
Abstract
Objective:
The contribution of genetic variants to body mass index (BMI) during adolescence across multiethnic samples is largely unknown. We selected genetic loci associated with BMI or obesity in European-descent samples and examined them in a multiethnic adolescent sample.
Design and Sample:
In 5103 European American (EA), 1748 African American (AfA), 1304 Hispanic American (HA) and 439 Asian American (AsA) participants of the National Longitudinal Study of Adolescent Health (Add Health; ages 12–21 years, 47.5% male), we assessed the association between 41 established obesity-related single-nucleotide polymorphisms (SNPs) with BMI using additive genetic models, stratified by race/ethnicity, and in a pooled meta-analysis sample. We also compared the magnitude of effect for BMI–SNP associations in EA and AfA adolescents to comparable effect estimates from 11 861 EA and AfA adults in the Atherosclerosis Risk in Communities study (ages 45–64 years, 43.2% male).
Results:
Thirty-five of 41 BMI–SNP associations were directionally consistent with published studies in European populations, 18 achieved nominal significance (P<0.05; effect sizes from 0.19 to 0.71 kg m−2 increase in BMI per effect allele), while 4 (FTO, TMEM18, TFAP2B, MC4R) remained significant after Bonferroni correction (P<0.0015). Of 41 BMI–SNP associations in AfA, HA and AsA adolescents, nine, three and five, respectively, were directionally consistent and nominally significant. In the pooled meta-analysis, 36 of 41 effect estimates were directionally consistent and 21 of 36 were nominally significant. In EA adolescents, BMI effect estimates were larger (P<0.05) for variants near TMEM18, PTER and MC4R and smaller for variants near MTIF3 and NRXN3 compared with EA adults.
Conclusion:
Our findings suggest that obesity susceptibility loci may have a comparatively stronger role during adolescence than during adulthood, with variation across race/ethnic subpopulation.
Similar content being viewed by others


Introduction
Adolescence is a period of high risk for weight gain1, 2 Adolescent obesity is highly predictive of adult-onset obesity and severe obesity (body mass index (BMI) ⩾40 kg m−2), particularly in minority populations.3, 4, 5 In the past 5 years, genome-wide association studies (GWAS) have identified nearly 40 common genetic loci associated with BMI and obesity in European middle-aged adults.6, 7, 8, 9 Studies in children have replicated some of these associations.8, 10, 11, 12, 13 Few studies examined these associations in adolescents and fewer have compared the magnitudes of effect during different phases of the life course. A few longitudinal studies of specific genetic variants have shown stronger genetic association during adolescence,14, 15, 16 and notable decline before14, 15 and following adolescence.15, 16 Another study suggested larger effect sizes for variants in/near TMEM18, SEC16B, KCTD15 and BNDF in European children and adolescents compared with adults.12
Recent GWAS in non-European ancestry populations have identified additional loci.17, 18, 19, 20 Targeted genotyping studies of selected variants have been undertaken in African Americans (AfA),20, 21 Hispanic Americans (HAs),22 and several ethnically diverse populations.23 However, these studies have been limited in scope of variants tested and have had relatively small sample sizes. The contribution of genetic loci for BMI on body mass during adolescence across different ancestral populations remains largely unknown.
To study the contributions during adolescence of BMI single-nucleotide polymorphisms (SNPs) identified in European adult populations, we used nationally representative data from the National Longitudinal Study of Adolescent Health (Add Health) aged 12–21 years. First, we tested the Add Health European-American adolescent sample for replication of published results from adults of European descent. Second, we examined consistency of directions of effect and significance in non-Hispanic European American (EA), non-Hispanic AfA, non-Hispanic Asian American (AsA), HA adolescents, and in the race/ethnic combined sample of adolescents with published results from adults of European descent. Third, we compared differences in the magnitude of the effect for BMI–SNP associations in EA Add Health adolescents relative to EA middle-age adults from the Atherosclerosis Risk in Communities (ARIC) study.
Materials and methods
Add Health
Participants
Add Health is a nationally representative cohort of adolescents (1994–95, n=20 745, aged 11–20 years, mean age 15.9 years) drawn from a probability sample of 80 US high schools and 52 US middle schools, representative of US schools in 1994–95 with respect to region, urban setting, school size, school type and race or ethnic background. Wave II (1996, n=14 738, aged 12–21 years, mean age 16.5 years) included by design Wave I adolescents still of school-age, including those currently in high school and high school drop outs. Oversampled subgroups include related and non-related adolescents sharing a Wave I household (n=5524 Wave I respondents living in 2639 households),24 and several race/ethnic subpopulations, including Chinese, Cubans, Puerto Ricans and Filipinos. Wave III (2001–2002, n=15 197, aged 18–27 years, mean age 22.3 years) and Wave IV (2008–2009, n=15 701, aged 23–32 years, mean age 28.9 years) followed all Wave I respondents, regardless of Wave II participation. The most recent data collection (Wave IV) included follow-up interviews from 15 701 respondents drawn from 19 962 of the original 20 745 Wave I respondents. Survey procedures have been described elsewhere25, 26, 27 and were approved by the Institutional Review Board, University of North Carolina at Chapel Hill.
Race/ethnicity
Genetic markers used to determine ancestry were unavailable. As a proxy for ancestry, we used a race/ethnicity variable constructed using survey data on ancestral background and family relationship status from participants and their parents (that is, country of origin, length of time in the US, ancestry and adoption). We use a four-category race/ethnicity classification: EA, AfA, HA and AsA, with control variables for subpopulation ancestry: Cuban, Puerto Rican, Central/South American, Mexican, Other Hispanic, Chinese, Filipino and Other Asian. In addition, we classified Hispanic and Asian individuals as non-US born (first generation immigrants), and US born (2nd or 3rd generation immigrants), given differences in BMI by immigrant status.28, 29
Literature-based SNPs
We initially selected 68 SNPs associated with BMI and waist circumference from the Genome-wide Investigation of ANThropometric measures (GIANT) consortium and other studies in European adults for genotyping.6, 7, 8, 30, 31 We assessed redundancy using the pattern of linkage disequilibrium (LD) among these 68 SNPs within a multiethnic HapMap reference panel (CEU, YRI and CHB populations, excluding SNPs with a pairwise r2>0.80), reducing the number of SNPs to be genotyped to 42 spanning 39 loci. We included two SNPs at three loci (near MC4R, SEC16B and SH2B1) to capture variation in LD for Asian and African genetic structure around these loci. Of these 42 SNPs, 38 have been associated (P<5 × 10−8) with BMI,6, 7, 8, 11, 17, 19 and 4 have been associated with obesity.9
Genotyping
DNA was isolated from Wave IV saliva samples at the Institute of Behavioral Genetics in Boulder, Colorado using the BuccalAmp DNA extraction kit (Epicenter, Madison, WI, USA), quantified by PicoGreen fluorescence and visualized on standard agarose gels. DNA quality was assessed and genotyping performed at the Biospeciman Processing Core and Mammalian Genotyping Core (University of North Carolina, Chapel Hill) using TaqMan assays (Applied Biosystems, Carlsbad, CA, USA) and standard protocols using the ABI Prism 7900-HT Sequence Detection System. Genotypes were called using SDS 2.2.2. Based on 277 duplicate sample pairs, the overall discordance rate across SNPs was 0.3%. Further assessment of quality control was performed using PLINK 1.07. The genotype call rate was >97.8% in each of the four race/ethnic groups. One SNP (rs2922763) inconsistent with Hardy–Weinberg equilibrium (P<0.001) was excluded, resulting in a 41 SNPs investigated in the analyses. Another two SNPs were excluded in specific race/ethnic groups: rs867559 (HA) and rs4712652 (AfA).
Outcome measure: BMI
Weight and height were measured during in-home surveys using standardized procedures. BMI (kilograms per meter squared) was calculated using measured height and weight assessed at Waves II or III when participants were 12–21 years, with priority for Wave II (n=14 646) unless the individual was only measured at Wave III (n=785). Self-reported height and weight were substituted for those refusing measurement and/or weighing more than the scale capacity (n=81), with sensitivity analyses to examine the effect of the substitution.
Using age- and sex-specific International obesity task force (IOTF) centile curves for individuals up to 18 years of age of age we classified overweight (BMI 25.0–29.9 kg m−2 equivalent) and obesity (BMI ⩾30 kg m−2 equivalent),20 and the adult cut points (BMI 25.0–29.9 kg m−2; BMI ⩾30 kg m−2 ) for individuals 18–21 years of age.21 As the BMI distributions within each race/ethnic group were slightly right skewed, natural-log transformed and inverse-normally transformed BMI were examined in sensitivity analyses, which indicated no differences in magnitude of effects. For simplicity, findings are reported for BMI in kg m−2.
Analytic sample
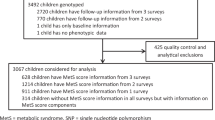
Of the 20 745 Wave I respondents, 15 701 (76%) were interviewed at Wave IV and 12 234 provided saliva with consent for banking and use in genetic studies. DNA was successfully extracted from 12 066 samples. Successful genotypes were obtained for at least 80% of the SNPs in 11 488 samples. Only individuals aged 12–21 years at either Waves II or III were eligible for this study. Among the 9129 eligible adolescents, we excluded the following participants: the twin with the fewest genotyped loci within a monozygotic twin pair (twin zygosity determined by matching 11 molecular genetic markers24, 32 or by full agreement of self-report measures, n=139); individuals of ‘other’ race (n=69), native American (n=57), pregnant (n=110) or disabled (n=47); and those with missing data for geographic region of residence (n=67), BMI (n=2), current smoking (n=1), or race/ethnicity (n=43). Our final sample consisted of 8594 adolescents (5103 EA, 1748 AfA, 1304 HA and 439 AsA) with complete data (Figure 1).

Add Health study population.
Statistical analysis
BMI–SNP association tests in Add Health were conducted using Stata software, version 12.1 (Stata Corp., College Station, TX, USA). All analyses were stratified by race/ethnicity, and results for BMI–SNP associations within each race/ethnic groups were combined and analyzed using pooled meta-analyses. We assumed an additive genetic model and used multivariable linear mixed models to regress each SNP on BMI, adjusting for age, sex, current smoking (at least one cigarette/day for 30 days), geographic region of residence and an indicator for self-reported (n=81) versus measured height and weight, and models for AfA included indicator variables for oversampling based on education level. In a sensitivity analysis, we ran all models without the participants with self-reported heights and weights (n=81) and found no difference in the magnitude and direction of estimated effects (see Supplementary Table 2). Hispanic and Asian models included indicator variables for subpopulation: Cuban, Puerto Rican, Central/South American, Mexican, other Hispanic, Chinese, Filipino and other Asian and for immigrant status: non-US born (first generation), and US born (2nd or 3rd generation). In all models, sample clustering and familial relatedness were accounted for using separate random effects for school and family. Effect estimates were combined and meta-analyzed in METAL software using the inverse s.e. weighted approach.33 For each SNP association, we evaluated heterogeneity between race/ethnic groups using Cochran’s Q. We considered evidence for heterogeneity when the χ2 P-value <0.10 or I2 index >50.34, 35
To assess generalization of results with published findings, we counted the number of SNPs with consistent direction of effect relative to published results from European populations by race/ethnicity and in the race/ethnic pooled sample. One-tailed P-values are reported based on a binomial draw with null expectation P=0.5 (random direction of effect compared with published result). We defined generalization as SNPs that were nominally significant (P<0.05) and directionally consistent with published results.
Power
Estimates of power to detect associations among each race/ethnic groups (EA: n=5103; AfA: n=1783; HA: n=1348; AsA: n=439) and all adolescents combined (n=8594) were calculated using QUANTO v1.2.4 (http://hydra.usc.edu/gxe/) and assumed additive coding, an independent sample size for each of the race/ethnic subpopulations and adolescents combined, a minor allele frequencies ranging from 0.05 to 0.5, and effect sizes from genome-wide associations identified in European descent populations from 0.05 to 0.4 kg m−2 (Supplementary Figures 1a–d).
Effect size comparisons
To compare the magnitude of the SNP and BMI effects in the EA Add Health adolescents with middle-aged adults, we used data available for EA participants of the ARIC Study. We chose the ARIC study for comparisons despite the availability of very large comparative consortia (for example, GIANT), as these studies present findings using inverse-normally transformed BMI, which is standardized and ranked and thus cannot be back-transformed to BMI units.36 Furthermore, the ARIC sample may be less heterogeneous than the GIANT consortia, which recruited across Europe and the United States, in the setting of both case–control and prospective data collection, as well as spanning the last 50 years, during rapid changes in the obesogenic environment. In contrast, the ARIC sample is a US sample that was initially measured within 6 years of our own sample (1989 versus 1995–1996), thus minimizing the potential impact of cohort effects. The other race/ethnic groups were not compared mainly because of lack of power and potentially lower LD among these groups for the SNPs selected.
ARIC
Participants
The ARIC Study is a prospective cohort study of cardiovascular disease risk in four US communities.37 Between 1987 and 1989, 11 478 EA men and women aged 45–64 years were recruited from Forsyth County, North Carolina; suburban Minneapolis, Minnesota; and Washington County, Maryland. The ARIC Study protocol was approved by the institutional review board of each participating university and written informed consent was obtained.
Genetic data
ARIC participants were genotyped using the Affymetrix Genome-Wide Human SNP Array 6.0 and the IBC chip array (Affymetrix, Santa Clara, CA, USA). Genotypes were excluded because of call rate <90%, MAF (minor allele frequency) <1%, Hardy–Weinberg equilibrium deviation <10−6, and genotype frequency different (P<10−6) from prior genotyped samples. Imputation to ∼2.5 million autosomal SNPs identified in HapMap Phase II CEU samples was performed using MACH v1.0.16. After filtering on SNPs with MAF ⩾1%, call rate ⩾95%, and Hardy–Weinberg equilibrium-P⩾10−5, 669 450 SNPs (79.5%) were used in the imputation. For the 41 SNPs used in the current study, 30 SNPs were imputed and 11 were genotyped.
Analytic sample
Participants with genetic information were included, n=9713. Of 9713, exclusions were made for scans from one problem plate of unintentional duplicates (n=7), discordance between phenotypic sex and X-chromosome analysis (n=30), race discrepancy, (n=4), genetic outliers based on average identity by state statistics (n=16) and principal components analysis using EIGENSTRAT (n=245), and suspected first‐degree relative of an included individual based on the genome-wide SNP data (n=341). After combining the genetic data and phenotypic data for individuals, we further excluded those missing current smoking (n=6), height or weight (n=8) and those with BMI <18.5 (n=70). The final analytic sample included 8986 EA adult participants.
Statistical analyses
Assuming an additive genetic model, we regressed each SNP on BMI, adjusting for age, sex, current smoking, study center and principal components for ancestry among each race group. We used Z-tests to compare BMI–SNP effect estimates (β) between Add Health adolescents (A) and ARIC adults (B) by race/ethnicity. The Z statistic:

where β=effect estimate, is asymptotically distributed as a standard normal variable.
Results
The Add Health adolescents were on average 16.4 years of age (ages 12–21 years, 47.5% male) and had a mean BMI of 23.4 kg m−2 (Table 1), ranging from 22.6 kg m−2 in AsA to 24.2 kg m−2 in AfA. Obesity ranged from 10.9% in AsA to 17.8% in AfA.
BMI–SNP associations
EA adolescents
In the Add Health EA adolescents, we observed 35 of 41 BMI–SNP effect estimates that were directionally consistent with published studies in European-descent adults (Figure 2, Table 2, and Supplementary Table 1), which is greater than expected, n=21.5, by chance (binomial test, P=2.04E-06). The six SNPs that were not directionally consistent included rs2444217 (near ADCY9), rs1424233 (near MAF), rs4771122 (near MTIF3), rs10146997 (near NRXN3), rs4929949 (near RPL27A) and rs1077393 (near NCR3). Eighteen of the 35 directionally consistent SNP–BMI associations achieved nominal significance (effect sizes of 0.22–0.71 kg m−2 BMI units per effect allele), four of these 18 (rs9939609 (near FTO), rs571312 (near MC4R), rs6548238 (near TMEM18) and rs987237 (near TFAP2B)), remained statistically significant after Bonferroni correction (P<0.002; effect sizes 0.46–0.70 kg m−2), 17 of the 18 nominally significant SNPs were previously reported to be associated with BMI and one (near PTER) with obesity in European adults.

Forty-one SNPs previously associated with obesity, BMI or central adiposity phenotypes in European-descent adults, are plotted according to directional consistency (compared with previous findings in adults of European descent) of effect estimates with BMI in each of the four race/ethnic groups within the National Longitudinal Study of Adolescent Health. BMI SNPs have shown in black and obesity SNPs shown in red. 1rs4788102, 2rs10913469, 3rs7359397, 4rs543874, 5rs571312, 6rs12970134.
AfA, HA and AsA adolescents
We observed directionally consistent effect estimates in AfA (25 SNPs), HA (30 SNPs) and AsA (24 SNPs) adolescents relative to published results from European-descent populations, with greater than expected number of nominally significant SNP associations in AfA (eight), marginal significance in AsA (five), but not HA (three) based on a binomial draw at P<0.05 (Figure 2, Table 2, and Supplementary Table 1). Of the associations that did not generalize in the AfA, HA and AsA populations, none had sufficient power (80%) to detect the previously reported effect sizes. Of note, 10 SNPs were directionally consistent across all four race/ethnic groups, with 9 previously reported to be associated with BMI (Table 2, Supplementary Table 1). Two of the BMI-associated SNPs, rs206936 (near NUDT3) and rs13107325 (near SLC39A8), were directionally inconsistent in AfA, HA and AsA adolescents.
Meta-analysis of BMI–SNP associations
In the weighted meta-analyzed sample of all adolescents, 36 SNPs were directionally consistent with the published literature for BMI and obesity; of these, 21 were nominally significant, and 8 remained significant after Bonferroni correction (P<0.002). (Supplementary Table 1). Yet we had power to detect literature reported effect sizes6, 7 in only three of these SNPs, rs9939609 (near FTO), rs6548238 (near TMEM18) and rs571312 (near MC4R). The majority of these 21 SNPs were selected for their association with BMI, the one notable exception being rs10508503 (near PTER), which was selected based on association with obesity. Heterogeneity of effect estimates (at χ2 P-value <0.1 and I2 value >50) across race/ethnic groups was observed for four of the nominally significant SNPs (near FTO, MC4R, MCP2K5, GPRC5B).
Comparison of BMI–SNP associations in adolescents and adults
In EA adolescents, effect estimates were compared with EA ARIC participants (Supplementary Table 3), who were on average aged 53.4 years. ARIC adults had a mean BMI of 27.1 kg m−2 with 22.5% prevalence of obesity.
In 41 comparisons of BMI–SNP point estimates in EA Add Health adolescents versus ARIC adults, rs6548238 (near TMEM18), rs571312 (near MC4R) and rs10508503 (near PTER), had larger effect sizes in adolescents than adults at P<0.05 (Supplementary Table 4, Figure 3). Four SNPs, rs887912 (near FANCL), rs1514175 (near TNNI3K), rs713586 (near POMC) and rs9939609 (near FTO), showed a trend (P<0.1) for larger effect sizes in adolescents compared with adults, but none showed a trend for larger effect sizes in adults compared with adolescents. Effect sizes were larger in adults compared with adolescents for rs1046997 (near NRXN3) and rs4771122 (near MTIF3), at P<0.05.

Effect sizes of BMI loci for European Americans in the Add Health adolescent cohort compared with the findings in adults. Z-test used to compare effect sizes of Add Health adolescents and ARIC adults:  . Of 41 comparisons, the SNPs displayed are those with P-value for differences <0.10, sorted from largest to smallest effect estimates in Add Health adolescents. *P<0.05 for difference, †P<0.1 for difference.
. Of 41 comparisons, the SNPs displayed are those with P-value for differences <0.10, sorted from largest to smallest effect estimates in Add Health adolescents. *P<0.05 for difference, †P<0.1 for difference.
Discussion
Although GWAS have identified several common genetic loci associated with BMI in European-descent, middle-aged adults6, 7, 8 the extent to which findings generalize to other lifecycle periods and other racial/ethnic groups is just beginning to be explored. Using ancestrally diverse, national data on adolescents, we evaluated 41 established BMI and obesity SNPs from European-descent adult studies to assess replication in EA adolescents, to measure generalization across EA, AfA, HA and AsA, and to compare effect estimates for EA adolescents to middle-aged adults. We observed evidence for generalization across all groups, especially in EA and AfA, which had the largest sample sizes. These data provide evidence for larger effect sizes in both EA adolescents compared with adults. In general, the observed effect estimates were larger, up to 3–5 times larger, indicating a stronger estimated effect on BMI in EA adolescents compared with middle-aged EA adults.
Generalization in EA adolescents
We observed 35 directionally consistent associations relative to published results in European-descent samples, of which 18 achieved nominal statistical significance (P<0.05). Only one of the four obesity SNPs achieved nominal significance. Most GWAS SNPs for BMI-related traits have replicated in EA adult populations.6, 7, 8, 30 Other periods in the life course have not been routinely interrogated, though studies in children have replicated findings for several of these same loci.8, 10, 11, 12, 13, 14, 15, 38, 39
Generalization across ancestrally diverse populations
In the meta-analyzed sample, we observed 21 of 41 nominally significant BMI–SNP associations, and 36 of 41 that were directionally consistent with published studies. Similarity in the genetic architecture of BMI across race-ethnic groups is of interest. Several of the loci are thought to have a role in the central nervous system, speficially with weight regulation. Of the 10 SNPs that were directionally consistent across all four race/ethnic groups, five are located near the genes NEGR1, MTCH2, SH2B1, MC4R and TMEM18 that are highly expressed in the brain. SH2B1 is implicated in leptin signaling,40 and disruption of MC4R and SH2B1 in mice results in hyperphagia and obesity.41, 42, 43 NEGR1 is thought to affect neuronal outgrowth in the developing brain.44 The SNP in NPC1 (rs1805081) is a nonsynonymous variant and may have implications for cholesterol trafficking in the central nervous system.45
In contrast, some findings were inconsistent across race/ethnic strata, likely due to insufficient power but also possibly due to differences in LD. For example, variants in FTO have been extensively studied,11, 14, 22, 30, 38, 46, 47, 48 and different SNPs have been highlighted, with rs1861867 most strongly associated in European-descent populations and rs8057044 in African descent populations.38 These SNPs exhibit low LD (<0.3) with each other in both the CEU and YRI HapMap Phase II populations and may exhibit LD with an unidentified causal variant. We tested one FTO variant (rs9939609) and found directional consistency with BMI in EA (P=4.16 × 10−12) and HA (not significant). No effect was detected in the AfA sample, likely because of differences in LD, nor in the AsA sample (although other Asian studies have confirmed the FTO–BMI association17, 18, 19 using the same SNP or one in high LD), likely because of the very small sample size. Therefore, additional SNPs are needed to further characterize these associations across race/ethnicity groups.
Effect size differences in adolescents versus adults
Our observation of comparatively stronger estimated genetic effects in adolescents than adults (Figure 3, Supplementary Tables 4) is supported by the literature. A study of the association between FTO and MC4R gene variants with body size in EAs from birth to age 53 years showed evidence for variation of effects that increased between birth and age 20, and then weakened during adulthood.15 A study of genetic susceptibility to obesity in European children, aged 9 years, and adolescents, aged 15 years, found effect sizes for BMI that were 1.4–2.8-fold higher in children/adolescents compared with adults for variants in/near TMEM18, SEC16B and KCTD15, but two-fold lower for the variant in/near BNDF.12 A meta-analysis comparison of BMI and obesity-related SNPs in children/adolescents versus adults in European cohorts demonstrated a larger, though not statistically significant, effect on BMI for five loci, including SEC16B, TMEM18, GNPDA2, MC4R and KCTD15; our study also identified significantly larger effect sizes for BMI in EA adolescents compared with EA adults for the SNPs near TMEM18 and MC4R.
Variants near MTIF3 and NRXN3 had larger effect sizes in EA adults compared with adolescents. The reasons for larger effect sizes during adolescence are unclear, however, we note that the adolescent MTIF3 and NRXN3 associations in EA were directionally inconsistent and non-significant. Other possible limitations include the relatively narrow age range marked by post-pubertal growth and body composition changes in Add Health, whereas the ARIC sample spanned almost 20 years (age 45–64 years), a comparatively stable period of weight change. Cohort differences between the two samples, such as the decade during which data were collected, may have contributed to observed differences. The ARIC adults matured before the obesity epidemic, whereas the Add Health cohort matured in a comparatively more obesogenic environment and may have experienced differential contributions of genetic influences on weight. Over time, and with age, the obesogenic environment may have a comparatively stronger influence on body weight than genetic factors. Finally, the Add Health sample is nationally representative, while the ARIC adults originated from four communities, which may have resulted in greater environmental homogeneity relative to Add Health. Other differences between Add Health and ARIC, including unmeasured environmental contexts, might influence BMI. These differences highlight the importance of replicating our findings in age diverse cohorts collected and analyzed with consistent sampling designs and methodology.
While our study capitalizes on an ancestrally diverse nationally representative cohort measured during a unique period of the lifecycle, there are study limitations. First, the lack of adult established obesity loci in all racial/ethnic groups is a limitation, particularly in the HA. Second, we were underpowered to detect effects for many of the subpopulations, particularly for the HA and AsA adolescents. Third, there may have been important limitations to our effect size comparisons, including contextual and sample size differences between the Add Health and ARIC samples.
Fourth, Add Health spans the period from post-puberty through young adulthood (only 6% of the girls had not attained menarche at measurement). However, most of the children were post pubertal, and therefore weight changes likely reflect changes in fat mass, Of greater relevance, however, is the fact that recent studies suggest pleiotropic effects for several loci associated with menarche and BMI.49 Without knowing which phenotype was influenced first by the genetic loci, we might diminish a true association on BMI by adjusting for menarche, thus we did not adjust for it.
Despite these limitations, our findings begin to fill a gap in the literature on the high risk period between adolescence and young adulthood and in racially/ethnically diverse samples. At the same time, these estimates can be used in future meta-analyses in these understudied race/ethnic groups. At present, our study is the only of its kind in this age group. Using these unique data, we find that many of the genetic variants known to be associated with BMI in European-descent adults have relatively larger effects during the adolescent period, with variation across race/ethnicity.
References
Flegal KM, Graubard BI, Williamson DF, Gail MH . Cause-specific excess deaths associated with underweight, overweight, and obesity. JAMA 2007; 298: 2028–2037.
Lamb MM, Ogden CL, Carroll MD, Lacher DA, Flegal KM . Association of body fat percentage with lipid concentrations in children and adolescents: United States, 1999-2004. Am J Clin Nutr 2011; 94: 877–883.
Flegal KM, Ogden CL, Yanovski JA, Freedman DS, Shepherd JA, Graubard BI et al. High adiposity and high body mass index-for-age in US children and adolescents overall and by race-ethnic group. Am J Clin Nutr 2010; 91: 1020–1026.
Gordon-Larsen P, Adair LS, Nelson MC, Popkin BM . Five-year obesity incidence in the transition period between adolescence and adulthood: the National Longitudinal Study of Adolescent Health. Am J Clin Nutr 2004; 80: 569–575.
The NS, Suchindran C, North KE, Popkin BM, Gordon-Larsen P . Association of adolescent obesity with risk of severe obesity in adulthood. JAMA 2010; 304: 2042–2047.
Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet 2010; 42: 937–948.
Thorleifsson G, Walters GB, Gudbjartsson DF, Steinthorsdottir V, Sulem P, Helgadottir A et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat Genet 2009; 41: 18–24.
Willer CJ, Speliotes EK, Loos RJ, Li S, Lindgren CM, Heid IM et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet 2009; 41: 25–34.
Meyre D, Delplanque J, Chèvre JC, Lecoeur C, Lobbens S, Gallina S et al. Genome-wide association study for early-onset and morbid adult obesity identifies three new risk loci in European populations. Nat Genet 2009; 41: 157–159.
Zhao J, Bradfield JP, Li M, Wang K, Zhang H, Kim CE et al. The role of obesity-associated loci identified in genome-wide association studies in the determination of pediatric BMI. Obesity 2009; 17: 2254–2257.
Zhao J, Grant SF . Genetics of childhood obesity. J Obes 2011; 2011: 845148.
den Hoed M, Ekelund U, Brage S, Grontved A, Zhao JH, Sharp SJ et al. Genetic susceptibility to obesity and related traits in childhood and adolescence: influence of loci identified by genome-wide association studies. Diabetes 2010; 59: 2980–2988.
Bradfield JP, Taal HR, Timpson NJ, Scherag A, Lecoeur C, Warrington NM et al. A genome-wide association meta-analysis identifies new childhood obesity loci. Nat Genet 2012; 44: 526–531.
Hallman DM, Friedel VC, Eissa MA, Boerwinkle E, Huber JC, Harrist RB et al. The association of variants in the FTO gene with longitudinal body mass index profiles in non-Hispanic white children and adolescents. Int J Obes 2012; 36: 61–68.
Hardy R, Wills AK, Wong A, Elks CE, Wareham NJ, Loos RJ et al. Life course variations in the associations between FTO and MC4R gene variants and body size. Hum Mol Genet 2010; 19: 545–552.
Elks CE, Loos RJ, Hardy R, Wills AK, Wong A, Wareham NJ et al. Adult obesity susceptibility variants are associated with greater childhood weight gain and a faster tempo of growth: the 1946 British Birth Cohort Study. Am J Clin Nutr 2012; 95: 1150–1156.
Okada Y, Kubo M, Ohmiya H, Takahashi A, Kumasaka N, Hosono N et al. Common variants at CDKAL1 and KLF9 are associated with body mass index in east Asian populations. Nat Genet 2012; 44: 302–306.
Wang J, Mei H, Chen W, Jiang Y, Sun W, Li F et al. Study of eight GWAS-identified common variants for association with obesity-related indices in Chinese children at puberty. Int J Obes 2012; 36: 542–547.
Wen W, Cho YS, Zheng W, Dorajoo R, Kato N, Qi L et al. Meta-analysis identifies common variants associated with body mass index in east Asians. Nat Genet 2012; 44: 307–311.
Ng MC, Hester JM, Wing MR, Li J, Xu J, Hicks PJ et al. Genome-wide association of BMI in African Americans. Obesity 2012; 20: 622–627.
Hester JM, Wing MR, Li J, Palmer ND, Xu J, Hicks PJ et al. Implication of European-derived adiposity loci in African Americans. Int J Obes 2012; 36: 465–473.
Wing MR, Ziegler J, Langefeld CD, Ng MC, Haffner SM, Norris JM et al. Analysis of FTO gene variants with measures of obesity and glucose homeostasis in the IRAS Family Study. Hum Genet 2009; 125: 615–626.
Fesinmeyer MD, North KE, Ritchie MD, Lim U, Franceschini N, Wilkens LR et al. Genetic risk factors for body mass index and obesity in an ethnically diverse population: results from the Population Architecture using Genomics and Epidemiology (PAGE) Study. Obesity 2012. ; e-pub ahead of print 22 June 2012; doi:10.1038/oby.2012.158.
Harris KM, Halpern CT, Smolen A, Haberstick BC . The National Longitudinal Study of Adolescent Health (Add Health) twin data. Twin Res Hum Genet 2006; 9: 988–997.
Resnick MD, Bearman PS, Blum RW, Bauman KE, Harris KM, Jones J et al. Protecting adolescents from harm. Findings from the National Longitudinal Study on Adolescent Health. JAMA 1997; 278: 823–832.
Miller WC, Ford CA, Morris M, Handcock MS, Schmitz JL, Hobbs MM et al. Prevalence of chlamydial and gonococcal infections among young adults in the United States. JAMA 2004; 291: 2229–2236.
Harris KM . An integrative approach to health. Demography 2010; 47: 1–22.
Bates LM, Acevedo-Garcia D, Alegría M, Krieger N . Immigration and generational trends in body mass index and obesity in the United States: results of the National Latino and Asian American Survey, 2002-2003. Am J Public Health 2008; 98: 70–77.
Harris KM, Perreira KM, Lee D . Obesity in the transition to adulthood: predictions across race/ethnicity, immigrant generation, and sex. Arch Pediatr Adolesc Med 2009; 163: 1022–1028.
Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 2007; 316: 889–894.
Heard-Costa NL, Zillikens MC, Monda KL, Johansson A, Harris TB, Fu M et al. NRXN3 is a novel locus for waist circumference: a genome-wide association study from the CHARGE Consortium. PLoS Genet 2009; 5: e1000539.
Team AHB In: Manhart LE (ed.). Biomarkers in Wave III of the Add Health Study 1999. pp 57.
Willer CJ, Li Y, Abecasis GR . METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010; 26: 2190–2191.
Higgins JP, Thompson SG . Quantifying heterogeneity in a meta-analysis. Stat Med 2002; 21: 1539–1558.
Huedo-Medina TB, Sánchez-Meca J, Marín-Martínez F, Botella J . Assessing heterogeneity in meta-analysis: Q statistic or I2 index? Psychol Methods 2006; 11: 193–206.
Beasley TM, Erickson S, Allison DB . Rank-based inverse normal transformations are increasingly used, but are they merited? Behav Genet 2009; 39: 580–595.
ARIC-investigators. The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. The ARIC investigators. Am J Epidemiol 1989; 129: 687–702.
Bollepalli S, Dolan LM, Deka R, Martin LJ . Association of FTO gene variants with adiposity in African-American adolescents. Obesity 2010; 18: 1959–1963.
Orkunoglu-Suer FE, Harmon BT, Gordish-Dressman H, Clarkson PM, Thompson PD, Angelopoulos TJ et al. MC4R variant is associated with BMI but not response to resistance training in young females. Obesity 2011; 19: 662–666.
Li Z, Zhou Y, Carter-Su C, Myers MG, Rui L . SH2B1 enhances leptin signaling by both Janus kinase 2 Tyr813 phosphorylation-dependent and -independent mechanisms. Mol Endocrinol 2007; 21: 2270–2281.
Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 1997; 88: 131–141.
Ren D, Zhou Y, Morris D, Li M, Li Z, Rui L et al. Neuronal SH2B1 is essential for controlling energy and glucose homeostasis. J Clin Invest 2007; 117: 397–406.
Morris DL, Cho KW, Rui L . Critical role of the Src homology 2 (SH2) domain of neuronal SH2B1 in the regulation of body weight and glucose homeostasis in mice. Endocrinology 2010; 151: 3643–3651.
Schafer M, Bräuer AU, Savaskan NE, Rathjen FG, Brümmendorf T . Neurotractin/kilon promotes neurite outgrowth and is expressed on reactive astrocytes after entorhinal cortex lesion. Mol Cell Neurosci 2005; 29: 580–590.
Ikonen E . Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol 2008; 9: 125–138.
Ng MC, Tam CH, So WY, Ho JS, Chan AW, Lee HM et al. Implication of genetic variants near NEGR1, SEC16B, TMEM18, ETV5/DGKG, GNPDA2, LIN7C/BDNF, MTCH2, BCDIN3D/FAIM2, SH2B1, FTO, MC4R, and KCTD15 with obesity and type 2 diabetes in 7705 Chinese. J Clin Endocrinol Metab 2010; 95: 2418–2425.
Cauchi S, Stutzmann F, Cavalcanti-Proença C, Durand E, Pouta A, Hartikainen AL et al. Combined effects of MC4R and FTO common genetic variants on obesity in European general populations. J Mol Med 2009; 87: 537–546.
Vasan SK, Fall T, Neville MJ, Antonisamy B, Fall CH, Geethanjali FS et al. Associations of variants in FTO and near MC4R with obesity traits in South Asian Indians. Obesity 2012. e-pub ahead of print 16 March 2012; doi:10.1038/oby.2012.164.
Elks CE, Perry JR, Sulem P, Chasman DI, Franceschini N, He C et al. Thirty new loci for age at menarche identified by a meta-analysis of genome-wide association studies. Nat Genet 2010; 42: 1077–1085.
Cole TJ, Bellizzi MC, Flegal KM, Dietz WH . Establishing a standard definition for child overweight and obesity worldwide: international survey. BMJ 2000; 320: 1240–1243.
NHLBI Obesity Education Initiative. Clinical guidelines on the identification, evaluation, and treatment of overweight and obesity in adults: the Evidence Report. US Department of Health and Human Services, Public Health Service, National Institutes of Health, National Heart, Lung, and Blood Institute: Bethesda, MD, 1998.
Acknowledgements
This work was funded by National Institutes of Health grant R01HD057194. This research uses data from Add Health, a program project directed by Kathleen Mullan Harris and designed by J Richard Udry, Peter S Bearman and Kathleen Mullan Harris at the University of North Carolina at Chapel Hill, and funded by grant P01-HD31921 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development, with cooperative funding from 23 other federal agencies and foundations. Special acknowledgment is due Ronald R Rindfuss and Barbara Entwisle for assistance in the original design. Information on how to obtain the Add Health data files is available on the Add Health website (http://www.cpc.unc.edu/addhealth). No direct support was received from grant P01-HD31921 for this analysis. We thank Amy Perou of the BioSpecimen Processing facility and Amanda Floyd Beaty and Michael Andre of the Mammalian Genotyping Core at University of North Carolina at Chapel Hill. We are grateful to the Carolina Population Center (R24 HD050924) for general support. The ARIC Study is carried out as a collaborative study supported by National Heart, Lung and Blood Institute contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C and HHSN268201100012C), R01HL087641, R01HL59367 and R01HL086694; National Human Genome Research Institute contract U01HG004402; and National Institutes of Health contract HHSN268200625226C. The authors thank the staff and participants of the ARIC study for their important contributions. Infrastructure was partly supported by Grant Number UL1RR025005, a component of the National Institutes of Health and NIH Roadmap for Medical Research.
Author contributions
PGL, KEN, EML, LAL, KLM and MG contributed to study design, MG and ASR performed the data analysis and PGL, KEN, EML, LAL, KLM, KMY and MG contributed to writing the manuscript, all other authors provided critical evaluation of the manuscript. MG, KEN and PGL had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Nutrition and Diabetes website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Graff, M., North, K., Mohlke, K. et al. Estimation of genetic effects on BMI during adolescence in an ethnically diverse cohort: The National Longitudinal Study of Adolescent Health. Nutr & Diabetes 2, e47 (2012). https://doi.org/10.1038/nutd.2012.20
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nutd.2012.20
Keywords
This article is cited by
-
BMI loci and longitudinal BMI from adolescence to young adulthood in an ethnically diverse cohort
International Journal of Obesity (2017)
-
Interaction of smoking and obesity susceptibility loci on adolescent BMI: The National Longitudinal Study of Adolescent to Adult Health
BMC Genetics (2015)
-
The bigger picture of FTO—the first GWAS-identified obesity gene
Nature Reviews Endocrinology (2014)
-
Adipose tissue depot specific promoter methylation of TMEM18
Journal of Molecular Medicine (2014)
-
Generalization of adiposity genetic loci to US Hispanic women
Nutrition & Diabetes (2013)
