) resulted in no change in the menin levels. STC cells treated with the MAPK inhibitor, UO126 (UO) alone or with GIP at 5 nM (UOg5), 20 nM (UOg20), respectively did not change menin expression (c). These results indicate GIP negatively regulates menin through pAKT pathway. A, one representative experiment out of three is shown; B and C, one representative experiment out of two is shown.
To further elucidate if phospho-p38 is a component of the GIP-mediated regulation of menin, we used the MAP kinase inhibitor UO126 (UO) to effectively downregulate the expression of phospho-p38 with or without GIP and analyzed menin levels. As previously shown, GIP effectively reduced menin expression at a concentration of 20 nM (g20) and phospho-p38 levels (Figure 7c). Interestingly, pretreatment of cells with UO126 prior to 20 nM GIP abrogated GIP’s ability to downregulate menin protein, even though phospho-p38 was still significantly inhibited by UO126 (Figure 7c) as previously described.37 This suggests that UO126-inhibition of GIP regulation on menin is independent of phospho-p38. Collectively, Figure 6 demonstrates that GIP inhibition of menin expression is in part via a PI3K-AKT-dependent signaling pathway independent of phospho-p38.
Discussion
The incretin effect mediated by GIP and GLP-1 account for 50–70% of total postprandial insulin release in healthy subjects.1 In addition to its physiological role in regulating endocrine pancreatic secretion, GIP also acts on the adipose tissue by regulating lipid metabolism. Notably the incretin effect in type 2 diabetes is markedly reduced with greater reduction in GIP’s insulinotropic effect on the pancreatic beta cells compared with GLP-1.9, 39
The homeostasis between anti-incretin factor(s) and incretins has been postulated to be disrupted most likely in the proximal foregut of diabetics.27 GIP is synthesized and released from K cells of the duodenum and is known to act as a principal mediator of the enteroinsular axis.11 GIP is also reported to possess insulin-mimetic properties and induce the activation of the AKT pathway40 resulting in the uptake of glucose by adipocytes. Inhibition of GIP signaling may also prevent obesity and metabolic syndrome.40
Although menin has recently been shown to inhibit AKT activation among its already known functions of interacting with transcriptional regulators such as NF-kB, JunD and Smad 3, little is known about factors that directly regulate this protein.18, 19, 20 Refeeding after fasting is known to increase serum GIP levels41 and leads to an increase in serum insulin that downregulates the expression of menin.29 We previously reported that menin is expressed in the duodenum22 and Ratineau et al.24 described higher menin expression in the duodenum of fasted animals compared with that in ad libitum-fed mice. The authors determined that MEN1 transcripts are expressed in the crypts of the intestine and dispersed on cells of the villi, but did not identify the specific cell-type that expressed menin. Since menin is a metabolic protein modulated by fasting and refeeding, and regulated by insulin,29 we hypothesized that menin is a regulator of GIP expressed in K cells. Although menin may has an antiproliferative role within the crypts of the intestine, this is the first report that identifies its expression in K cells and describes the negative regulation of GIP by menin.
In this study, we demonstrate that menin is expressed in K cells of the proximal duodenum by colocalizing with GIP. We also present evidence that food intake and HF diet downregulate menin expression in the proximal duodenum and is inversely correlated with changes in GIP expression. We show for the first time that menin is a negative regulator of GIP at the promoter level and GIP has a negative feedback effect on menin by directly downregulating menin protein levels via a phosphatidylinositol-3-kinase/protein kinase B (AKT) (PI3K-AKT)-dependent pathway.
STC-1 cells have been used as a model for studies on GIP expression secretion and K cell function.28 Using STC-1 cells, we describe a functional interplay between menin and GIP to explain how diet and feeding regulate menin and GIP expression in the proximal duodenum through the PI3K-AKT signaling cascades. However, MAPK signaling may be partially implicated in GIP regulation of menin, albeit independent of phospho-p38, as UO126 abrogated GIP-induced reduction of menin expression without a change in phospho-p38 levels. As a feedback mechanism, we propose that GIP downregulates menin levels to relieve its repression. Additionally, since insulin is a direct transcriptional regulator of menin expression, changes in serum insulin as a result of fasting and refeeding or diet may also contribute to the modulation of menin levels in the proximal duodenum. The physiological response to this cascade of events is a decrease in serum insulin as a result of the fasted state, which causes an increase in menin levels. Refeeding again decreases menin expression and increases GIP.
This study is the first to correlate physiologically induced changes in menin levels as a result of fasting and refeeding and diet with GIP. Indeed, menin is expressed in other cells of the duodenum such as in the crypts along with K cells; therefore, changes in expression as shown in Figures 2, 3, 4 cannot be attributed to levels in K cells alone. However, it is intriguing that the overall levels of menin and GIP are inversely modulated by physiological events in the proximal duodenum, as menin is also expressed in K cells (Figure 1). The inverse role of these proteins have also been described in the pancreas, where menin has an anti-proliferative effect on beta cells,42 while GIP expression is associated with increased cell proliferation and insulin secretion.43, 44
In summary, we show that food intake and diet modulate menin in the proximal duodenum in an inverse correlation with GIP expression. We demonstrate that menin is a negative regulator of GIP expression and inhibition of PI3K-AKT signaling by LY294002 causes a decrease in GIP expression and is reversed with menin siRNA. Furthermore, GIP downregulates menin protein via the PI3K-AKT pathway, where activation of AKT appears to be a key component in maintaining the expression of menin levels. Our findings provide evidence of an anti-incretin effect exerted by menin in response to food intake and diet involving the PI3-AKT signaling cascade.
References
Meier JJ, Nauck MA, Schmidt WE, Gallwitz B . Gastric inhibitory polypeptide: the neglected incretin revisited. Regul Pept 2002; 107: 1–13.
Eissele R, Goke R, Willemer S, Harthus HP, Vermeer H, Arnold R et al. Glucagon-like peptide-1 cells in the gastrointestinal tract and pancreas of rat, pig and man. Eur J Clin Invest 1992; 22: 283–291.
Fehmann HC, Goke R, Goke B . Cell and molecular biology of the incretin hormones glucagon-like peptide-I and glucose-dependent insulin releasing polypeptide. Endocr Rev 1995; 16: 390–410.
Bray GA, Popkin BM . Dietary fat intake does affect obesity!. Am J Clin Nutr 1998; 68: 1157–1173.
Baggio LL, Drucker DJ . Biology of incretins: GLP-1 and GIP. Gastroenterology 2007; 132: 2131–2157.
Takemura J, Seino Y, Yamamura T, Yoshiya K, Ishikawa Y, Itoh N et al. The evidence for the regulatory role of endogenous GIP as a glucose dependent insulinotropic hormone in patients with duodenal ulcer. Clin Endocrinol 1983; 19: 345–353.
Hansen EN, Tamboli RA, Isbell JM, Saliba J, Dunn JP, Marks-Shulman PA et al. Role of the foregut in the early improvement in glucose tolerance and insulin sensitivity following Roux-en-Y gastric bypass surgery. Am J Physiol Gastrointest Liver Physiol 2011; 300: G795–G802.
Dupre J, Ross SA, Watson D, Brown JC . Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J Clin Endocrinol Metab 1973; 37: 826–828.
Mentis N, Vardarli I, Kothe LD, Holst JJ, Deacon CF, Theodorakis M et al. GIP does not potentiate the antidiabetic effects of GLP-1 in hyperglycemic patients with type 2 diabetes. Diabetes 2011; 60: 1270–1276.
Nauck MA, El-Ouaghlidi A, Gabrys B, Hucking K, Holst JJ, Deacon CF et al. Secretion of incretin hormones (GIP and GLP-1) and incretin effect after oral glucose in first-degree relatives of patients with type 2 diabetes. Regul Pept 2004; 122: 209–217.
Cheung AT, Dayanandan B, Lewis JT, Korbutt GS, Rajotte RV, Bryer-Ash M et al. Glucose-dependent insulin release from genetically engineered K cells. Science 2000; 290: 1959–1962.
Jessen L, D’Alessio D . The incretins and beta-cell health: contrasting glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1as a path to understand islet function in diabetes. Gastroenterology 2009; 137: 1891–1894.
Hansotia T, Maida A, Flock G, Yamada Y, Tsukiyama K, Seino Y et al. Extrapancreatic incretin receptors modulate glucose homeostasis, body weight, and energy expenditure. J Clin Invest 2007; 117: 143–152.
Harada N, Yamada Y, Tsukiyama K, Yamada C, Nakamura Y, Mukai E et al. A novel GIP receptor splice variant influences GIP sensitivity of pancreatic beta-cells in obese mice. Am J Physiol Endocrinol Metab 2008; 294: E61–E68.
Saxena R, Hivert M-F, Langenberg C, Tanaka T, Pankow JS, Vollenweider P et al. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat Genet 2010; 42: 142–148.
Flock G, Holland D, Seino Y, Drucker DJ . GPR119 regulates murine glucose homeostasis through incretin receptor-dependent and independent mechanisms. Endocrinology 2011; 152: 374–383.
Miyawaki K, Yamada Y, Ban N, Ihara Y, Tsukiyama K, Zhou H et al. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med 2002; 8: 738–742.
Agarwal SK, Guru SC, Heppner C, Erdos MR, Collins RM, Park SY et al. Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell 1999; 96: 143–152.
Tsukada T, Nagamura Y, Ohkura N . MEN1 gene and its mutations: Basic and clinical implications. Cancer Sci 2009; 100: 209–215.
Wang Y, Ozawa A, Zaman S, Prasad NB, Chandrasekharappa SC, Agarwal SK et al. The tumor suppressor protein menin inhibits AKT activation by regulating its cellular localization. Cancer Res 2011; 71: 371–382.
Gallo A, Cuozzo C, Esposito I, Maggiolini M, Bonofiglio D, Vivacqua A et al. Menin uncouples Elk-1, JunD and c-Jun phosphorylation from MAP kinase activation. Oncogene 2002; 21: 6434–6445.
Mensah-Osman E, Zavros Y, Merchant JL . Somatostatin stimulates menin gene expression by inhibiting protein kinase A. Am J Physiol Gastrointest Liver Physiol 2008; 295: G843–G854.
Karnik SK, Chen H, McLean GW, Heit JJ, Gu X, Zhang AY et al. Menin controls growth of pancreatic beta-cells in pregnant mice and promotes gestational diabetes mellitus. Science 2007; 318: 806–809.
Ratineau C, Bernard C, Poncet G, Blanc M, Josso C, Fontaniere S et al. Reduction of menin expression enhances cell proliferation and is tumorigenic in intestinal epithelial cells. J Biol Chem 2004; 279: 24477–24484.
Ayala JE, Bracy DP, Hansotia T, Flock G, Seino Y, Wasserman DH et al. Insulin action in the double incretin receptor knockout mouse. Diabetes 2008; 57: 288–297.
Brazil DP, Yang ZZ, Hemmings BA . Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci 2004; 29: 233–242.
Rubino F . Is type 2 diabetes an operable intestinal disease? A provocative yet reasonable hypothesis. Diabetes Care 2008; 31 (Suppl 2): S290–S296.
Fujita Y, Chui JW, King DS, Zhang T, Seufert J, Pownall S et al. Pax6 and Pdx1 are required for production of glucose-dependent insulinotropic polypeptide in proglucagon-expressing L cells. Am J Physiol Endocrinol Metab 2008; 295: E648–E657.
Wuescher L, Angevine K, Hinds T, Ramakrishnan S, Najjar SM, Mensah-Osman EJ . Insulin regulates Menin expression, cytoplasmic localization and interaction with FOXO 1. Am J Physiol Endocrinol Metab 2011; 301: E474–E483.
Han J, Lee JD, Bibbs L, Ulevitch RJ . AMAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 1994; 265: 808–811.
Ye DZ, Kaestner KH . Foxa1 and Foxa2 control the differentiation of goblet and enteroendocrine L- and D-cells in mice. Gastroenterology 2009; 137: 2052–2062.
Wuescher L AK, Patel P, Mensah-Osman E . Menin liver-specific hemizygous mice challenged with high fat diet show increased weight gain and markers of metabolic impairment. Nutr Diabetes 2012; 2: e34.
Vilsboll T, Krarup T, Sonne J, Madsbad S, Volund A, Juul AG et al. Incretin secretion in relation to meal size and body weight in healthy subjects and people with type 1 and type 2 diabetes mellitus. J Clin Endocrinol Metab 2003; 88: 2706–2713.
Cho YM, Kieffer TJ . K-cells and glucose-dependent insulinotropic polypeptide in health and disease. Vitam Horm 2010; 84: 111–150.
Lodefalk M, Carlsson-Skwirut C, Holst JJ, Aman J, Bang P . Effects of fat supplementation on postprandial GIP, GLP-1, ghrelin and IGFBP-1 levels: a pilot study on adolescents with type 1 diabetes. Horm Res Paediatr 2010; 73: 355–362.
Creutzfeldt W, Talaulicar M, Ebert R, Willms B . Inhibition of gastric inhibitory polypeptide (GIP) release by insulin and glucose in juvenile diabetes. Diabetes 1980; 29: 140–145.
Widenmaier SB, Ao Z, Kim SJ, Warnock G, McIntosh CH . Suppression of p38 MAPK and JNK via Akt-mediated inhibition of apoptosis signal-regulating kinase 1 constitutes a core component of the beta-cell pro-survival effects of glucose-dependent insulinotropic polypeptide. J Biol Chem 2009; 284: 30372–30382.
Gupta D, Peshavaria M, Monga N, Jetton TL, Leahy JL . Physiologic and pharmacologic modulation of glucose-dependent insulinotropic polypeptide (GIP) receptor expression in beta-cells by peroxisome proliferator-activated receptor (PPAR)-gamma signaling: possible mechanism for the GIP resistance in type 2 diabetes. Diabetes 2010; 59: 1445–1450.
Meier JJ, Gallwitz B, Kask B, Deacon CF, Holst JJ, Schmidt WE et al. Stimulation of insulin secretion by intravenous bolus injection and continuous infusion of gastric inhibitory polypeptide in patients with type 2 diabetes and healthy control subjects. Diabetes 2004; 53 (Suppl 3): S220–S224.
Song DH, Getty-Kaushik L, Tseng E, Simon J, Corkey BE, Wolfe MM . Glucose-dependent insulinotropic polypeptide enhances adipocyte development and glucose uptake in part through Akt activation. Gastroenterology 2007; 133: 1796–1805.
Clements RH, Gonzalez QH, Long CI, Wittert G, Laws HL . Hormonal changes after Roux-en Y gastric bypass for morbid obesity and the control of type-II diabetes mellitus. Am Surg 2004; 70: 1–4 discussion 4-5.
Lu J, Herrera PL, Carreira C, Bonnavion R, Seigne C, Calender A et al. Alpha cell-specific Men1 ablation triggers the transdifferentiation of glucagon-expressing cells and insulinoma development. Gastroenterology 2010; 138: 1954–1965.
Fujita Y, Wideman RD, Asadi A, Yang GK, Baker R, Webber T et al. Glucose-dependent insulinotropic polypeptide is expressed in pancreatic islet alpha-cells and promotes insulin secretion. Gastroenterology 2010; 138: 1966–1975.
Portela-Gomes GM, Stridsberg M . Chromogranin A in the human gastrointestinal tract: an immunocytochemical study with region-specific antibodies. J Histochem Cytochem 2002; 50: 1487–1492.
Acknowledgements
We acknowledge the contributions of Sadeesh Ramakrishnan, Jennifer Kailsz, Mats Fernstrom and Sonia Najjar. University of Toledo Health System Institutional Start-up Funding provided to Edith Mensah-Osman.
Author Contributions
KRA, LMW, KA, LAA and MFM contributed to the acquisition of data; MFM and TJK contributed to the critical revision of the manuscript; EMO contributed to study concept and design, acquisition of data, analysis and interpretation, study supervision, funding and writing of manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Angevine, K., Wuescher, L., Andrews, K. et al. Menin and GIP are inversely regulated by food intake and diet via PI3/AKT signaling in the proximal duodenum. Nutr & Diabetes 2, e55 (2012). https://doi.org/10.1038/nutd.2012.30
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nutd.2012.30
