
Key Points
-
Genome-wide association studies of very common variants have neither identified associations that explain a large portion of the heritability for most traits studied nor identified the causal variants behind the associations seen.
-
Although few common variants that cause a disease have been securely identified, rare variants have been found that have strong influences on common diseases: for example, a SNP in type 1 diabetes and copy-number variants in schizophrenia.
-
It seems likely that rare variants, similar in some ways to those identified in Mendelian diseases, will be found that influence common diseases. It is also likely that these rare variants will often influence the coding regions of genes in a manner that is readily recognizable, and will be of large enough effect size to be identified despite their low frequencies.
-
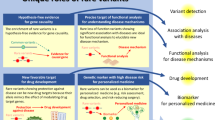
Whole-genome sequencing will provide the best means of identifying rare causal variants. We propose two strategies for studies: resequencing the genomes of individuals with extreme phenotypes and resequencing the genomes of individuals with a familial disease.
-
We predict that whole-genome sequencing will identify rare variants with large effects on many diseases and traits in the coming years. The knowledge that could potentially be gained about these traits, such as the type of mutation and the gene that influences each trait, could provide information for new drug targets.
Abstract
Although genome-wide association (GWA) studies for common variants have thus far succeeded in explaining only a modest fraction of the genetic components of human common diseases, recent advances in next-generation sequencing technologies could rapidly facilitate substantial progress. This outcome is expected if much of the missing genetic control is due to gene variants that are too rare to be picked up by GWA studies and have relatively large effects on risk. Here, we evaluate the evidence for an important role of rare gene variants of major effect in common diseases and outline discovery strategies for their identification.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$189.00 per year
only $15.75 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


References
Maher, B. Personal genomes: the case of the missing heritability. Nature 456, 18–21 (2008). One of the first articles to explicitly recognize that GWA studies explain a small part of the genetic components of many diseases.
Kasowski, M. et al. Variation in transcription factor binding among humans. Science 328, 232–235 (2010).
Pickrell, J. K. et al. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 464, 768–772 (2010).
Heinzen, E. L. et al. Tissue-specific genetic control of splicing: implications for the study of complex traits. PLoS Biol. 6, e1 (2008).
Stankiewicz, P. & Lupski, J. R. Structural variation in the human genome and its role in disease. Annu. Rev. Med. 61, 437–455 (2010).
Dickson, S. P., Wang, K., Krantz, I., Hakonarson, H. & Goldstein, D. B. Rare variants create synthetic genome-wide associations. PLoS Biol. 8, e1000294 (2010).
Bodmer, W. & Bonilla, C. Common and rare variants in multifactorial susceptibility to common diseases. Nature Genet. 40, 695–701 (2008).
Schork, N. J., Murray, S. S., Frazer, K. A. & Topol, E. J. Common vs. rare allele hypotheses for complex diseases. Curr. Opin. Genet. Dev. 19, 212–219 (2009).
Pritchard, J. K. Are rare variants responsible for susceptibility to complex diseases? Am. J. Hum. Genet. 69, 124–137 (2001).
International HapMap Consortium. The International HapMap Project. Nature 426, 789–796 (2003).
Pritchard, J. K. & Cox, N. J. The allelic architecture of human disease genes: common disease-common variant.or not? Hum. Mol. Genet. 11, 2417–2423 (2002).
Stephens, J. W. & Humphries, S. E. The molecular genetics of cardiovascular disease: clinical implications. J. Intern. Med. 253, 120–127 (2003).
Plomin, R., Haworth, C. M. & Davis, O. S. Common disorders are quantitative traits. Nature Rev. Genet. 10, 872–878 (2009).
Reich, D. E. & Lander, E. S. On the allelic spectrum of human disease. Trends Genet. 17, 502–510 (2001).
Tandon, R., Keshavan, M. S. & Nasrallah, H. A. Schizophrenia, 'just the facts' what we know in 2008. 2. Epidemiology and etiology. Schizophr. Res. 102, 1–18 (2008).
Crow, T. J. How and why genetic linkage has not solved the problem of psychosis: review and hypothesis. Am. J. Psychiatry 164, 13–21 (2007).
Serretti, A. & Mandelli, L. The genetics of bipolar disorder: genome 'hot regions', genes, new potential candidates and future directions. Mol. Psychiatry 13, 742–771 (2008).
Risch, N. & Merikangas, K. The future of genetic studies of complex human diseases. Science 273, 1516–1517 (1996).
Steinberg, M. H. & Adewoye, A. H. Modifier genes and sickle cell anemia. Curr. Opin. Hematol. 13, 131–136 (2006).
Thein, S. L. & Menzel, S. Discovering the genetics underlying foetal haemoglobin production in adults. Br. J. Haematol. 145, 455–467 (2009).
Manolio, T. A. et al. Finding the missing heritability of complex diseases. Nature 461, 747–753 (2009).
Zeggini, E. et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nature Genet. 40, 638–645 (2008).
Shi, J. et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 460, 753–757 (2009).
SEARCH Collaborative Group et al. SLCO1B1 variants and statin-induced myopathy — a genomewide study. N. Engl. J. Med. 359, 789–799 (2008).
Tanaka, Y. et al. Genome-wide association of IL28B with response to pegylated interferon-α and ribavirin therapy for chronic hepatitis C. Nature Genet. 41, 1105–1109 (2009).
Daly, A. K. et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nature Genet. 41, 816–819 (2009).
Fellay, J. et al. ITPA gene variants protect against anemia in patients treated for chronic hepatitis C. Nature 464, 405–408 (2010).
Ge, D. et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 461, 399–401 (2009).
Need, A. C. et al. A genome-wide study of common SNPs and CNVs in cognitive performance in the CANTAB. Hum. Mol. Genet. 18, 4650–4661 (2009).
Cirulli, E. T. et al. Common genetic variation and performance on standardized cognitive tests. Eur. J. Hum. Genet. 3 Feb 2010 (doi:10.1038/ejhg.2010.2).
Bhattacharjee, S. et al. Using principal components of genetic variation for robust and powerful detection of gene–gene interactions in case–control and case-only studies. Am. J. Hum. Genet. 86, 331–342 (2010).
Kong, A. et al. Parental origin of sequence variants associated with complex diseases. Nature 462, 868–874 (2009).
Meyer, K. B. et al. Allele-specific up-regulation of FGFR2 increases susceptibility to breast cancer. PLoS Biol. 6, e108 (2008).
Chang, B. L. et al. Fine mapping association study and functional analysis implicate a SNP in MSMB at 10q11 as a causal variant for prostate cancer risk. Hum. Mol. Genet. 18, 1368–1375 (2009).
Hughes, A. E. et al. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nature Genet. 38, 1173–1177 (2006).
Hageman, G. S. et al. Extended haplotypes in the complement factor H (CFH) and CFH-related (CFHR) family of genes protect against age-related macular degeneration: characterization, ethnic distribution and evolutionary implications. Ann. Med. 38, 592–604 (2006).
Spencer, K. L. et al. Deletion of CFHR3 and CFHR1 genes in age-related macular degeneration. Hum. Mol. Genet. 17, 971–977 (2008).
Frayling, T. M. Genome-wide association studies provide new insights into type 2 diabetes aetiology. Nature Rev. Genet. 8, 657–662 (2007).
McCarthy, M. I. & Hirschhorn, J. N. Genome-wide association studies: potential next steps on a genetic journey. Hum. Mol. Genet. 17, R156–R165 (2008).
Bouatia-Naji, N. et al. A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nature Genet. 41, 89–94 (2009).
Frayling, T. M. et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316, 889–894 (2007).
Todd, J. A. et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nature Genet. 39, 857–864 (2007).
Pillai, S. G. et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet. 5, e1000421 (2009).
Nejentsev, S., Walker, N., Riches, D., Egholm, M. & Todd, J. A. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 324, 387–389 (2009). This study showed that rare variants in the same region as a GWA signal for diabetes were associated with the disease.
Sanna, S. et al. Common variants in the GDF5–UQCC region are associated with variation in human height. Nature Genet. 40, 198–203 (2008).
Stefansson, H. et al. Large recurrent microdeletions associated with schizophrenia. Nature 455, 232–236 (2008). One of the first studies to identify rare CNVs associated with a common disease.
Gruber, S. B. et al. Genetic variation in 8q24 associated with risk of colorectal cancer. Cancer Biol. Ther. 6, 1143–1147 (2007).
Tomlinson, I. et al. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nature Genet. 39, 984–988 (2007).
Zanke, B. W. et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nature Genet. 39, 989–994 (2007).
Prokunina-Olsson, L. & Hall, J. L. No effect of cancer-associated SNP rs6983267 in the 8q24 region on co-expression of MYC and TCF7L2 in normal colon tissue. Mol. Cancer 8, 96 (2009).
Sotelo, J. et al. Long-range enhancers on 8q24 regulate c-Myc. Proc. Natl Acad. Sci. USA (2010).
Weedon, M. N. et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nature Genet. 40, 575–583 (2008).
Goldstein, D. B. Common genetic variation and human traits. N. Engl. J. Med. 360, 1696–1698 (2009).
Need, A. C. et al. A genome-wide investigation of SNPs and CNVs in schizophrenia. PLoS Genet. 5, e1000373 (2009).
Kumar, R. A. et al. Recurrent 16p11.2 microdeletions in autism. Hum. Mol. Genet. 17, 628–638 (2008).
Metzker, M. L. Sequencing technologies — the next generation. Nature Rev. Genet. 11, 31–46 (2010).
Dean, M. et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science 273, 1856–1862 (1996).
Liu, R. et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 86, 367–377 (1996).
Samson, M. et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382, 722–725 (1996).
Huang, Y. et al. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nature Med. 2, 1240–1243 (1996).
Mallal, S. et al. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet 359, 727–732 (2002).
Martin, A. M. et al. Predisposition to abacavir hypersensitivity conferred by HLA-B*5701 and a haplotypic Hsp70-Hom variant. Proc. Natl Acad. Sci. USA 101, 4180–4185 (2004).
Young, B. et al. First large, multicenter, open-label study utilizing HLA-B*5701 screening for abacavir hypersensitivity in North America. AIDS 22, 1673–1675 (2008).
Ng, S. B. et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 461, 272–276 (2009). The first study to show that next-generation sequencing can be used to identify disease-causing variants.
Choi, M. et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl Acad. Sci. USA 106, 19096–19101 (2009). The first study to diagnose a disease using next-generation sequencing.
Ng, S. B. et al. Exome sequencing identifies the cause of a mendelian disorder. Nature Genet. 42, 30–35 (2010).
Yang, S. et al. Genomic landscape of a three-generation pedigree segregating affective disorder. PLoS ONE 4, e4474 (2009).
Sobreira, N. L. M. et al. Whole genome sequencing of a single proband together with linkage analysis identifies a Mendelian disease gene. PLoS Genet. (in the press).
Manolio, T. A., Brooks, L. D. & Collins, F. S. A HapMap harvest of insights into the genetics of common disease. J. Clin. Invest. 118, 1590–1605 (2008).
Verlaan, D. J. et al. Targeted screening of cis-regulatory variation in human haplotypes. Genome Res. 19, 118–127 (2009).
Barrett, J. C. et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nature Genet. 40, 955–962 (2008).
Stenson, P. D. et al. The Human Gene Mutation Database: 2008 update. Genome Med. 1, 13 (2009).
Botstein, D. & Risch, N. Discovering genotypes underlying human phenotypes: past successes for Mendelian disease, future approaches for complex disease. Nature Genet. 33, 228–237 (2003). A thoughtful overview of the kinds of mutations responsible for Mendelian disease that provides many insights about appropriate designs for studying common disease.
Caskey, C. T. The drug development crisis: efficiency and safety. Annu. Rev. Med. 58, 1–16 (2007).
Roach, J. C. et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science 10 Mar 2010 (doi:10.1126/science.1186802).
Drmanac, R. et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327, 78–81 (2010).
Clayton, D. G. Prediction and interaction in complex disease genetics: experience in type 1 diabetes. PLoS Genet. 5, e1000540 (2009).
Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678 (2007). A technically important early study providing well-powered GWA tests for multiple conditions.
Diabetes Genetics Initiative. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 316, 1331–1336 (2007).
Scott, L. J. et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 316, 1341–1345 (2007).
Hamming, K. S. et al. Coexpression of the type 2 diabetes susceptibility gene variants KCNJ11 E23K and ABCC8 S1369A alter the ATP and sulfonylurea sensitivities of the ATP-sensitive K+ channel. Diabetes 58, 2419–2424 (2009).
Nicolson, T. J. et al. Insulin storage and glucose homeostasis in mice null for the granule zinc transporter ZnT8 and studies of the type 2 diabetes-associated variants. Diabetes 58, 2070–2083 (2009).
Gaulton, K. J. et al. A map of open chromatin in human pancreatic islets. Nature Genet. 42, 255–259 (2010).
Motulsky, A. G. Drug reactions enzymes, and biochemical genetics. JAMA 165, 835–837 (1957).
Ingelman-Sundberg, M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J. 5, 6–13 (2005).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Hormozdiari, F., Alkan, C., Eichler, E. E. & Sahinalp, S. C. Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes. Genome Res. 19, 1270–1278 (2009).
Yoon, S., Xuan, Z., Makarov, V., Ye, K. & Sebat, J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 19, 1586–1592 (2009).
Simpson, J. T., McIntyre, R. E., Adams, D. J. & Durbin, R. Copy number variant detection in inbred strains from short read sequence data. Bioinformatics 26, 565–567 (2010).
Milne, I. et al. Tablet — next generation sequence assembly visualization. Bioinformatics 26, 401–402 (2010).
Bao, H. et al. MapView: visualization of short reads alignment on a desktop computer. Bioinformatics 25, 1554–1555 (2009).
Manske, H. M. & Kwiatkowski, D. P. LookSeq: a browser-based viewer for deep sequencing data. Genome Res. 19, 2125–2132 (2009).
Arner, E., Hayashizaki, Y. & Daub, C. O. NGSView: an extensible open source editor for next-generation sequencing data. Bioinformatics 26, 125–126 (2010).
Schuster, S. C. et al. Complete Khoisan and Bantu genomes from southern Africa. Nature 463, 943–947 (2010).
Bentley, D. R. et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456, 53–59 (2008). One of the first studies to sequence an entire human genome using next-generation sequencing.
Wang, J. et al. The diploid genome sequence of an Asian individual. Nature 456, 60–65 (2008).
Ng, P. C. et al. Genetic variation in an individual human exome. PLoS Genet. 4, e1000160 (2008).
Axelrod, N. et al. The HuRef Browser: a web resource for individual human genomics. Nucleic Acids Res. 37, D1018–D1024 (2009).
Kirov, G. et al. Comparative genome hybridization suggests a role for NRXN1 and APBA2 in schizophrenia. Hum. Mol. Genet. 17, 458–465 (2008).
Friedman, J. M. et al. Oligonucleotide microarray analysis of genomic imbalance in children with mental retardation. Am. J. Hum. Genet. 79, 500–513 (2006).
Autism Genome Project Consortium et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nature Genet. 39, 319–328 (2007).
Acknowledgements
We thank D. Ge, E. L. Heinzen, A. C. Need, J. C. Fellay, J. M. Maia, E. K. Ruzzo and H. F. Willard for helpful comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
Glossary
- Minor allele frequency
-
Ranging from 0 to 50%, this is the proportion of alleles at a locus that consists of the less frequent allele. This number does not take genotype into account.
- Effect size
-
The increase in risk (or proportion of population variation) that is conferred by a given causal variant.
- Heritability
-
The proportion of phenotypic variation in a trait that is due to underlying genetic variation. In studies of humans, this value is usually calculated by comparing trait correlations in individuals of varying degrees of relatedness.
- Mendelian disease
-
A disease that is carried in families in either a dominant or recessive manner and that is typically controlled by variants of large effect in a single gene.
- Imputation
-
Based on the known linkage disequilibrium structure in fully genotyped individuals, the genotype of untyped variants can be inferred in individuals who are genotyped for a smaller number of variants.
- Exome
-
The exome is the collection of known exons in our genome: this is the portion of the genome that is translated into proteins. As exons comprise only 1% of the genome and contain the most easily understood, functionally relevant information, sequencing of only the exome is a cheaper method of identifying most of the variants that are most likely to affect a trait.
- Linkage disequilibrium
-
A nonrandom association between alleles at different loci.
- Endophenotype
-
An intermediate phenotype that is heritable and associated with a disease but is not itself a symptom of the disease. Although there is little evidence to support the theory, it has been argued that endophenotypes would be a more tractable target for genetic analysis than the relevant disease state itself.
- Haploinsufficiency
-
This occurs when a diploid organism only has one copy of a gene and both copies are required for correct function. This is one way that a protein-truncating mutation can influence predisposition to a disease.
- Haplotype
-
A combination of alleles that are inherited together.
- 1000 Genomes Project
-
An international research consortium that will sequence the genomes of 1,200 individuals of various ethnicities. Most individuals will be sequenced to low coverage, or in exons only. The goals are to catalogue human variation with minor allele frequencies of ∼1% or greater and to refine and optimize strategies for sequencing large numbers of genomes.
- Coverage
-
The number of sequence reads that have alignments that overlap a certain position. Because current sequencing strategies produce random reads, resulting in an uneven distribution of reads across the genome, a high average coverage is required to assure that most bases in the genome are covered by multiple reads.
- Indel
-
A small insertion or deletion of nucleotides. If it occurs in an exon and is not a multiple of three in length, it results in a frameshift and usually the loss of gene function.
- Splice-site variant
-
A variant, usually found at the intron–exon boundary, that alters the splicing of an exon to its surrounding exons.
- Non-synonymous variant
-
A genetic variant that changes a codon for one amino acid to another amino acid. Many non-synonymous variants are well-tolerated, but others can cause a disease.
- Co-segregation
-
In the pedigree of a family with a condition, the segregation pattern shows how often the putative causal variant is found to coincide with the condition. When a variant coincides with the condition in a family, the condition and the variant are said to co-segregate.
- Compound heterozygote
-
When an individual inherits two different recessive mutations, one from each parent, in the same gene that cause the same phenotype. An example would be a single-nucleotide variant causing a codon for an amino acid to be changed into a stop codon in one allele and a 4-bp deletion in the other allele: each of these variants knock out their respective allele, resulting in neither copy functioning.
Rights and permissions
About this article
Cite this article
Cirulli, E., Goldstein, D. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet 11, 415–425 (2010). https://doi.org/10.1038/nrg2779
Issue Date:
DOI: https://doi.org/10.1038/nrg2779
This article is cited by
-
Whole genome sequencing identifies genetic variants associated with neurogenic inflammation in rosacea
Nature Communications (2023)
-
Common and rare variant associations with latent traits underlying depression, bipolar disorder, and schizophrenia
Translational Psychiatry (2023)
-
Detecting disease association with rare variants using weighted entropy
Journal of Genetics (2023)
-
A novel missense compound heterozygous variant in TLR1 gene is associated with susceptibility to rheumatoid arthritis — structural perspective and functional annotations
Clinical Rheumatology (2023)
-
Noninvasive prenatal diagnosis targeting fetal nucleated red blood cells
Journal of Nanobiotechnology (2022)
