
Abstract
Little is known about the extent to which individual microRNAs (miRNAs) regulate common processes of tumor biology across diverse cancer types. Using molecular profiles of >3,000 tumors from 11 human cancer types in The Cancer Genome Atlas, we systematically analyzed expression of miRNAs and mRNAs across cancer types to infer recurrent cancer-associated miRNA-target relationships. As we expected, the inferred relationships were consistent with sequence-based predictions and published data from miRNA perturbation experiments. Notably, miRNAs with recurrent target relationships were frequently regulated by genetic and epigenetic alterations across the studied cancer types. We also identify new examples of miRNAs that coordinately regulate cancer pathways, including the miR-29 family, which recurrently regulates active DNA demethylation pathway members TET1 and TDG. The online resource http://cancerminer.org allows exploration and prioritization of miRNA-target interactions that potentially regulate tumorigenesis.
Similar content being viewed by others

Main
miRNAs are small RNAs that regulate gene expression by binding partially complementary sites in target mRNAs1. Dysregulation of miRNAs can contribute to tumor formation and progression2,3. For example, genetic and epigenetic alterations target miRNA loci in cancer2,3, tumor tissues show distinctive miRNA expression signatures compared with normal tissue4,5, and studies in mice show that malignant tumors can form by and depend on dysregulation of a single miRNA6. Notably, miRNAs can be both antagonized and mimicked by therapeutic oligonucleotides, potentially offering new targeted approaches to cancer treatment7.
Individual miRNAs can target hundreds or thousands of mRNAs on the basis of sequence complementarity, but a substantial fraction of these predicted interactions may depend on cell type and context1 and on the binding of additional cofactors8. Furthermore, an even smaller subset of target interactions is expected to affect tumor development and progression in vivo. It is therefore challenging to nominate functionally relevant target genes and pathways on the basis of dysregulated miRNA expression profiles in tumor samples. Common approaches to studying miRNA target genes and function in cancer involve experimental perturbation of miRNA expression in cell lines and mouse models of cancer2,3. Although these model systems have yielded important mechanistic insights into cancer cell biology, they may not fully capture the complexity of tumorigenesis in patients9. More recently, comprehensive multidimensional genetic and molecular profiles of large tumor populations generated by research consortia such as The Cancer Genome Atlas (TCGA) have enabled integrated analysis of genetic and molecular alterations associated with individual human cancer types10,11. These data sets enable tracking of miRNA and mRNA expression across a population of tumors. As miRNAs commonly destabilize and degrade their target mRNAs12,13, we expect that miRNAs have inverse expression relationships with their target mRNAs. Variations of this principle have been used to predict miRNA-target interactions on the basis of miRNA and mRNA expression profiles14,15. Such approaches have also shown functionally relevant miRNA-target interactions in individual cancer types (for example, in TCGA glioblastoma multiforme16,17,18 and serous ovarian carcinoma data sets19,20). However, systematic studies that evaluate miRNA-mRNA associations across multiple cancer types are needed to explore the hypothesis that individual miRNAs regulate common processes of tumorigenesis that are independent of organ or tissue of origin.
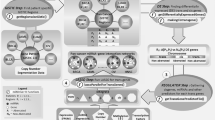
We developed a method and statistical score, the association recurrence (REC) score, that uses miRNA and mRNA expression profiles across many cancer types to infer miRNA-target interactions that could be active and functional in many different cancer types (Fig. 1). Using this approach, we inferred recurrent cancer-associated miRNA-target relationships from miRNA and mRNA expression profiles of >3,000 tumors and 11 cancer types profiled by TCGA. We further analyzed these recurrent target relationships using sequence- and conservation-based predictions, experimentally validated target interactions curated from the literature, and published data from miRNA perturbation experiments. We derived a high-confidence pan-cancer network of 143 recurrent target relationships, and we show that these relationships include new examples of miRNAs that are likely to coordinately regulate multiple members of pathways across many cancer types. All predictions are available through an online resource, http://cancerminer.org, which allows exploration and visualization of candidate miRNA-target interactions in TCGA data.

Statistical method used to evaluate recurrence of miRNA-mRNA expression association across cancer types. In individual cancer types, pairwise miRNA-mRNA relationships are evaluated using a multivariate linear model, which also factors in variation (noise) in mRNA expression induced by changes in DNA copy number and promoter methylation at the mRNA gene locus. Associations are rank transformed in individual cancer types, and the method subsequently evaluates the null hypothesis that no association exists between the miRNA-mRNA pair in all cancer types.
Results
Inferring miRNA targets in individual cancer types
We used comprehensive molecular data sets for ten epithelial cancer types and glioblastoma multiforme in TCGA (Table 1; this included 11 TCGA cancer types, but colon and rectal cancer data sets were merged). Each cancer type had miRNA and mRNA expression profiles measured for 94−671 tumor samples (patients), and the combined data set comprised 3,290 samples. miRNA expression was profiled by microarrays or small RNA sequencing, and mRNA expression by microarrays or mRNA sequencing (depending on cancer type; see Online Methods).
Our method first evaluates expression relationships of miRNAs and mRNAs in individual cancer types. For each miRNA-mRNA pair, we measured the association between miRNA and mRNA expression across the set of tumors using a multivariate linear model that also factors in variation (noise) in mRNA expression induced by changes in DNA copy number and promoter methylation at the mRNA gene locus (Fig. 1 and Online Methods). This multivariate linear model could more accurately evaluate miRNA-mRNA expression associations in the presence of DNA copy-number and promoter methylation aberrations that extensively influence mRNA expression (see Supplementary Fig. 1 for examples).
In all individual cancer types, we found that miRNA-mRNA pairs with negative expression association had markedly more predicted miRNA-target interactions (determined by intersection of miRanda and TargetScan predictions21,22, using thresholds of −0.5 and −0.2, respectively) compared with weakly or positively associated pairs (Fig. 2a and Supplementary Fig. 2). Using the same approach, all cancer types were significantly enriched for predicted target interactions in the percentile of pairs with strongest negative association (P < 1 × 10−20 in each cancer type, two-tailed Fisher's exact test, n ~ 15,000 in each cancer type). Consistent with earlier analyses of miRNA target determinants, negatively associated pairs were enriched in predicted target interactions with high repressive efficacy and in binding sites confined to mRNA 3′ untranslated regions (UTRs; Fig. 2b). Together these observations indicate that miRNA and mRNA expression associations can be used to infer probable active and functional target interactions in tumors of all individual cancer types.

(a) Enrichment of predicted miRNA-target interactions as a function of miRNA-mRNA expression association in the ten cancer types (using 100 equally sized bins; cancer types are color coded as in Fig. 1). (b) Enrichment for predicted target interactions in the percentile of miRNA-mRNA pairs with strongest negative association, evaluated using different thresholds for miRNA target prediction methods: miRanda-miRSVR score (−0.15 versus −1.2), TargetScan context score (−0.1 versus −0.45) and presence of heptamer in mRNA 5′ UTR, coding sequence and 3′ UTR. Enrichment was evaluated using Fisher's exact test. (c) The top ten inferred recurring negative miRNA-mRNA associations. Left, inferred association rank (negative to positive) in each cancer type. Predicted target interactions with bars corresponding to (absolute) scores of three target prediction methods: miRanda-miRSVR, TargetScan context score and TargetScan conservation. (d) Results are available at http://cancerminer.org.
Recurrence of target associations across cancer types
To explore the hypothesis that individual miRNA-target relationships are active in multiple cancer types and may regulate common cancer traits, we developed a method and rank-based statistical score, the REC score. The method ranks miRNA-mRNA expression associations in the context of miRNA and cancer type and evaluates the null hypothesis that no association exists between the miRNA-mRNA pair in all cancer types (Fig. 1 and Online Methods). The rank-based approach ensures that individual cancer types are weighted equally, and limits bias from cancer data sets with large sample sizes or from strong associations measured in only a single cancer type. Furthermore, the REC statistic allows different types of cross-cancer relationships to achieve high scores: a miRNA-mRNA pair with very strong association in only four cancer types (let-7b:LIN28B, REC = −6.76) and a pair with less strong but consistent association in all cancer types (miR-21:PDCD4, REC = −6.49) may each achieve a high REC score.
We computed REC scores for all miRNA-mRNA pairs in which the miRNA and mRNA were expressed simultaneously in at least five of the ten cancer types. Of the top ten pairs with the strongest negative REC scores, eight had evolutionarily conserved target interactions or target interactions predicted by both miRanda or TargetScan (Fig. 2c). At least two of the top ten target interactions have previously been studied and are likely to be functionally relevant in a cancer context. A target interaction between miR-18a, a member of the mir-17-92 cluster, and the transcription factor ZBTB4 has been reported in breast and prostate cancer23,24. Our analysis suggests that this interaction could be functionally relevant in all analyzed cancer types. The target relationship between miR-141, a member of the miR-200 family, and ZEB1 has been widely studied in many cancer types and is a critical component of the epithelial-mesenchymal transition25. Similarly, the other recurring miRNA-mRNA relationships in the top ten might represent unappreciated functional miRNA-target relationships with a general role in tumorigenesis.
We present all predictions in an online resource that allows rapid exploration and visualization of candidate miRNA-target interactions in TCGA cancer types. The user may query inferred target relationships using an miRNA, gene or pathway identifier, and relationships can be scored for recurrence across all or selected subsets of cancer types (Fig. 2d).
Global analysis of interactions using public data sets
To further analyze whether recurrent pan-cancer miRNA-mRNA associations capture miRNA regulatory relationships, we evaluated the extent to which the REC score could predict mRNA expression changes induced by experimental perturbation of miRNAs in vitro. For six miRNAs with many strong negative associations (miR-106b, miR-29, miR-30d, miR-200b, miR-16 and miR-21), we obtained public data sets of mRNA expression changes after miRNA perturbation. These data sets captured both miRNA inhibition and overexpression experiments, and were all done in cancer cell lines (see Online Methods). For each miRNA, we defined, independent of sequence-based predictions, a set of putative target mRNAs using a set slightly less conservative REC score threshold (REC < −5.7, corresponding to a false discovery rate (FDR) < 0.001). In all analyzed miRNA perturbation experiments, we found that these REC target mRNAs were significantly downregulated or upregulated after miRNA overexpression or inhibition, respectively (Fig. 3, range of P values: 0.06–1.9 × 10−13, one-tailed Wilcoxon's rank-sum test, 7 < n < 179), consistent with the hypothesis that the recurrent pan-cancer miRNA-mRNA associations capture miRNA regulatory relationships.

For six miRNAs in the inferred pan-cancer network (miR-106b, miR-29c, miR-30d, miR-200b, miR-16 and miR-21), we obtained public data sets measuring mRNA expression changes after miRNA perturbation (inhibition or overexpression) in different cancer cell lines. In each data set, we compared the distribution of expression changes for inferred target mRNAs (REC < −5.7, FDR < 0.001) with expression changes for all other mRNAs measured in the given experiment (Wilcoxon's rank-sum test, one-tailed).
A pan-cancer network of recurring miRNA-target interactions
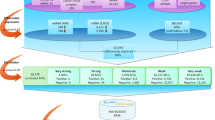
We extracted miRNA-mRNA pairs with strong negative REC score (REC < −6.2, FDR < 2 × 10−4, 4,584 pairs with less than one estimated false positive) and evidence for target interaction as predicted by miRanda (score < −0.5), TargetScan (context score < −0.2) and evolutionary conservation (TargetScan probability of conserved targeting, PCT, >0.5). These thresholds were chosen to obtain a high-confidence list of candidate miRNA-target interactions with possible functional roles across a range of cancer types. The combination of the REC score and target prediction filters yielded 143 miRNA-mRNA pairs (Fig. 4a), significantly more than was expected by chance (P = 3.1 × 10−85, two-tailed binomial test, k = 143, n = 4,584, r = 3.4 × 10−3 = 22,589 predicted targets / 6,642,349 total pairs), consistent with the hypothesis that the REC score can be used to augment sequence-based miRNA-target predictions and infer functionally relevant target interactions in vivo. These 143 putative recurring target interactions formed a network of 40 evolutionarily conserved miRNAs and 72 target mRNAs (Fig. 4b and Supplementary Table 1). At least 61 of the 143 putative target interactions have experimental support, and 23 interactions (comprising 16 miRNAs and 8 genes) have functional relevance in cancer on the basis of earlier studies (Supplementary Table 2). Interactions with strong functional evidence include pairs such as let-7b:LIN28B, miR-21:PDCD4, miR-16:RECK, miR-19a:ZBTB4 and miR-106:TGFBR2, and the interactions between the miR-200 family and ZEB1, ZEB2 and ZFPM2. The network also showed several possible and less studied target interactions with genes frequently studied in cancer, such as those encoding estrogen receptor α (miR-18a:ESR1), BLIMP-1 (miR-30c:PRDM1) and Janus kinase 1 (miR-106:JAK1).

(a) Pan-cancer network defined by intersection of miRNA-mRNA pairs with strong negative REC scores and strong evidence for conservation-based target interaction. (b) Inferred pan-cancer network comprising 143 putative target interactions between 40 evolutionarily conserved miRNAs and 72 target mRNAs. Edge width represents strength of the REC score for a given miRNA-mRNA pair, and miRNAs are color coded by seed family relationships (singletons in white). (c) Genetic and epigenetic alterations regulating miRNAs. For the 40 miRNAs in the pan-cancer network and all other miRNAs also expressed across all the studied cancer types (n = 180), we estimated the extent (third highest R2 measured in the ten cancer types) to which changes in copy number (left) or promoter DNA methylation (right) at the miRNA locus could explain the variation in miRNA expression.
In summary, these results suggest that the REC score, in combination with sequence- and conservation-based predicted target interactions, can be used to infer candidate target interactions with functional roles across many cancer types.
Genetic and epigenetic alterations regulating miRNAs
We explored the possibility that a subset of miRNAs, and thereby also target mRNAs, in the inferred pan-cancer network could be regulated by somatic genetic or epigenetic alterations, a common property of cancer driver genes. We first considered DNA copy-number alterations targeting miRNA loci. For each miRNA, we estimated the extent to which changes in DNA copy number at the miRNA locus could explain the variation in miRNA expression measured for a given cancer type (R2, Supplementary Table 3). We then compared these copy-number correlations (selecting the third highest R2 in the ten cancer types) measured for the 40 miRNAs in the pan-cancer network with correlations for all other miRNAs also expressed in the studied cancer types (n = 180). This test showed that miRNAs in the pan-cancer network were more often regulated by DNA copy-number alterations across the different cancer types (P = 1.2 × 10−8, two-tailed Wilcoxon rank-sum test, n = 40 versus 180; Fig. 4c), suggesting that dysregulation of these particular miRNAs and target mRNAs could be under clonal selection in multiple cancer types. For example, mir-30d is encoded in a frequently amplified region (8q24, ~7 megabases (Mb) from MYC), and its expression was strongly regulated by DNA copy-number alterations in breast, ovarian and bladder carcinoma (R2 of 0.41, 0.31 and 0.25, respectively).
We then analyzed the influence of promoter DNA methylation on transcription of miRNA loci (Supplementary Table 3). A similar statistical approach showed that changes in promoter DNA methylation more often influenced expression of miRNAs in the pan-cancer network than other expressed miRNAs (P = 3.6 × 10−5, Wilcoxon rank-sum test; Fig. 4c). The miR-200 family members, which are encoded at two different loci (1p36 and 12p13), showed the most marked evidence for regulation by promoter DNA methylation across multiple cancer types. Expression of miR-200b (1p36) and miR-200c (12p13) was strongly correlated with DNA methylation (R2 > 0.2) in six cancer types (LUSC, BRCA, UCEC, BLCA, HNSC and LUAD; defined in Table 1), and changes in DNA methylation could explain >50% of expression variance of miR-200b (R2 = 0.50) and miR-200c (R2 = 0.67) in bladder cancer. In summary, these data are consistent with the hypothesis that the inferred recurrent miRNA-target relationships have a role in tumorigenesis of many different cancer types.
miR-106 family modulation of TGF-β signaling
Several miRNA families that have been widely studied in cancer were represented by many putative target interactions in the inferred pan-cancer network (such as the miR-200, miR-30, miR-29 and miR-106 families), and we hypothesized that selection for miRNA dysregulation would be particularly advantageous to tumors when miRNAs coordinately regulate multiple components of a tumorigenic pathway or process. The miR-106 family of miRNAs was represented with several putative target relationships in the network. At least two of these targets, TGFBR2 and DAB2, encode known components of the transforming growth factor (TGF)-β signaling pathway (TGF-β type II receptor and disabled homolog 2, respectively), and our analysis shows consistent negative associations among miR-106 family members and the two pathway components in all cancer types except for colorectal cancer (Fig. 5a). Furthermore, members of the miR-106 family directly target TGFBR2 and attenuate TGF-β signaling in cancer cells5,26,27. The function of TGF-β signaling may be context dependent during tumorigenesis by inhibiting cell growth at early tumor stages and promoting tumor progressive processes at later stages28. Epigenetic silencing of DAB2, which encodes an adaptor protein that facilitates interaction of Smad proteins with the activated TGF-β receptor complex29, is one mechanism by which cancer cells can switch TGF-β signaling from growth suppressive to tumor progressive30.

(a) Association between miR-106 family members and predicted target genes TGFBR2 and DAB2 across cancer types. Cancer types are color coded as in Figure 1, and REC scores are listed for each pair. (b) The relationship between miR-106b expression and TGFBR2 and DAB2 mRNA expression in tumor and representative normal samples in four different cancer types. (c) Endometrioid tumors have a recurrent focal genomic amplification spanning the mir-106b locus. Copy-number alterations in the region for 18 samples with focal amplification of the mir-106b locus. Samples are sorted by copy-number amplification level. (d) Expression of miR-106b in endometrioid tumors with diploid mir-106b and tumors with focal mir-106b amplification. (e) Expression of miR-106b and TGFBR2 in endometrioid tumors; tumor samples with focal mir-106b amplification are highlighted, and expression in normal samples is included for comparison.
The miR-106 family members are encoded at three different genomic loci (represented by miR-17, miR-106a and miR-106b in Fig. 5a), yet miRNAs from each of these loci frequently showed negative association with TGBFBR2 and DAB2 expression in the same cancer types, indicating transcriptional or post-transcriptional co-regulation of the three miR-106 loci. miR-106b showed the strongest negative association with the two target genes across cancer types, and a comparison of tumor and representative normal samples showed that miR-106b was generally upregulated, whereas TGFBR2 and DAB2 were downregulated, in tumors of most cancer types (Fig. 5b). We analyzed DNA copy-number alterations targeting miRNA loci and found that mir-106b showed moderate and consistent regulation by copy-number alterations across most cancer types (R2 > 0.08 in nine of ten cancer types). Although cases of significantly recurring focal alterations targeting miRNA loci were generally rare (as inferred by the Gistic algorithm31), we identified a significantly recurring focal amplification targeting mir-106b in endometrial cancer (FDR < 0.18, Gistic; Fig. 5c). In endometrioid tumor samples for which both DNA copy-number and miRNA expression profiles were available, 4% (18 of 479) of samples had evidence of focal chromosomal amplification of the mir-106b locus, and these tumors showed significant miR-106b upregulation compared with diploid mir-106b tumors (two-fold on average, P = 1.9 × 10−5, two-tailed Wilcoxon rank-sum test, n = 16; Fig. 5d). The focally amplified region contained six additional genes, but only the host gene encoding the intronic mir-106b, MCM7, and the neighboring gene, COPS6, showed consistent mRNA expression upregulation in amplified samples (P = 3.0 × 10−5 and 2.0 × 10−5, respectively, two-tailed Wilcoxon rank-sum test, n = 16). Finally, the tumors with focal amplification and overexpression of miR-106b also tended to have lower mRNA levels of DAB2 and TGFBR2 when compared with tumors lacking focal miR-106b amplification (Fig. 5e). In summary, these data suggest that the miR-106 family targets and modulates the TGF-β pathway at multiple levels in many cancer types.
miR-29 regulates active DNA demethylation pathway
The miR-29 family had multiple inferred target interactions in the pan-cancer network, and two of these genes, TET1 and TDG, encode critical components of the active DNA demethylation pathway in mammals32,33,34,35. In this pathway, TET proteins recognize and successively oxidize methylated cytosine nucleotides, and thymine DNA glycosylase (TDG) subsequently converts these modified bases to unmethylated cytosine through base-excision repair (Fig. 6a). We observed a very strong inverse correlation between miR-29a and TDG across all cancer types except kidney cancer, and TET1 expression was strongly negatively correlated with miR-29 family members in all cancer types (Fig. 6a). Furthermore, two earlier studies provide experimental support for the direct miR-29 target interaction with TET1 (ref. 36) and TDG37 in cancer cells. The three miR-29 family miRNAs are encoded at two different genomic loci, yet miRNAs from each of these loci (miR-29a and miR-29c) frequently showed anticorrelation with TET1 and TDG in the same cancer types (Fig. 6a), suggesting strong co-regulation of the miR-29 loci. miR-29a was generally downregulated, and TET1 and TDG were generally upregulated, in tumors of most cancer types as compared with representative normal samples (Fig. 6b).

(a) Association between the three miR-29 family members and predicted target genes TET1 and TDG across cancer types. Cancer types are color coded as in Figure 1a, and REC scores are listed for each pair. (b) Relationship between miR-29a expression and TET1 and TDG expression in tumor and representative normal samples in four different cancer types. (c) Global association between TDG expression and gene promoter hypomethylation. In each cancer type, we computed the fraction of gene promoters that were hypomethylated with TDG overexpression among the top 100 promoters showing strongest correlation (Spearman) of DNA methylation and TDG mRNA expression. The average fraction of TDG-associated hypomethylated gene promoters across all cancer types was compared with an empirical null distribution computed from 1,000 sample permutations of the DNA methylation data set in each cancer type.
Given the observation that TDG and TET1 were probably targeted and coordinately upregulated by miR-29 family downregulation in many cancer types, we hypothesized that the upregulation of these target genes is associated with patterns of DNA demethylation in the tumors. To test this hypothesis, we identified gene promoters with strong correlation (positive or negative Spearman correlation coefficient) of DNA methylation and TDG mRNA expression across tumor samples. In nine of ten cancer types we found that the majority of TDG-associated promoters (top 100) were hypomethylated with TDG mRNA upregulation (Fig. 6c), and the average fraction (0.85) of hypomethylated promoters across all cancer types was significantly higher than expected by chance (P < 0.001, sample permutation test, n = 1,000; Fig. 6c). These data are consistent with earlier observations that TDG and TET1 regulate active DNA demethylation, and they support a functional role for the miR-29 family as a possible master regulator of this process in many cancer types.
miR-29b and NREP form a double-negative feedback loop
The pan-cancer network included known examples of regulatory miRNA-mRNA double-negative feedback loops, such as let-7b:LIN28, ref. 38, and miR-200:ZEB1 (ref. 25). We hypothesized that miR-29 family expression is regulated by such a relationship with NREP, which showed the strongest recurrent negative association of all mRNAs; we observed strong anticorrelation of miR-29b and NREP expression in all cancer types studied (REC = −19.6; Fig. 7a). miR-29b had a single evolutionarily conserved and unusually highly complementary predicted target site in the NREP 3′ UTR, with base pairing potential for 20 of 23 miRNA bases (Fig. 7a). miR-29a and miR-29c also showed extensive complementarity with the NREP target site (18–19 bases with pairing), and both of these miRNAs also had strong recurring negative association with NREP expression (REC < −8.4). NREP was generally upregulated in tumors compared with normal samples, but unlike miR-29a, miR-29b showed less consistent downregulation in tumor samples (Fig. 7b). We experimentally tested the putative target interaction between miR-29b and NREP in cancer cell lines. Overexpression of miR-29b caused at least 40% reduction of NREP mRNA expression in HeLa and U251 glioma cell lines relative to experiments with control siRNAs (P = 0.03 and P = 0.07 respectively, one-tailed t-test, ntreatment = 2, ncontrol = 4, mean ± range of two measurements for treatment and mean ± s.e.m. for control groups; Fig. 7c). Inhibition of miR-29b expression using antisense oligonucleotides resulted in less potent but consistent upregulation of NREP mRNA expression across the two cell lines relative to experiments with control antisense oligonucleotides (P = 0.03 and P = 0.2 respectively, one-tailed t-test, ntreatment = 2, ncontrol = 4, mean ± range of two measurements for treatment and mean ± s.e.m. for control groups; Fig. 7d). These data strongly suggest that miR-29b targets and destabilizes NREP mRNA. NREP encodes a small protein (P311, 68 amino acids) that is associated with wound healing and glioma migration39,40 but whose specific biological function is largely unknown. Knockdown of NREP mRNA expression (using two different small interfering RNAs (siRNAs), yielding 35–70% NREP mRNA reduction) led to strong (1.6- to 2.8-fold) upregulation of miR-29b expression in the two cancer cell lines (P = 0.002 and P = 0.02, respectively, one-tailed t-test, ntreatment = 4, ncontrol = 4, mean ± s.e.m.; Fig. 7e), suggesting that NREP could directly or indirectly repress miR-29b expression. In summary, these data indicate that miR-29b and NREP expression could in part be regulated through a novel double-negative feedback loop that might also involve the other miR-29 family members, and this could provide conditions for a bistable system balancing miR-29 activity and active DNA demethylation (Fig. 7f).

(a) Association between miR-29b and NREP across cancer types. Bottom, conserved predicted miR-29 target site in NREP, Watson-Crick and wobble (G•U) base pairs are highlighted. (b) Relationship between miR-29b and NREP expression in tumor and representative normal samples in four different cancer types. (c) NREP mRNA expression 24 h after transfection with miR-29b mimic (n = 2 biological replicates, mean ± range) and two different control hairpins (n = 4 biological replicates, mean ± s.e.m.) in cancer cell lines. (d) Relative NREP mRNA expression 24 h after transfection with miR-29b locked nucleic acid (LNA) anti-miR (n = 2, mean ± range) and two control LNAs (n = 4 biological replicates, mean ± s.e.m.). (e) miR-29b expression 24 h after transfection with two different NREP siRNAs (n = 4 biological replicates, mean ± s.e.m.) and two different random control siRNAs (n = 4 biological replicates, mean ± s.e.m.). (f) Putative double-negative feedback loop between the miR-29 family of miRNAs and NREP, which is predicted to impact the active DNA demethylation pathway.
Discussion
In this study, we demonstrate that miRNA-mRNA expression covariation in patient tumors can augment sequence-based miRNA target predictions to infer probable active and functional miRNA-target interactions in vivo. We used this observation to develop a robust rank-based statistical approach that infers recurrent miRNA-target relationships across multiple cancer types. By applying this method to transcriptomes of >3,000 tumors in 11 different cancer types, we have inferred a pan-cancer network of 143 evolutionarily conserved miRNA-target interactions, comprising 40 miRNAs and 72 target mRNAs. These candidate interactions show strong evidence of regulatory activity across many cancer types, and miRNAs in the network were more likely to be dysregulated by genetic and epigenetic alterations than were other miRNAs also expressed in the studied cancer types, consistent with the hypothesis that these interactions could be implicated in tumorigenesis. Furthermore, several miRNA families that have been widely studied in cancer were represented by many target interactions in the pan-cancer network, and there is functional evidence for at least 23 interactions from earlier experimental studies in cancer.
The association patterns we observe between miRNA and target mRNA expression may reflect a trace left on target mRNA expression by perturbation of miRNA expression through multiple and varying genetic, epigenetic and regulatory alterations across the set of tumors. We acknowledge that our approach cannot be used to infer target mRNAs for miRNAs with very low expression in tumors because variation in expression for such miRNAs would in most cases not have a strong impact on target mRNA expression. Furthermore, some miRNA-target interactions primarily influence mRNA translation efficiency, and our approach may miss such interactions if there is not an associated change in mRNA stability. Our pan-cancer network of candidate miRNA-target relationships also sacrifices sensitivity in favor of specificity by applying stringent sequence- and conservation-based filters, and it may also miss interactions that are functional only in a few cancer types. However, our online resource allows rapid exploration and visualization of any miRNA-mRNA association independent of sequence-based filters, and it can also explore relationships specific to individual cancer types.
Our analysis highlights at least two cases in which miRNA families are predicted to coordinately target and regulate multiple members of a cancer-related pathway across many cancer types. In the first case, our method predicts that the miR-106 family directly targets and regulates TGFBR2 and DAB2, two genes encoding components of the TGF-β signaling pathway. Consistent with the hypothesis that miR-106 miRNAs are oncogenic, we identified a novel recurring focal amplification targeting the mir-106b loci in endometrial cancer, and tumor samples with focally amplified mir-106b showed significant miR-106b upregulation in combination with TGFBR2 and DAB2 downregulation. Although earlier studies have shown that members of the miR-106 family directly target TGFBR2 and attenuate TGF-β signaling in cancer cells5,26,27, DAB2 has not been reported as a functional target of the miR-106 family. Moreover, our analysis suggests that all three miR-106 family loci, and in particular mir-106b (at 7q), contribute to TGFBR2 and DAB2 mRNA repression in vivo. In summary, these results suggest that activation of miR-106 family expression is a potent mechanism by which cancer cells can target the TGF-β pathway at multiple levels to switch TGF-β signaling from growth suppressive to tumor progressive.
We also identified a strong recurring negative correlation between members of the miR-29 family and two genes encoding recently discovered core components of the active DNA demethylation pathway, TET1 and TDG32,33,34,35. Active DNA demethylation is important for embryonic development and tissue differentiation41. Although somatic mutations in genes encoding TET protein family members (TET1 and TET2) have been reported in various hematologic malignancies42, it is currently unknown to what extent dysregulation of active DNA demethylation pathways has a general role in cancer development. Two studies experimentally support direct miR-29 target interaction with TET1 (ref. 36) and TDG37 in acute myeloid leukemia and nasopharyngeal carcinoma, respectively. Furthermore, miR-29 miRNAs directly regulate expression of TDG and TET proteins with downstream effects on DNA 5-hydroxymethylcytosine (5hmC) levels in noncancer cells43. In this study we show that both these genes are probably potent miR-29 targets in a wide range of cancer types, suggesting that miR-29 dysregulation may have profound consequences for active DNA demethylation processes in cancer. Additionally, miR-29 miRNAs target genes encoding DNA methyltransferases (DNMT3A and DNMT3B) in cancer44,45 (as supported by our analysis, for example, miR-29a:DNMT3A, REC = −4.36), and in combination these data suggest a model in which miR-29 dysregulation in cancer induces a phenotype of DNA methylation instability that could facilitate tumorigenesis. Our analysis also shows that miR-29 dysregulation in tumors cannot generally be attributed to changes in DNA copy number or promoter methylation at the two miR-29 loci. Instead, we found that the top recurring miRNA-mRNA association in our analysis, miR-29b:NREP, represents a novel double-negative feedback loop that could impose a bistable system for miR-29 regulatory activity and active DNA demethylation activity.
Finally, we present our predictions in an online resource, http://cancerminer.org. The resource will continuously evolve as the TCGA consortium profiles additional cancer types, and we think these in vivo miRNA target predictions will be important for future efforts to unravel the role of miRNAs in tumorigenesis and for the design of miRNA-targeted therapeutics in human cancers.
Methods
Statistical evaluation of miRNA-mRNA association.
To avoid measuring associations related to cis regulation of neighboring miRNAs and mRNAs (for example through regional epigenetic regulation or DNA copy-number aberrations), we evaluated expression association only for pairs of miRNAs and mRNAs that were on different chromosomes or that were >10 Mb apart on the same chromosome. Alterations in gene DNA copy number and promoter DNA methylation often alter the mRNA expression of a given gene and may introduce noise into the evaluation of a possible post-transcriptional regulatory interaction between a given pair of miRNA μ and mRNA j. To account for such effects, we used a multivariate linear regression model in which mRNA j expression (log2) changes as a linear function of DNA copy number (log2 tumor/normal ratio), DNA methylation (beta value, [0,1]) and miRNA μ expression (log2) across tumor samples of a given cancer type (see Supplementary Note for details).
To evaluate the recurrence of a given miRNA-mRNA association across multiple cancer types, we had to combine the associations measured in each individual cancer data set. The P value computed for individual cancer types using the linear regression model above might strongly bias associations found in single studies with large sample sizes. We also observed that the distribution of associations found for individual ubiquitously and highly expressed miRNAs varied notably among different cancer studies. This could, for example, be due to study-dependent confounding effects such as differences in tumor heterogeneity between cancer types or the purity of tumor samples used for a given study. To account for these types of bias, we used a rank-based statistic to evaluate the relative strength of associations in the context of a specific miRNA and cancer type, and we evaluated the null hypothesis that that no negative association exists between miRNA μ and mRNA j across all n cancer types (see Supplementary Note for details).
TCGA data.
All miRNA expression data sets were obtained from the TCGA open access data directory (https://tcga-data.nci.nih.gov/tcgafiles/ftp_auth/distro_ftpusers/anonymous/tumor/, December 2012). miRNA expression was profiled by Agilent microarrays in the GBM and OVA studies, and by small RNA sequencing in the remaining studies. For microarray data sets, TCGA level 1 microarray expression data were processed and normalized using the AgiMicroRna R package (using between-array quantile normalization)46. For miRNA sequencing data sets, miRNA-mapped reads (level 3) were used to quantify miRNA expression by computing the ratio of mature miRNA reads (adding a pseudo count) relative to the total number of reads mapping to annotated miRNAs in the given sample. To filter miRNAs with very low expression across most samples in a cancer-type data set, we removed miRNAs that were detected in <5% of samples (using the 'detected' flag in the microarray data sets and a read count threshold of 10 in the sequencing data sets). The microarray and sequencing data expression values were log2 transformed for subsequent analysis. Mature and precursor miRNA sequences, coordinates and relationships were obtained through miRMaid (http://170.mirmaid.org/)47. For global target interaction enrichment analysis (Fig. 1b,c), we defined a set of highly expressed miRNAs in each tumor type. This set was defined by miRNAs highly expressed (top 100) in ≥2% of the samples for a given tumor type. This threshold led to selection of ~150 (actual number depends on tumor type) mature miRNAs in each cancer type for the statistical evaluation.
All mRNA expression data sets were obtained from the TCGA open access data directory (https://tcga-data.nci.nih.gov/tcgafiles/ftp_auth/distro_ftpusers/anonymous/tumor/, December 2012). Normalized TCGA level 3 Agilent microarray mRNA expression profiles were used for GBM, OVA and COAD/READ studies. For the remaining tumor types, mapped and gene-level-summarized (level 3, RPKM) RNA-seq data sets were used. To filter mRNAs not expressed across most samples in RNA-seq data sets, we removed mRNAs with <20 reads in >95% of samples. To allow log transformation, mRNA RPKM expression values of 0 were set to the minimum nonzero RPKM in the given sample. The microarray and RNA-seq mRNA expression values were log2 transformed for all subsequent analysis.
DNA copy-number (aCGH) data sets were obtained from Firehose (http://gdac.broadinstitute.org/runs/analyses__2012_12_21/). We used level 4 nondiscretized gene-summarized log2-transformed aCGH copy-number calls (tumor / normal ratio) computed by the Gistic2 algorithm31.
DNA methylation data sets profiled by either Illumina HumanMethylation27 (for GBM, OVA and COAD/READ) or HumanMethylation450 (for the remaining cancer types) platforms were obtained from Firehose (http://gdac.broadinstitute.org/runs/analyses__2012_12_21/). We used level 4 data with methylation probes mapped to gene promoters, and selected for each gene data corresponding to the methylation probe showing strongest negative correlation (Pearson correlation coefficient) of methylation beta-value and gene mRNA expression across all samples in a cancer type. We similarly analyzed methylation probes mapping to known miRNA promoters (±2 kb of annotated transcription start sites) using a manually curated database of miRNA gene transcription start sites48.
miRNA target predictions.
miRanda-miRSVR (August 2010 release) human miRNA target predictions were obtained from http://microrna.org22. We used miRanda-miRSVR scores aggregated per gene and miRNA. TargetScan version 5.2 human miRNA target predictions were obtained from http://targetscan.org21. We used TargetScan context score and evolutionary conservation scores aggregated per gene and miRNA. Throughout the manuscript, predicted miRNA targets are defined by the intersection of miRanda (score < −0.5) and TargetScan (context-score < −0.2) unless otherwise stated. miRNA targets were also predicted by matching the miRNA seed (position 2−8) complement to the 5′ UTR, coding region and 3′ UTR sequences of individual mRNAs. Sequences were obtained from Ensembl (version 63), and the longest sequence was selected if a gene had multiple sequences defined for a given mRNA region.
Public miRNA perturbation data sets.
We obtained public miRNA perturbation data sets from the Gene Expression Omnibus: miR-106b and miR-16 overexpression and inhibition in HeLa cervical cancer cells (GSE6838), miR-29c overexpression in MKN45 gastric cancer cells (GSE38581), miR-30d overexpression in 5B1 melanoma cells (GSE27718), miR-200b overexpression in A498 kidney cancer cells (GSM911073) and miR-21 inhibition in MCF7 breast cancer cells (supplementary data in ref. 49).
Experimental assays.
U251 glioma cells and HeLa cells were cultured under 5% CO2 at 37 °C in DMEM (ATCC: 30-2002) with 10% heat-inactivated calf serum (Colorado Serum Co.). miRIDIAN miRNA mimic negative control oligonucleotides (n = 2), miRIDIAN miRNA mimics (hsa-miR-29b), NREP targeting ON-TARGETplus siRNAs (n = 2) and controls (n = 2) were purchased from ThermoFisher Scientific. miRCURY LNA microRNA Power Inhibitors (hsa-miR-29b) and control LNA inhibitors (n = 2) were purchased from Exiqon. Oligonucleotides were transfected to a final concentration of 100 nM using Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions. Total RNA was extracted using the miR-Vana RNA isolation system (Life Technologies). Expression of miR-29b and NREP was measured using TaqMan qPCR assays (Life Technologies) according to the manufacturer's instructions, and RNU6B and ACTB were used as endogenous controls, respectively. All experimental assays were done with two biological replicates, and different control compounds were also treated as control biological replicates in the statistical analysis.
Accession codes.
miRNA and mRNA expression data sets: TCGA open access data directory, https://tcga-data.nci.nih.gov/tcgafiles/ftp_auth/distro_ftpusers/anonymous/tumor/. DNA copy number and DNA methylation data sets: The Broad Institute, Firehose, http://gdac.broadinstitute.org/runs/analyses__2012_12_21/.
References
Bartel, D.P. MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233 (2009).
Croce, C.M. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 10, 704–714 (2009).
Lujambio, A. & Lowe, S.W. The microcosmos of cancer. Nature 482, 347–355 (2012).
Lu, J. et al. MicroRNA expression profiles classify human cancers. Nature 435, 834–838 (2005).
Volinia, S. et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 103, 2257–2261 (2006).
Medina, P.P., Nolde, M. & Slack, F.J. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature 467, 86–90 (2010).
Garzon, R., Marcucci, G. & Croce, C.M. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat. Rev. Drug Discov. 9, 775–789 (2010).
Jacobsen, A., Wen, J., Marks, D.S. & Krogh, A. Signatures of RNA binding proteins globally coupled to effective microRNA target sites. Genome Res. 20, 1010–1019 (2010).
Domcke, S., Sinha, R., Levine, D.A., Sander, C. & Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 4, 2126 (2013).
Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008).
Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 (2011).
Baek, D. et al. The impact of microRNAs on protein output. Nature 455, 64–71 (2008).
Selbach, M. et al. Widespread changes in protein synthesis induced by microRNAs. Nature 455, 58–63 (2008).
Huang, J.C. et al. Using expression profiling data to identify human microRNA targets. Nat. Methods 4, 1045–1049 (2007).
Muniategui, A., Pey, J., Planes, F.J. & Rubio, A. Joint analysis of miRNA and mRNA expression data. Brief. Bioinform. 14, 263–278 (2013).
Silber, J. et al. miR-34a Repression in proneural malignant gliomas upregulates expression of its Target PDGFRA and promotes tumorigenesis. PLoS ONE 7, e33844 (2012).
Genovese, G. et al. microRNA regulatory network inference identifies miR-34a as a novel regulator of TGF-β signaling in glioblastoma. Cancer Discov. 2, 736–749 (2012).
Setty, M. et al. Inferring transcriptional and microRNA-mediated regulatory programs in glioblastoma. Mol. Syst. Biol. 8, 605 (2012).
Creighton, C.J. et al. Integrated analyses of microRNAs demonstrate their widespread influence on gene expression in high-grade serous ovarian carcinoma. PLoS ONE 7, e34546 (2012).
Yang, D. et al. Integrated analyses identify a master microRNA regulatory network for the mesenchymal subtype in serous ovarian cancer. Cancer Cell 23, 186–199 (2013).
Grimson, A. et al. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell 27, 91–105 (2007).
Betel, D., Koppal, A., Agius, P., Sander, C. & Leslie, C. Comprehensive modeling of microRNA targets predicts functional nonconserved and noncanonical sites. Genome Biol. 11, R90 (2010).
Kim, K. et al. Identification of oncogenic microRNA-17–92/ZBTB4/specificity protein axis in breast cancer. Oncogene 31, 1034–1044 (2012).
Kim, K. et al. Induction of the transcriptional repressor ZBTB4 in prostate cancer cells by drug-induced targeting of microRNA-17–92/106b-25 clusters. Mol. Cancer Ther. 11, 1852–1862 (2012).
Brabletz, S. & Brabletz, T. The ZEB/miR-200 feedback loop—a motor of cellular plasticity in development and cancer? EMBO Rep. 11, 670–677 (2010).
Dews, M. et al. The Myc-miR-17 92 axis blunts TGF signaling and production of multiple TGF-dependent antiangiogenic factors. Cancer Res. 70, 8233–8246 (2010).
Mestdagh, P. et al. The miR-17–92 microRNA cluster regulates multiple components of the TGF-β pathway in neuroblastoma. Mol. Cell 40, 762–773 (2010).
Akhurst, R.J. & Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 11, 790–811 (2012).
Hocevar, B.A., Smine, A., Xu, X.X. & Howe, P.H. The adaptor molecule Disabled-2 links the transforming growth factor beta receptors to the Smad pathway. EMBO J. 20, 2789–2801 (2001).
Hannigan, A. et al. Epigenetic downregulation of human disabled homolog 2 switches TGF-beta from a tumor suppressor to a tumor promoter. J. Clin. Invest. 120, 2842–2857 (2010).
Mermel, C.H. et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41 (2011).
Tahiliani, M. et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935 (2009).
Ito, S. et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303 (2011).
He, Y.-F. et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307 (2011).
Cortellino, S. et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 146, 67–79 (2011).
Garzon, R. et al. MicroRNA 29b functions in acute myeloid leukemia. Blood 114, 5331–5341 (2009).
Sengupta, S. et al. MicroRNA 29c is down-regulated in nasopharyngeal carcinomas, up-regulating mRNAs encoding extracellular matrix proteins. Proc. Natl. Acad. Sci. USA 105, 5874–5878 (2008).
Nam, Y., Chen, C., Gregory, R.I., Chou, J.J. & Sliz, P. Molecular Basis for Interaction of let-7 MicroRNAs with Lin28. Cell 147, 1080–1091 (2011).
Mariani, L. et al. Identification and validation of P311 as a glioblastoma invasion gene using laser capture microdissection. Cancer Res. 61, 4190–4196 (2001).
Penn, J.W., Grobbelaar, A.O. & Rolfe, K.J. The role of the TGF-β family in wound healing, burns and scarring: a review. Int. J. Burns Trauma 2, 18 (2012).
Bhutani, N., Burns, D.M. & Blau, H.M. DNA demethylation dynamics. Cell 146, 866–872 (2011).
Dawson, M.A. & Kouzarides, T. Cancer epigenetics: from mechanism to therapy. Cell 150, 12–27 (2012).
Zhang, P., Huang, B., Xu, X. & Sessa, W.C. Ten-eleven translocation (Tet) and thymine DNA glycosylase (TDG), components of the demethylation pathway, are direct targets of miRNA-29a. Biochem. Biophys. Res. Commun. 437, 368–373 (2013).
Fabbri, M. et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc. Natl. Acad. Sci. USA 104, 15805–15810 (2007).
Garzon, R. et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood 113, 6411–6418 (2009).
López-Romero, P. Pre-processing and differential expression analysis of Agilent microRNA arrays using the AgiMicroRna Bioconductor library. BMC Genomics 12, 64 (2011).
Jacobsen, A., Krogh, A., Kauppinen, S. & Lindow, M. miRMaid: a unified programming interface for microRNA data resources. BMC Bioinformatics 11, 29 (2010).
Bhattacharyya, M., Das, M. & Bandyopadhyay, S. miRT: a database of validated transcription start sites of human microRNAs. Genomics Proteomics Bioinformatics 10, 310–316 (2012).
Frankel, L.B. et al. Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J. Biol. Chem. 283, 1026–1033 (2008).
Acknowledgements
We acknowledge contributions from the TCGA Research Network and its TCGA Pan-Cancer Analysis Working Group (contributing consortium members are listed in Supplementary Table 4). The TCGA Pan-Cancer Analysis Working Group is coordinated by J.M. Stuart, C.S. and I. Shmulevich. We thank M.L. Miller for reviewing the manuscript and for valuable comments. We acknowledge these sources of funding: The Danish Research Council (A.J.), Lundbeck Foundation (J.S.), the Carlsberg Foundation (J.S.), Christian og Ottilia Brorsons Rejselegat for Yngre Videnskabsmænd og kvinder (J.S.), the Sidney Kimmel Foundation (J.T.H.), the Sontag Foundation (J.T.H.) and a TCGA Genome Data Analysis Center Grant (U24 CA143840) (A.J., N.S. and C.S.).
Author information
Authors and Affiliations
Contributions
A.J. designed the statistical method and carried out the analysis. N.S. and C.S. supervised the study. J.S. and G.H. did experiments in cell lines. J.T.H. supervised experiments in cell lines. A.J. designed and implemented the online resource. A.J., N.S. and C.S. wrote the manuscript with contributions from J.S. All authors commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1 and 2, Supplementary Tables 1–3 and Supplementary Note (PDF 6903 kb)
Supplementary Table 4
Supplementary Table 4 (XLSX 33 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/.
About this article
Cite this article
Jacobsen, A., Silber, J., Harinath, G. et al. Analysis of microRNA-target interactions across diverse cancer types. Nat Struct Mol Biol 20, 1325–1332 (2013). https://doi.org/10.1038/nsmb.2678
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nsmb.2678
This article is cited by
-
Non-coding RNAs in disease: from mechanisms to therapeutics
Nature Reviews Genetics (2024)
-
Decreased cell proliferation and induced apoptosis in human B-chronic lymphocytic leukemia following miR-221 inhibition through modulation of p27 expression
Egyptian Journal of Medical Human Genetics (2022)
-
MiR-29b may suppresses peritoneal metastases through inhibition of the mesothelial–mesenchymal transition (MMT) of human peritoneal mesothelial cells
Scientific Reports (2022)
-
Pan-cancer analysis reveals cooperativity of both strands of microRNA that regulate tumorigenesis and patient survival
Nature Communications (2020)
-
An analysis about heterogeneity among cancers based on the DNA methylation patterns
BMC Cancer (2019)
