
Key Points
-
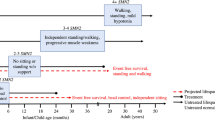
Spinal muscular atrophy (SMA) is the most common cause of infant death resulting from genetic defect
-
Children affected by SMA present with various degrees of muscular wasting, along with a complex profile of accompanying symptoms
-
No effective therapy for SMA is currently available in clinical practice
-
An increasing comprehension of SMA pathophysiology, including the characterization of SMN1 and SMN2 genes and SMN protein functions, has led to the development of multiple experimental therapeutic strategies
-
Therapeutic approaches aim to replace or correct the faulty SMN1 gene, promote exon 7 inclusion in SMN2, increase SMN2 promoter activity, or stabilize and protect full-length and Δ7 SMN proteins
-
Worldwide, several clinical trials evaluating the efficacy of these approaches are ongoing
Abstract
In the past decade, improved understanding of spinal muscular atrophy (SMA) aetiopathogenesis has brought us to a historical turning point: we are at the verge of development of disease-modifying treatments for this hitherto incurable disease. The increasingly precise delineation of molecular targets within the survival of motor neuron (SMN) gene locus has led to the development of promising therapeutic strategies. These novel avenues in treatment for SMA include gene therapy, molecular therapy with antisense oligonucleotides, and small molecules that aim to increase expression of SMN protein. Stem cell studies of SMA have provided an in vitro model for SMA, and stem cell transplantation could be used as a complementary strategy with a potential to treat the symptomatic phases of the disease. Here, we provide an overview of established data and novel insights into SMA pathogenesis, including discussion of the crucial function of the SMN protein. Preclinical evidence and recent advances from ongoing clinical trials are thoroughly reviewed. The final remarks are dedicated to future clinical perspectives in this rapidly evolving field, with a broad discussion on the comparison between the outlined therapeutic approaches and the remaining open questions.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

References
Werdnig, G. Two early infantile hereditary cases of progressive muscular atrophy simulating dystrophy, but on a neural basis. 1891. Arch. Neurol. 25, 276–278 (1971).
Brzustowicz, L. M. et al. Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q11.2–133. Nature 344, 540–541 (1990).
Arnold, W. D., Kassar, D. & Kissel, J. T. Spinal muscular atrophy: diagnosis and management in a new therapeutic era. Muscle Nerve 51, 157–167 (2014).
Darras, B. T. Non-5q spinal muscular atrophies: the alphanumeric soup thickens. Neurology 77, 312–314 (2011).
Rossor, A. M. et al. Phenotypic and molecular insights into spinal muscular atrophy due to mutations in BICD2. Brain 138, 293–310 (2014).
Sugarman, E. A. et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur. J. Hum. Genet. 20, 27–32 (2012).
Prior, T. W. et al. Newborn and carrier screening for spinal muscular atrophy. Am. J. Med. Genet. A. 152A, 1608–1616 (2010).
Finkel, R. S. et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology 83, 810–817 (2014).
Munsat, T. L. & Davies, K. E. International SMA consortium meeting. (26–28 June 1992, Bonn, Germany). Neuromuscul. Disord. 2, 423–428 (1992).
Feldkötter, M., Schwarzer, V., Wirth, R., Wienker, T. F. & Wirth, B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 70, 358–368 (2002).
Cho, S. & Dreyfuss, G. A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity. Genes Dev. 24, 438–442 (2010).
Mercuri, E., Bertini, E. & Iannaccone, S. T. Childhood spinal muscular atrophy: controversies and challenges. Lancet Neurol. 11, 443–452 (2012).
Wang, C. H. et al. Consensus statement for standard of care in spinal muscular atrophy. J. Child. Neurol. 22, 1027–1049 (2007).
Shababi, M. et al. Cardiac defects contribute to the pathology of spinal muscular atrophy models. Hum. Mol. Genet. 19, 4059–4071 (2010).
Rudnik-Schöneborn, S. et al. Congenital heart disease is a feature of severe infantile spinal muscular atrophy. J. Med. Genet. 45, 635–638 (2008).
Palladino, A. et al. Cardiac involvement in patients with spinal muscular atrophies. Acta Myol. 30, 175–178 (2011).
Iannaccone, S. T. Modern management of spinal muscular atrophy. J. Child. Neurol. 22, 974–978 (2007).
Durkin, E. T., Schroth, M. K., Helin, M. & Shaaban, A. F. Early laparoscopic fundoplication and gastrostomy in infants with spinal muscular atrophy type I. J. Pediatr. Surg. 43, 2031–2037 (2008).
Joyce, N. C., Hache, L. P. & Clemens, P. R. Bone health and associated metabolic complications in neuromuscular diseases. Phys. Med. Rehabil. Clin. N. Am. 23, 773–799 (2012).
Verrillo, E. et al. Sleep architecture in infants with spinal muscular atrophy type 1. Sleep Med. 15, 1246–1250 (2014).
Kaufmann, P. et al. Observational study of spinal muscular atrophy type 2 and 3: functional outcomes over 1 year. Arch. Neurol. 68, 779–786 (2011).
Yuan, P. & Jiang, L. Clinical characteristics of three subtypes of spinal muscular atrophy in children. Brain Dev. 37, 537–541 (2014).
Piepers, S. et al. A natural history study of late onset spinal muscular atrophy types 3b and 4. J. Neurol. 255, 1400–1404 (2008).
Prior, T. W., Nagan, N., Sugarman, E. A., Batish, S. D. & Braastad, C. Technical standards and guidelines for spinal muscular atrophy testing. Genet. Med. 13, 686–694 (2011).
Rudnik–Schöneborn, S. et al. Clinical utility gene card for: proximal spinal muscular atrophy. Eur. J. Hum. Genet. 20, (2012).
D'Amico, A., Mercuri, E., Tiziano, F. D. & Bertini, E. Spinal muscular atrophy. Orphanet J. Rare Dis. 6, 71 (2011).
Vilchis, Z. et al. The high frequency of genetic diseases in hypotonic infants referred by neuropediatrics. Am. J. Med. Genet. A. 164A, 1702–1705 (2014).
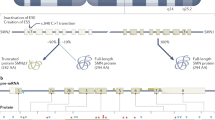
Lefebvre, S. et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165 (1995).
Ogino, S. & Wilson, R. B. Genetic testing and risk assessment for spinal muscular atrophy (SMA). Hum. Genet. 111, 477–500 (2002).
Scully, M. A., Farrell, P. M., Ciafaloni, E., Griggs, R. C. & Kwon, J. M. Cystic fibrosis newborn screening: a model for neuromuscular disease screening? Ann. Neurol. 77, 189–197 (2014).
Swoboda, K. J. SMN-targeted therapeutics for spinal muscular atrophy: are we SMArt enough yet? J. Clin. Invest. 124, 487–490 (2014).
Kolb, S. J. NeuroNEXT SMA biomarkers study. Ann. Neurol. 74, A8 (2013).
Castro, D. & Iannaccone, S. T. Spinal muscular atrophy: therapeutic strategies. Curr. Treat. Options Neurol. 16, 316 (2014).
Monani, U. R. et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 8, 1177–1183 (1999).
Lorson, C. L., Hahnen, E., Androphy, E. J. & Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl Acad. Sci. USA 96, 6307–6311 (1999).
Burnett, B. G. et al. Regulation of SMN protein stability. Mol. Cell. Biol. 29, 1107–1115 (2009).
Elsheikh, B. et al. An analysis of disease severity based on SMN2 copy number in adults with spinal muscular atrophy. Muscle Nerve 40, 652–656 (2009).
Prior, T. W., Swoboda, K. J., Scott, H. D. & Hejmanowski, A. Q. Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am. J. Med. Genet. A. 130A, 307–310 (2004).
Schrank, B. et al. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc. Natl Acad. Sci. USA 94, 9920–9925 (1997).
Monani, U. R. et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(-/-) mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet. 9, 333–339 (2000).
Le, T. T. et al. SMNΔ7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 14, 845–857 (2005).
Bevan, A. K. et al. Early heart failure in the SMNΔ7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Hum. Mol. Genet. 19, 3895–3905 (2010).
Butchbach, M. E. R., Edwards, J. D., Schussler, K. R. & Burghes, A. H. A novel method for oral delivery of drug compounds to the neonatal SMNΔ7 mouse model of spinal muscular atrophy. J. Neurosci. Methods 161, 285–290 (2007).
Foust, K. D. et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 28, 271–274 (2010).
Dominguez, E. et al. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum. Mol. Genet. 20, 681–693 (2011).
Cherry, J. J. et al. Enhancement of SMN protein levels in a mouse model of spinal muscular atrophy using novel drug-like compounds. EMBO Mol. Med. 5, 1035–1050 (2013).
Ebert, A. D. et al. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 457, 277–280 (2009).
Sareen, D. et al. Inhibition of apoptosis blocks human motor neuron cell death in a stem cell model of spinal muscular atrophy. PLoS ONE 7, e39113 (2012).
Corti, S. et al. Genetic correction of human induced pluripotent stem cells from patients with spinal muscular atrophy. Sci. Transl. Med. 4, 165ra162 (2012).
Vitte, J. et al. Refined characterization of the expression and stability of the SMN gene products. Am. J. Pathol. 171, 1269–1280 (2007).
Wang, J. & Dreyfuss, G. A cell system with targeted disruption of the SMN gene: functional conservation of the SMN protein and dependence of Gemin2 on SMN. J. Biol. Chem. 276, 9599–9605 (2001).
Pellizzoni, L., Kataoka, N., Charroux, B. & Dreyfuss, G. A novel function for SMN, the spinal muscular atrophy disease gene product, in pre-mRNA splicing. Cell 95, 615–624 (1998).
Eggert, C., Chari, A., Laggerbauer, B. & Fischer, U. Spinal muscular atrophy: the RNP connection. Trends Mol. Med. 12, 113–121 (2006).
Pellizzoni, L. Chaperoning ribonucleoprotein biogenesis in health and disease. EMBO Rep. 8, 340–345 (2007).
Chari, A. et al. An assembly chaperone collaborates with the SMN complex to generate spliceosomal SnRNPs. Cell 135, 497–509 (2008).
Battle, D. J. et al. The SMN complex: an assembly machine for RNPs. Cold Spring Harb. Symp. Quant. Biol. 71, 313–320 (2006).
Kolb, S. J., Battle, D. J. & Dreyfuss, G. Molecular functions of the SMN complex. J. Child. Neurol. 22, 990–994 (2007).
Gabanella, F. et al. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS ONE 2, e921 (2007).
Gabanella, F., Carissimi, C., Usiello, A. & Pellizzoni, L. The activity of the spinal muscular atrophy protein is regulated during development and cellular differentiation. Hum. Mol. Genet. 14, 3629–3642 (2005).
Burghes, A. H. & Beattie, C. E. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 10, 597–609 (2009).
Boulisfane, N. et al. Impaired minor tri-snRNP assembly generates differential splicing defects of U12-type introns in lymphoblasts derived from a type I SMA patient. Hum. Mol. Genet. 20, 641–648 (2011).
Pellizzoni, L., Yong, J. & Dreyfuss, G. Essential role for the SMN complex in the specificity of snRNP assembly. Science 298, 1775–1779 (2002).
Zhang, Z. et al. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 133, 585–600 (2008).
Li, D. K., Tisdale, S., Lotti, F. & Pellizzoni, L. SMN control of RNP assembly: from post-transcriptional gene regulation to motor neuron disease. Semin. Cell Dev. Biol. 32, 22–29 (2014).
Kariya, S. et al. Requirement of enhanced survival motoneuron protein imposed during neuromuscular junction maturation. J. Clin. Invest. 124, 785–800 (2014).
Oprea, G. E. et al. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 320, 524–527 (2008).
McWhorter, M. L., Monani, U. R., Burghes, A. H. & Beattie, C. E. Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J. Cell Biol. 162, 919–931 (2003).
Rossoll, W. et al. Specific interaction of Smn, the spinal muscular atrophy determining gene product, with hnRNP-R and gry-rbp/hnRNP-Q: a role for Smn in RNA processing in motor axons? Hum. Mol. Genet. 11, 93–105 (2002).
Rossoll, W. & Bassell, G. J. Spinal muscular atrophy and a model for survival of motor neuron protein function in axonal ribonucleoprotein complexes. Results Probl. Cell Differ. 48, 289–326 (2009).
Hubers, L. et al. HuD interacts with survival motor neuron protein and can rescue spinal muscular atrophy-like neuronal defects. Hum. Mol. Genet. 20, 553–579 (2011).
Martinez, T. L. et al. Survival motor neuron protein in motor neurons determines synaptic integrity in spinal muscular atrophy. J. Neurosci. 32, 8703–8715 (2012).
Goulet, B. B., Kothary, R. & Parks, R. J. At the “junction” of spinal muscular atrophy pathogenesis: the role of neuromuscular junction dysfunction in SMA disease progression. Curr. Mol. Med. 13, 1160–1174 (2013).
Fayzullina, S. & Martin, L. J. Skeletal muscle DNA damage precedes spinal motor neuron DNA damage in a mouse model of spinal muscular atrophy (SMA). PLoS ONE 9, e93329 (2014).
Boyer, J. G. et al. Early onset muscle weakness and disruption of muscle proteins in mouse models of spinal muscular atrophy. Skelet. Muscle 3, 24 (2013).
Hunter, G., Aghamaleky Sarvestany, A., Roche, S. L., Symes, R. C. & Gillingwater, T. H. SMN-dependent intrinsic defects in Schwann cells in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 23, 2235–2250 (2014).
Imlach, W. L. et al. SMN is required for sensory-motor circuit function in Drosophila. Cell 151, 427–439 (2012).
Prior, T. W. et al. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am. J. Hum. Genet. 85, 408–413 (2009).
Jarecki, J. et al. Diverse small-molecule modulators of SMN expression found by high-throughput compound screening: early leads towards a therapeutic for spinal muscular atrophy. Hum. Mol. Genet. 14, 2003–2018 (2005).
Cherry, J. J. et al. Assays for the identification and prioritization of drug candidates for spinal muscular atrophy. Assay Drug Dev. Technol. 12, 315–341 (2014).
Lorson, M. A. & Lorson, C. L. SMN-inducing compounds for the treatment of spinal muscular atrophy. Future Med. Chem. 4, 2067–2084 (2012).
Thurmond, J. et al. Synthesis and biological evaluation of novel 2,4-diaminoquinazoline derivatives as SMN2 promoter activators for the potential treatment of spinal muscular atrophy. J. Med. Chem. 51, 449–469 (2008).
Yuo, C.-Y., Lin, H.-H., Chang, Y.-S., Yang, W.-K. & Chang, J.-G. 5-(N-ethyl-N-isopropyl)-amiloride enhances SMN2 exon 7 inclusion and protein expression in spinal muscular atrophy cells. Ann. Neurol. 63, 26–34 (2008).
Bowerman, M., Murray, L. M., Boyer, J. G., Anderson, C. L. & Kothary, R. Fasudil improves survival and promotes skeletal muscle development in a mouse model of spinal muscular atrophy. BMC Med. 10, 24 (2012).
Bowerman, M., Beauvais, A., Anderson, C. L. & Kothary, R. Rho-kinase inactivation prolongs survival of an intermediate SMA mouse model. Hum. Mol. Genet. 19, 1468–1478 (2010).
Miller, R. G. et al. A placebo-controlled trial of gabapentin in spinal muscular atrophy. J. Neurol. Sci. 191, 127–131 (2001).
Wirth, B., Garbes, L. & Riessland, M. How genetic modifiers influence the phenotype of spinal muscular atrophy and suggest future therapeutic approaches. Curr. Opin. Genet. Dev. 23, 330–338 (2013).
Swoboda, K. J. et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann. Neurol. 57, 704–712 (2005).
Passini, M. A. et al. CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J. Clin. Invest. 120, 1253–1264 (2010).
Valori, C. F. et al. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci. Transl. Med. 2, 35ra42 (2010).
Mitrpant, C. et al. Improved antisense oligonucleotide design to suppress aberrant SMN2 gene transcript processing: towards a treatment for spinal muscular atrophy. PLoS ONE 8, e62114 (2013).
Duque, S. I. et al. A large animal model of spinal muscular atrophy and correction of phenotype. Ann. Neurol. 77, 399–414 (2014).
Gray, S. J., Nagabhushan Kalburgi, S., McCown, T. J. & Jude Samulski, R. Global CNS gene delivery and evasion of anti-AAV-neutralizing antibodies by intrathecal AAV administration in non-human primates. Gene Ther. 20, 450–459 (2013).
Foust, K. D. et al. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 27, 59–65 (2009).
Bevan, A. K. et al. Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol. Ther. 19, 1971–1980 (2011).
Meyer, K. et al. Improving single injection CSF delivery of AAV9-mediated gene therapy for SMA: a dose-response study in mice and nonhuman primates. Mol. Ther. 23, 477–487 (2014).
Asokan, A., Schaffer, D. V. & Jude Samulski, R. The AAV vector toolkit: poised at the clinical crossroads. Mol. Ther. 20, 699–708 (2012).
Singh, N. K., Singh, N. N., Androphy, E. J. & Singh, R. N. Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol. Cell. Biol. 26, 1333–1346 (2006).
Williams, J. H. et al. Oligonucleotide-mediated survival of motor neuron protein expression in CNS improves phenotype in a mouse model of spinal muscular atrophy. J. Neurosci. 29, 7633–7638 (2009).
Passini, M. A. et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med. 3, 72ra18 (2011).
Hua, Y. et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 24, 1634–1644 (2010).
Keil, J. M. et al. A short antisense oligonucleotide ameliorates symptoms of severe mouse models of spinal muscular atrophy. Mol. Ther. Nucleic Acids 3, e174 (2014).
Mitrpant, C. et al. Improved antisense oligonucleotide design to suppress aberrant SMN2 gene transcript processing: towards a treatment for spinal muscular atrophy. PLoS ONE 8, e62114 (2013).
Hua, Y. et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 478, 123–126 (2011).
Zhou, H. et al. A novel morpholino oligomer targeting ISS-N1 improves rescue of severe spinal muscular atrophy transgenic mice. Hum. Gene Ther. 24, 331–342 (2013).
Osman, E. Y. et al. Morpholino antisense oligonucleotides targeting intronic repressor Element1 improve phenotype in SMA mouse models. Hum. Mol. Genet. 23, 4832–4845 (2014).
Rigo, F. et al. Pharmacology of a central nervous system delivered 2′-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J. Pharmacol. Exp. Ther. 350, 46–55 (2014).
Nizzardo, M. et al. Effect of combined systemic and local morpholino treatment on the spinal muscular atrophy Δ7 mouse model phenotype. Clin. Ther. 36, 340–356.e5 (2014).
Titus, S. et al. High throughput screening for SMA. Probe Reports from the NIH Molecular Libraries Program [online], (2010).
Makhortova, N. R. et al. A screen for regulators of survival of motor neuron protein levels. Nat. Chem. Biol. 7, 544–552 (2011).
Hastings, M. L. et al. Tetracyclines that promote SMN2 exon 7 splicing as therapeutics for spinal muscular atrophy. Sci. Transl. Med. 1, 5ra12 (2009).
Naryshkin, N. A. et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 345, 688–693 (2014).
Acknowledgements
The financial support from Ministry of Health (GR-2009-1483560) and Cariplo grant (2012-0513) to S.C., and Telethon grant (GGP14025) to M.N. are gratefully acknowledged. All authors gratefully acknowledge the support from Associazione Amici del Centro Dino Ferrari.
Author information
Authors and Affiliations
Contributions
S.C. and I.F. researched data for the article. S.C., I.F. and G.P.C. wrote the manuscript. All authors substantially contributed to discussion of content and reviewing, editing and revising of manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Faravelli, I., Nizzardo, M., Comi, G. et al. Spinal muscular atrophy—recent therapeutic advances for an old challenge. Nat Rev Neurol 11, 351–359 (2015). https://doi.org/10.1038/nrneurol.2015.77
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrneurol.2015.77
This article is cited by
-
Optimized MLPA workflow for spinal muscular atrophy diagnosis: identification of a novel variant, NC_000005.10:g.(70919941_70927324)del in isolated exon 1 of SMN1 gene through long-range PCR
BMC Neurology (2024)
-
Multi-omics profiling of CSF from spinal muscular atrophy type 3 patients after nusinersen treatment: a 2-year follow-up multicenter retrospective study
Cellular and Molecular Life Sciences (2023)
-
Quality of life of children with spinal muscular atrophy and their caregivers from the perspective of caregivers: a Chinese cross-sectional study
Orphanet Journal of Rare Diseases (2021)
-
Comprehensive Mutation Analysis and Report of 12 Novel Mutations in a Cohort of Patients with Spinal Muscular Atrophy in Iran
Journal of Molecular Neuroscience (2021)
-
Current understanding of and emerging treatment options for spinal muscular atrophy with respiratory distress type 1 (SMARD1)
Cellular and Molecular Life Sciences (2020)
