
Key Points
-
Covalent drugs block protein function by forming a specific bond between the ligand and target protein.
-
A covalent mechanism of action can provide many pharmacological advantages over a reversible mechanism of action; these advantages include enhanced potency, selectivity and prolonged duration of action.
-
As a therapeutic class, covalent drugs have made a major impact on human health, as indicated by the many examples of US Food and Drug Administration (FDA)-approved covalent drugs in various indications.
-
Many of the approved covalent drugs were discovered through serendipity. Computer-assisted drug design has led to a predictable means of creating a new generation of covalent drugs that have been termed 'targeted covalent inhibitors'.
-
Several targeted covalent inhibitors are in late-stage clinical development and are showing encouraging efficacy.
-
This Review surveys the prevalence and pharmacological advantages of covalent drugs, and discusses how potential risks and challenges may be addressed through innovative design, as well as the broad opportunities presented by targeted covalent inhibitors.
Abstract
Covalent drugs haveproved to be successful therapies for various indications, but largely owing to safety concerns, they are rarely considered when initiating a target-directed drug discovery project. There is a need to reassess this important class of drugs, and to reconcile the discordance between the historic success of covalent drugs and the reluctance of most drug discovery teams to include them in their armamentarium. This Review surveys the prevalence and pharmacological advantages of covalent drugs, discusses how potential risks and challenges may be addressed through innovative design, and presents the broad opportunities provided by targeted covalent inhibitors.
Similar content being viewed by others

Main
Drugs that form a covalent attachment to their target have traditionally been considered as conceptually distinct from conventional non-covalent drugs. In particular, the fact that such drugs derive part of their affinity by forming a covalent bond with their target has engendered anxiety concerning their potential for off-target reactivity and has led to these drugs being disfavoured as a drug class. These concerns largely stem from pioneering work that was carried out in the early 1970s on the hepatotoxic properties of compounds such as bromobenzene and acetaminophen, which undergo metabolism to form highly reactive intermediates that covalently bind to liver proteins. Although there has been much controversy over the years on the role of covalent binding in the pathogenesis of idiosyncratic drug-related toxicity, the formation of chemically reactive drug metabolites has been viewed as a risk factor in drug development, either through direct tissue damage or through haptenization of proteins that may elicit an immune response.
Consequently, despite many examples of successful and effective drugs that function through a covalent mechanism (for example, esomeprazole (Nexium; AstraZeneca) and clopidogrel (Plavix; Sanofi-Aventis/Bristol-Myers Squibb); see Supplementary information S1 (table)), there has been a reluctance to apply a covalent mode of action in drug discovery programmes. Indeed, essentially all existing first-in-class covalent drugs were initially discovered through screening in biological assays, and their covalent molecular mechanisms were elucidated afterwards.
In recent years, it has been recognized that the distinct strengths of covalent and non-covalent modes of drug action may be combined by designing compounds that combine carefully tuned reactivity with specific complementarity to the target. This concept has a long track-record in the form of mechanism-based or suicide inhibitors that directly target a catalytic nucleophile within the active site of the enzyme. However, current covalent drug discovery programmes take a different approach by instead targeting a non-catalytic nucleophile that is poorly conserved across the target protein family. Such compounds, herein referred to as 'targeted covalent inhibitors' (TCIs), possess distinct selectivity profiles compared to reversible inhibitors or mechanism-based inactivators. Moreover, unlike these earlier approaches that are mostly specific to enzymes, the TCI approach is quite general and can be applied to many druggable proteins.
In this Review, we briefly examine the clinical utility of covalent drugs and their potential pharmacological advantages compared to conventional agents. We discuss the potential risks and challenges associated with covalent drugs and how they may be mitigated by careful optimization of binding and reactivity using structure-based drug design. We summarize mechanistic and design considerations for TCIs, and some of the ways in which the discovery and optimization of such compounds differ from the methods used for more traditional agents.
Prevalence of covalent drugs
Despite generally being avoided by the pharmaceutical industry, covalent drugs have been approved as treatments for diverse clinical indications and have made a major positive impact on human health1,2. In addition, covalent drugs have proved to be a highly profitable class of therapeutics for the pharmaceutical industry. Indeed, three of the ten top-selling drugs in the United States in 2009 (clopidogrel, lansoprazole and esomeprazole) are covalent inhibitors of their targets, and all three have achieved blockbuster status. We estimate that together, the 26 covalent drugs for which data are available account for over US$33 billion in annual worldwide sales (Supplementary information S1 (table)). Moreover, this figure probably underestimates the pharmacoeconomic impact of covalent drugs, as many are now available as generics.
Another metric that may be used to assess the impact of covalent drugs is to examine target patient populations. In 2008, clopidogrel and esomeprazole together accounted for approximately 60 million prescriptions in the United States3, and aspirin (another covalent drug) is the most widely used medication in the world, with an estimated 80 billion tablets being consumed annually in the United States alone4.
An analysis of 39 covalent drugs that have been approved by the US Food and Drug Administration (FDA) shows that approximately 33% of these drugs are anti-infectives (most notably the β-lactam class of antibiotics), 20% treat cancer, 15% treat gastrointestinal disorders, and ∼15% are used to treat central nervous system and cardiovascular indications (Fig. 1). In oncology, important covalent drugs include inhibitors of aromatase5, thymidylate synthetase6 and ribonucleotide reductase7. Clopidogrel — a drug that covalently inhibits P2Y purinergic receptor 12 — represents a breakthrough treatment for vascular disorders8. Proton pump inhibitors (PPIs) such as omeprazole, esomeprazole and lansoprazole have been shown to be safe and effective in millions of patients9. Covalent drugs have also made an impact in the treatment of central nervous system disorders, with monoamine oxidase inhibitors such as rasagiline (Azilect; Teva/Lundbeck) and selegiline (Zelapar; Valeant Pharmaceuticals) being used to treat depression and Parkinson's disease10. Several of these successful covalent drugs are used as long-term therapies (for example, proton pump and 5α-reductase inhibitors), which indicates that a covalent mechanism of action may be efficacious for the chronic treatment of serious diseases.

Pie chart of 39 covalent drugs as detailed in Supplementary information S1 (table). Anti-infective (33%), cancer (20%), gastrointestinal (15%), central nervous system (10%), cardiovascular (5%), inflammation (3%) and other areas (13%).
The scope of molecular targets that can be addressed by covalent drugs is broad. Approximately one-third of all enzyme targets for which there is an FDA-approved inhibitor have an example of an approved covalent drug2. Although the majority of covalent drugs target enzymes, there are examples of agents that target other protein classes — for example, clopidogrel, which modifies a G protein-coupled receptor11.
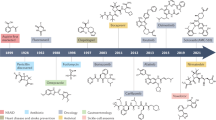
However, a common element in the discovery of covalent drugs is that they were identified not by design but by serendipity, with their covalent mechanism of action becoming apparent only after their clinical utility had been well established. The earliest example is aspirin (Fig. 2), which was first marketed over 100 years ago; aspirin covalently modifies cyclooxygenase by causing acetylation of a serine residue that is proximal to the active site12. Penicillin antibiotics represent another class of covalent inhibitors, as the penicillin β-lactam binds covalently to the active site serine of penicillin binding protein 1B (also known as bacterial DD-transpeptidase) to form inactive penicilloyl enzymes that are unable to catalyse a key step in cell-wall synthesis13.

Over the past 100 years there have been many examples of medicines that are covalent drugs.
PPIs are another example of a major class of covalent drugs that was discovered by pharmacological screening, and the PPI omeprazole has revolutionized the treatment of gastrointestinal reflux disease. PPIs are activated by acid in the gastric compartment; this locally converts the drug into a covalent modifier of the proton pump, thus minimizing systemic exposure to the active form. This covalent modification leads to a long duration of action, which makes PPIs effective at treating acid reflux diseases. Although this class of compounds was discovered through a rational screening process, the covalent mechanism of action was discovered post hoc rather than designed14. Similarly, the anticlotting drug clopidogrel was discovered through a pharmacological screen for antiplatelet drugs11.
Over recent decades, the growing reliance on target-based and structure-guided approaches to drug discovery, combined with concerns about potential toxicity risks, led to a bias against covalent inhibitors throughout the discovery pipeline. Rather than identifying active compounds in cell-based assays first and establishing their target and mechanism of action afterwards, target-based discovery focuses on a specific protein and mechanism of action. Not only the molecular target, but typically also the particular binding site and desired mode of action must be specified at the start of the project. Accordingly, there is a reluctance to pursue covalent mechanisms of action, owing to concerns over nonspecific or off-target activity, or lack of appreciation of the clinical precedent for this approach15. Hence, there has been a growing divergence between the historical and current success of covalent drugs and a reluctance to include covalent mechanisms of action in the drug discovery armamentarium.
Safety
The scepticism over the potential of covalent drugs appears to stem largely from safety concerns, owing to adverse reactions that have been associated with two major classes of therapeutic agents that covalently modify cellular proteins. These classes are: drugs that contain a pre-existing reactive electrophilic functionality in the parent structure (for example, penicillin); and compounds that undergo biotransformation in vivo to yield chemically reactive, electrophilic metabolites (for example, acetaminophen). The second group encompasses numerous therapeutic categories in which the parent compound is subjected to one or more metabolic reactions that lead to the formation of chemically reactive intermediates that covalently modify cellular proteins16.
Random, covalent binding of highly reactive drug metabolites to cellular macromolecules can sometimes result in acute tissue injury, or it can activate the immune system through haptenization of proteins. This can result in the generation of antibodies that target elements of the drug molecule or autoantibodies that recognize epitopes on proteins that are now rendered 'foreign'17. The molecular mechanisms by which biologically reactive intermediates cause their characteristic toxicities are complex and poorly understood, and the role of covalent modification of proteins by electrophilic metabolites in mediating the adverse effects associated with their parent drugs remains controversial.
However, recent advances in analytical techniques have provided insights into the structures of reactive drug metabolites18,19. Compilations of 'toxicophores' — functional groups that can be metabolized into reactive electrophiles — are available as guides for medicinal chemists who are engaged in drug design, and most pharmaceutical companies have now taken steps to minimize this potential liability in drug candidates20. The dose of a drug also appears to be important, as the majority of approved drugs that have been withdrawn from the market owing to safety concerns were administered at doses greater than 100 mg per day, whereas most drugs that are given at doses lower than 10 mg per day have acceptable safety profiles21.
It is important to recognize two fundamental differences between TCI drugs that are the subject of this article and compounds that are highly reactive per se and/or generate electrophilic metabolites. First, the reactive functionality present in highly reactive compounds, including the electrophilic intermediates they generate through metabolic processes (for example, quinones or acyl halides), typically exhibits much higher chemical reactivity than the weakly electrophilic warheads that are incorporated into TCIs. As a result, these electrophiles will alkylate or acylate a broad range of proteins with which they come into contact22. Second, these highly reactive compounds and drug metabolites typically modify those nucleophilic residues on proteins that are most accessible from the medium, as opposed to requiring a specific, non-covalent orientation within the enzyme active site, as in the case of TCIs. As a consequence, chemically reactive drugs and reactive drug metabolites exhibit a low degree of molecular selectivity in their covalent interactions with cellular proteins. Thus, it may be argued that there should be a lower chance of a covalent protein modification leading to an adverse reaction with a TCI than with a reactive electrophile that modifies biological macromolecules on a relatively indiscriminate basis1.
The demonstrated safety profiles of many approved covalent drugs seem to support this expectation, although it must be stressed that as generally applicable animal models for the human immune system do not exist, there remains a possibility that haptenization of proteins by TCIs may — in rare cases — lead to an adverse immune response in humans; the risk of such a phenomenon cannot be accurately assessed using current technology. Clearly, the safety of TCI drugs will need to be evaluated on a case-by-case basis, through conventional preclinical and clinical studies, as with any other new chemical entity.
Importantly, TCI development is currently focusing on life-threatening indications for which there are no effective therapies, so the benefits of the mechanism of action of TCIs can be justified relative to the risk of potential immunotoxicities. This benefit–risk analysis has informed the widespread clinical utility of β-lactam antibiotics. Several TCIs have generated encouraging efficacy data with safety profiles that have allowed them to advance into late-stage clinical testing. As TCIs advance towards the market, important information about their safety profiles and perhaps a better understanding of their potential to exert immunotoxicity will become available.
Mechanistic and pharmacological features
General mechanistic features. There are important issues to consider in the discovery and optimization of covalent drugs compared to reversible drugs (Fig. 3). These include: whether a target is amenable to specific covalent inhibition, and the characterization of the covalent mechanism of action at the in vitro as well as the in vivo level.

For each area, specific activities that are important for designing and optimizing covalent drugs are highlighted. PK/PD, pharmacokinetics/pharmacodynamics.
The action of a target-specific covalent inhibitor can be described by the generic mechanism shown in equation 1.

Inhibition occurs in at least two steps: the compound must first bind non-covalently to the target protein, placing its moderately reactive electrophile close to a specific nucleophile on the protein. The resulting complex then undergoes specific bond formation, which gives rise to the inhibited complex. In cases in which bond formation is effectively irreversible, k−2 will essentially be zero. TCIs are distinct from mechanism-based inactivators in that the latter use the catalytic machinery of an enzyme target to convert an unreactive ligand into a highly reactive intermediate, which then leads to covalent, irreversible inhibition of the enzyme target.
It is apparent from equation 1 that in considering the impact of covalent bond formation on inhibition, it is appropriate to think in terms of a continuum or range of covalent effects. One extreme is defined by fully irreversible inhibitors, for which k−2 = 0. If they are given sufficient time to react, irreversible covalent inhibitors will provide complete and permanent blockade of the target protein, with their concentration and time-dependent inhibition governed by Ki and k2 (Box 1). The other extreme is defined by fully non-covalent compounds, for which the final covalent complex, E – I, does not form and thus k2 = 0.
Reversible covalent inhibitors have finite values for both k2 and k−2, and they encompass a range of behaviours between these two extremes, in which covalent bond formation increases the degree of inhibition and thus renders the inhibitor more potent, and in which the lifetime of the inhibited complex is typically governed by k−2. Some non-covalent inhibitors have binding mechanisms that involve slow conformational changes subsequent to initial binding, similar to that shown in equation 1 but without the formation of a covalent complex. These so-called 'slow, tight-binding inhibitors' share some of the favourable properties of covalent drugs, such as a long target residence time (vide infra).
It is important to note that even though a protein target is subjected to irreversible inhibition, activity will be restored following re-synthesis of that enzyme or receptor, once the unbound drug has been cleared from the body. Thus, inhibition can be considered to be mechanistically irreversible if the kinetic half-life of the covalent complex is long compared to re-synthesis of the target protein. The time taken for the activity of the target protein to recover after withdrawal or elimination of the compound depends on both the residence time of the inhibitor–protein complex and also on the protein re-synthesis rate.
Owing to the kinetic considerations outlined above, the approaches required to identify and optimize covalent inhibitors differ in important ways from the methods used for reversible inhibitors. The potency and selectivity of conventional reversible inhibitors are typically defined in terms of the equilibrium binding affinity for the target, or the concentration of the compound that is required to achieve 50% inhibition in a biochemical or cellular assay (IC50). However, the potency of irreversible or slowly reversible covalent inhibitors must be considered quite differently, as described in Box 1. If the reaction is allowed to proceed for a sufficient period of time, any inhibitor concentration (provided it is higher than the target concentration) would be expected to result in essentially complete inhibition. Explicit consideration of the time-dependence of inhibition is thus essential to any assessment of the absolute or relative activity of covalent inhibitors. The kinetics of inhibition for reversible inhibitors, particularly the target residence time, has also started to receive increased attention for reversible inhibitors in recent years23,24,25,26.
Importantly, the potency and selectivity of an irreversible inhibitor can be optimized by altering the structure of the compound to modulate either its non-covalent binding to the target (Ki), or the rate at which it reacts with the target nucleophile after it is bound (k2). These dual parameters allow the potency and selectivity of covalent inhibitors to be fine-tuned, and they have been used to characterize structure–activity relationships (SARs) for irreversible inhibitors of several drug targets, including dopamine hydroxylase27, caspases28 and the serine hydrolase fatty acid amide hydrolase (FAAH)29.
Potency. A major challenge in drug discovery is achieving high potency and selectivity in a compound without increasing its molecular mass to the point at which beneficial pharmaceutical properties are jeopardized. It seems that covalent inhibitors may have the potential to address this challenge. There is a limit to the binding affinity that can generally be achieved for a ligand of a given size using non-covalent interactions30. This concept is quantified in terms of 'ligand efficiency', defined as the free energy of binding per heavy atom of the ligand31. Thus, it has been shown — across a set of drug optimization projects — that the maximum non-covalent binding energy that can be achieved by conventional medicinal chemistry is, on average, ∼0.3 kcal per mol per heavy atom32. However, this level of ligand efficiency can be difficult to attain.
Houk and colleagues33 have reported that protein–ligand complexes that rely on covalent interactions routinely exceed these ligand efficiency limits. Inhibitors that rely on covalent bonding dramatically favour the bound form, which leads to potencies and ligand efficiencies that are either exceptionally high or, for irreversible covalent interactions, even essentially infinite. Covalent bonding thus allows high potency to be routinely achieved in compounds of low molecular mass, along with all the beneficial pharmaceutical properties that are associated with small size.
Moreover, for some applications it has been shown that exceptionally high affinities are desirable. The TCI approach provides a means for reliably improving potency in situations in which this cannot always be achieved with non-covalent compounds. This approach places considerably fewer constraints on the size and structure of the remainder of the molecule, thus giving greater scope for the optimization of other important properties such as absorption, distribution, metabolism and excretion (ADME)33.
Selectivity. Covalent inhibitors have two drivers for achieving selectivity towards their protein target: the initial binding step, Ki, and the subsequent chemical step, k2 (equation 1). For high selectivity, the non-covalent affinity (Ki) of the inhibitor must be high enough to ensure that the compound binds selectively to the desired target and achieves a residence time that is sufficient for a covalent reaction. Similarly, the reaction rate of the bound inhibitor (k2) must be high enough to give a high probability that the reaction will occur within the lifetime of the non-covalent complex that is formed in the initial step of the reaction. However, because highly reactive electrophiles must be avoided, this reaction rate must be achieved primarily by optimal positioning of the electrophile relative to the nucleophile on the target. Thus, achieving desirable selectivity requires optimization of both Ki and k2. The effect of this strategy on the acquisition of SAR data and its use in optimizing covalent inhibitors is discussed in Box 1.
The factors that affect Ki are, in terms of the structural recognition of the target by the drug, generally quite distinct from those that affect k2. Non-covalent binding typically correlates with overall sequence and structural conservation at the binding site, which often makes it difficult to achieve high degrees of discrimination between closely related members of the same protein family. By contrast, k2 depends on the presence of an appropriate nucleophilic residue at a specific position on the target, which — depending on the residue chosen — may or may not be conserved across a protein family. The availability of these two orthogonal drivers of drug–target interaction allows exceptional potency and selectivity to be achieved for carefully designed compounds34.
Achieving high selectivity against off-target reactions clearly requires that the intrinsic reactivity of the electrophilic warhead on the inhibitor must be low, such that reaction with thiols is only appreciable when it is preceded by specific non-covalent binding, which holds the reacting groups at a mutual distance and orientation that is highly favourable for reaction. It is well known that an intimate proximity between reacting groups can accelerate the rate of reaction by many orders of magnitude35,36, and this can allow efficient bonding between reactants for which the intrinsic bimolecular reaction rate is negligible. In the case of covalent drugs, the electrophilic reactivity must be sufficiently low so that no appreciable irreversible reaction occurs with other thiol-containing molecules, even if they are present at a high concentration in vivo.
Even with an electrophile of low intrinsic reactivity, achieving selectivity can be challenging if the compound targets a nucleophilic residue that is highly conserved across a protein family, as is the case for suicide inhibitors. This is because each family member can be expected to present an appropriately positioned nucleophile. For example, many covalent inhibitors have been described for the serine protease and cysteine protease families37, in which the inhibitor covalently modifies the catalytically essential nucleophile. Consequently, selectivity has typically been difficult to achieve, because even relatively weak binding to the active site of an off-target protease within the same family could result in a covalent interaction with the conserved catalytic nucleophile and lead to irreversible inhibition38. Similarly, in the protein kinase family, covalent inhibitors (such as wortmannin) that target a conserved, catalytic lysine residue have substantial selectivity challenges that are compounded by the intrinsic reactivity of the electrophile. However, there are rare examples in which high selectivity has been achieved; for example, an irreversible inhibitor of FAAH exhibits excellent potency and selectivity in vitro and in vivo29.
An alternative strategy, which is embodied in the TCI approach, is to develop inhibitors that covalently target a nucleophile that is unique or rare across a protein family, thereby ensuring that covalent bond formation cannot occur with most other family members. This approach can lead to high selectivity against closely related proteins because although the inhibitor might bind transiently to the active sites of such proteins, it will not covalently label them if they lack the targeted nucleophilic residue in the appropriate position. Only inhibitors that possess favourable values for both Ki and k2 will bind to and react with the target. Proteomic analysis confirms the selectivity of covalent inhibitors towards off-target proteins in complex biological mixtures and supports their potential for high specificity at therapeutic concentrations29,39,40.
Pharmacodynamics. Irreversible inhibition has important and potentially advantageous consequences for drug pharmaco-dynamics in which the level and frequency of dosing relates to the extent and duration of the resulting pharmacological effect. In particular, when covalent modification of a drug target is irreversible, the restoration of pharmacological activity requires re-synthesis of the protein target. Schramm and colleagues41 have termed the situation in which the rate of inhibitor dissociation is negligible compared to the lifetime of the protein target as the “ultimate physiological goal” of inhibitor design41.
The prolonged duration of drug action on the target effectively uncouples the pharmacodynamics of the drug from the pharmacokinetics of exposure, as target inhibition persists after the drug has been cleared. This property of covalent drugs enables less frequent dosing and the potential for lower drug doses. For example, benzimidazole-based PPIs — such as omeprazole — have relatively short pharmacokinetic half-lives (1–2 hours), but these covalent agents can be dosed daily because the (H++K+)ATPase has a slow re-synthesis half-life (∼54 hours) and, consequently, the half-life for the recovery of gastric acid secretion is as long as 28 hours after treatment with omeprazole42.
The prolonged duration of inhibition for the covalently modified drug target also contributes to the selectivity that can be achieved with TCIs in vivo. This is because even in cases in which the inhibitor is capable of binding non-covalently to an off-target, unless binding is exceptionally strong (that is, much stronger than the non-covalent interactions with the intended target), the resulting complex will be short-lived and inhibition will be relieved as the drug is cleared. Thus, sustained inhibition will only be achieved for targets that have both non-covalent and covalent complementarity with the drug.
Importantly, after the target protein has been inactivated by the irreversible reaction, the drug should preferably be cleared rapidly to minimize off-target interactions (both covalent and non-covalent), which could conceivably reduce non-mechanism-based toxicity. Although reversible inhibitors with long residence times could potentially achieve a prolonged duration of action as well, the use of a covalent inhibitor-based strategy is a reliable and rational way to confer this important property.
Drug resistance
A major challenge for the treatment of cancer and infectious diseases is the emergence of drug resistance owing to mutations in the binding site of a drug target. It appears that irreversible inhibitors may maintain activity against drug-resistant mutations that are acquired after treatment with reversible inhibitors43.
For example, approximately 50% of patients with non-small-cell lung carcinoma (NSCLC) who initially responded to reversible EGFR inhibitors relapsed owing to the emergence of tumour cells that express EGFR with mutations at T790M and/or L858R in the ATP binding site43,44. Screening of a panel of known inhibitors for activity against an NSCLC cell line expressing the T790M–L858R double-mutant form of EGFR showed that the irreversible inhibitors were all effective at inhibiting cell proliferation43,45. By contrast, none of the reversible EGFR inhibitors tested was effective against the mutant cell line.
One reason why irreversible inhibitors might be more effective against resistance mutants is that they do not affect the extent of inhibition; they only affect the rate at which the inhibited complex forms. Thus, given sufficient exposure to the compound, even mutants that react considerably more slowly will become fully inhibited. An irreversible mechanism of action also helps to mitigate against competition by high intracellular concentrations of ATP46. Another possible advantage of irreversible inhibitors in this regard is their sustained duration of inhibition of the target, as repeated periods of incomplete target coverage could promote the development of resistance mutations.
Beyond oncology, it seems that irreversible inhibitors may have applications in the treatment of infectious diseases that have developed resistance to existing therapies. For example, numerous mutations in hepatitis C virus (HCV) protease have been reported that render HCV resistant to the emerging protease inhibitors that are currently in clinical development. However, an irreversible HCV protease inhibitor has recently been described that is active against clinical mutations and therefore may provide an alternative strategy to overcome the substantial challenge of drug resistance40.
Although targeting rare amino acids is a strategy for conferring selectivity of covalent inhibitors, evasion of inhibition may arise by mutational resistance in indications such as cancer and infectious diseases. In such situations, the rarity of the targeted amino acid may indicate that it is not essential to the function of the targeted protein, thus offering a facile escape from TCI therapy. However, mutations in tumours typically exist before therapy and only emerge clonally following inhibition, so it may be possible to anticipate this mechanism of resistance through tumour proteomics analysis. With pathogens, therapy-induced mutation is often observed, regardless of the mechanism of action of the inhibitor. It should be noted, however, that many non-catalytic residues are well-conserved, which implies that they have important yet obscure roles in the fitness of the organism.
In practice, when designing a therapeutic agent with the maximum potential for clinical success, TCIs offer the possibility of dual inhibitory mechanisms: irreversible modification of the intended amino acid and reversible occupancy of the binding site. Thus, should a pathogen or tumour attempt to evade covalent inhibition by mutation of the targeted nucleophile, the retention of high-affinity non-covalent inhibition provides a second mechanism for inhibition.
Design and optimization of TCIs
The design of covalent drugs requires careful optimization of both the non-covalent binding affinity (which is reflected in Ki) and the reactivity of the electrophilic warhead (which is reflected in k2). The initial design of TCIs involves three key steps. First, bioinformatics analysis is used to identify a nucleophilic amino acid (for example, cysteine) that is either inside or near to a functionally relevant binding site on a drug target, but is rare in that protein family. Next, a reversible inhibitor is identified for which the binding mode is known. Finally, structure-based computational methods are used to guide the design of modified ligands that have electrophilic functionality, and are positioned to react specifically with the nucleophilic amino acid in the target protein.
From the initial designs, a small set of candidate compounds can be synthesized and tested for their ability to modify the target compound. The most active compounds are then characterized to determine Ki and k2 (Box 1), thus allowing their activity to be optimized in rational, data-driven ways. Small modifications to the inhibitor structure that introduce minor changes in the orientation of the electrophilic warhead with respect to the nucleophilic reaction partner on the target protein can be used to optimize potency and selectivity, the success of these efforts being monitored through increases in k2. The potency and selectivity of the inhibitor can also be modulated by tuning the non-covalent affinity of the compound to optimize Ki.
The opportunity to start with reversible inhibitors that display inherent affinity enables less reactive electrophiles to be used for TCIs, thus minimizing off-target reactivity. Because the attachment of the electrophilic warhead does not generally increase either the molecular mass or the lipophilicity of the compound, the ability to start from a drug-like, non-covalent scaffold increases the likelihood of quickly achieving a drug candidate with desired pharmaceutical characteristics.
An early example of the TCI approach targeted the kinase domain of EGFR, which is a major oncology drug target47. The majority of EGFR inhibitors target the ATP binding site, which is highly conserved across the 471 human protein kinases, thus making it difficult to achieve high levels of selectivity. Bioinformatics analysis has identified a cysteine residue (Cys797) in the ATP binding site of the EGFR kinase that is rare in the protein kinase family47. Starting with the natural co-factor, ATP, structure-based methods were used to design thioadenosine, which was predicted to bind to EGFR and covalently modify Cys797; this prediction was confirmed experimentally. Subsequently, this strategy was applied to more drug-like EGFR inhibitors such as quinazolines48,49. The design strategy involved modelling the quinazoline inhibitor into the binding site of EGFR and identifying suitable positions on the scaffold to append an electrophile to allow efficient covalent bonding with Cys797 (Fig. 4a). This TCI approach was originally applied to EGFR in the early 1990s47 and subsequently to other protein kinases50,51,52,53, proteases40 (Fig. 4b) and recently to transthyretin (TTR) to achieve highly selective irreversible inhibitors52.

a | Epidermal growth factor receptor (EGFR) with neratinib (HKI-72) (PDB code: 2JIV). b | Hepatitis C virus (HCV) protease with ligand 3 (PDB code: 3OYP). The protein backbone is shown in green, and the small molecule and the cysteine side chain of the protein are shown in a stick representation and coloured by atom type. For both the EGFR and the HCV protein, a covalent bond exists between the targeted covalent inhibitor and the cysteine side chain.
Various electrophilic functionalities have been tried with TCIs, with the constraint that the groups show minimal nonspecific reactivity towards other thiols but react efficiently with the target cysteine residue when held in proximity. Ultimately, an acrylamide or substituted acrylamide proved to be the electrophile of choice across several scaffolds; acrylamides imparted potency and selectivity against the enzyme in both biochemical assays and in cells, and showed strong activity in vivo54.
Several publications have reported on TCIs that use substituted acrylamides as warheads and demonstrated that they are relatively poor electrophiles and require proximity to their target proteins for reaction. For example, incorporation of an acrylamide onto a quinazoline core at position six completely inactivated EGFR signalling in cells in less than a minute, whereas substitution of the quinazoline core at position seven took hours55. In addition, an acrylamide-containing HCV protease inhibitor specifically labelled HCV protease on only one out of seven possible cysteines, and this inhibitor exhibited low reactivity towards off-target proteins in cells. Beyond acrylamides, chemoselective agents that covalently modify the abundant plasma protein TTR in preference to the large number of other human plasma proteins have also been described52.
These data are encouraging as they demonstrate that specificity can be achieved with the TCI approach; however, it is important to carefully assess their potential for off-target reactivity. A number of papers have described the assessment of the inherent reactivity of TCIs towards nonspecific thiols, such as glutathione34,56. For more advanced compounds, the potential for performing radiolabelling studies to assess the off-target reactivity of irreversible inhibitors is feasible. Radiolabelling studies have demonstrated high specificity of irreversible EGFR inhibitors in cellular lysates55,57. These studies were also useful in demonstrating that neratinib (HKI-272) forms a reversible covalent adduct with K190 of human serum albumin but not with mouse, rat or rabbit serum albumin58,59. In addition, proteomics-based approaches have been suggested as a way to evaluate off-target reactivity and 'de-risk' the safety issues of irreversible inhibitors60. The opportunity to use these assays to minimize the off-target reactivity enables a data-driven means to reduce the potential for toxicities.
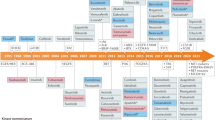
The advancement of several rationally engineered covalent inhibitors into late-stage clinical trials validates the TCI approach as a new path for the expansion of this therapeutic class. To date, only a small fraction of the targets for which the TCI approach could be applied has been exploited clinically. For example, bioinformatics analysis of the protein kinase ATP-binding site has shown that there is a broad opportunity for TCIs to target uncommon cysteines (∼100 kinases)50. However, so far only three of these kinases have been pursued clinically with TCIs, in contrast to over 50 kinases that have been pursued with reversible inhibitors (see the ChEMBL-og website).
As selectivity and drug resistance remain serious issues for many kinase inhibitors, there is a need for further exploration of the potential of the TCI approach to overcome these limitations. Importantly, the general applicability of the approach beyond the kinase family is also validated by examples of recently designed covalent inhibitors that have been reported for TTR52 and HCV protease40. Efforts are underway to analyse the human genome using structural bioinformatics and chemogenomics to assess the breadth of targets amenable to the TCI opportunity (J.S. and R.P., unpublished observations).
Clinical progress with TCIs
Despite the apparent lack of attention towards covalent inhibitor drug discovery by most pharmaceutical companies, there are several examples of covalent drugs that are progressing to late-stage clinical development (Table 1). Afatinib (BIBW-2992)61 and PF-00299804 (Ref. 62) are potent TCIs of both EGFR and the receptor tyrosine-protein kinase ERBB2, and have advanced to Phase III trials for NSCLC. There has also been progress in the treatment of metastatic breast cancer with the irreversible EGFR/ERBB2 inhibitor neratinib49. Recently, results from a Phase II trial showed that neratinib had substantial clinical activity and was well tolerated as a form of monotherapy63. This trial involved patients with breast cancer tumours that were positive for ERBB2, who either had or had not previously undergone therapy with trastuzumab (Herceptin; Genentech/Roche)63.
There has also been progress in targeting Bruton's tyrosine kinase (BTK) with a TCI-based approach for the treatment of haematological cancers. Two covalent BTK inhibitors — AVL-292 and PCI-32765 — have advanced into clinical trials and they demonstrate potent inhibition of the target and prolonged duration of action, with exciting early clinical activity observed with PCI-32765 in a small trial of haematological cancers39,64. In addition, the cytochrome P450 17A1 inhibitor abiraterone has advanced to Phase III clinical trials in prostate cancer65. Beyond cancer, the reversible covalent HCV protease inhibitors telapravir and boceprevir are currently awaiting approval for the treatment of HCV infection.
Summary
Despite the many examples of successful covalent drugs, principles for the rational design of these molecules have only recently emerged, thus enabling the expansion of this therapeutic class. It is now apparent that structural bioinformatics approaches, coupled with structure-based drug design, may enable the engineering of highly selective covalent drugs.
As TCIs advance through clinical development, important insights into their safety and efficacy profiles will emerge, which will enable a better understanding of the benefit–risk balance of this mechanism of drug action. Also, for cases in which TCIs can be compared clinically with reversible drugs against the same target, it will be important to assess the benefits and/or disadvantages of these different mechanisms of action to both the efficacy and safety of TCIs.
The purpose of this Review is to encourage the investigation and promote an informed assessment of the advantages and limitations of a covalent approach, and to demonstrate that a covalent strategy is compatible with a target-directed, structure-guided drug discovery paradigm. We anticipate that the next decade will see a resurgence of interest in this important class of therapeutics.

References
Potashman, M. H. & Duggan, M. E. Covalent modifiers: an orthogonal approach to drug design. J. Med. Chem. 52, 1231–1246 (2009). An excellent historical perspective on the prevalence of covalent drugs.
Robertson, J. G. Mechanistic basis of enzyme-targeted drugs. Biochemistry 44, 5561–5571 (2005).
Bartholow, M. Top 200 Prescription Drugs of 2009. Pharmacy Times [online], (2010).
Jeffreys, D. Aspirin: The Remarkable Story of a Wonder Drug (Bloomsbury, New York, 2004).
Dixon, J. M. Exemestane: a potent irreversible aromatase inactivator and a promising advance in breast cancer treatment. Expert. Rev. Anticancer Ther. 2, 267–275 (2002).
Thomas, D. M. & Zalcberg, J. R. 5-fluorouracil: a pharmacological paradigm in the use of cytotoxics. Clin. Exp. Pharmacol. Physiol. 25, 887–895 (1998).
Xu, H., Faber, C., Uchiki, T., Racca, J. & Dealwis, C. Structures of eukaryotic ribonucleotide reductase I define gemcitabine diphosphate binding and subunit assembly. Proc. Natl Acad. Sci. USA 103, 4028–4033 (2006).
Yusuf, S. et al. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N. Engl. J. Med. 345, 494–502 (2001).
Esplugues, J. V. & Marti-Cabrera, M. Safety and interactions of proton pump inhibitors: lessons learned in millions of patients. Gastroenterol. Hepatol. 33 (Suppl. 1), 15–21 (2010).
Oldfield, V., Keating, G. M. & Perry, C. M. Rasagiline: a review of its use in the management of Parkinson's disease. Drugs 67, 1725–1747 (2007).
Savi, P. et al. Identification and biological activity of the active metabolite of clopidogrel. Thromb. Haemost. 84, 891–896 (2000).
Warner, T. D. & Mitchell, J. A. Cyclooxygenase-3 (COX-3): filling in the gaps toward a COX continuum? Proc. Natl Acad. Sci. USA 99, 13371–13373 (2002).
Waxman, D. J. & Strominger, J. L. Penicillin-binding proteins and the mechanism of action of β-lactam antibiotics. Annu. Rev. Biochem. 52, 825–869 (1983).
Olbe, L., Carlsson, E. & Lindberg, P. A proton-pump inhibitor expedition: the case histories of omeprazole and esomeprazole. Nature Rev. Drug Discov. 2, 132–139 (2003).
Lipinski, C. & Hopkins, A. Navigating chemical space for biology and medicine. Nature 432, 855–861 (2004).
Erve, J. C. Chemical toxicology: reactive intermediates and their role in pharmacology and toxicology. Expert. Opin. Drug Metab. Toxicol. 2, 923–946 (2006).
Uetrecht, J. Immune-mediated adverse drug reactions. Chem. Res. Toxicol. 22, 24–34 (2009).
Baillie, T. A. Future of toxicology — metabolic activation and drug design: challenges and opportunities in chemical toxicology. Chem. Res. Toxicol. 19, 889–893 (2006). A comprehensive overview of the toxicological implications of biotransformation of drugs.
Liebler, D. C. Protein damage by reactive electrophiles: targets and consequences. Chem. Res. Toxicol. 21, 117–128 (2008).
Kumar, S., Kassahun, K., Tschirret-Guth, R. A., Mitra, K. & Baillie, T. A. Minimizing metabolic activation during pharmaceutical lead optimization: progress, knowledge gaps and future directions. Curr. Opin. Drug Discov. Devel. 11, 43–52 (2008).
Lammert, C. et al. Relationship between daily dose of oral medications and idiosyncratic drug-induced liver injury: search for signals. Hepatology 47, 2003–2009 (2008).
Zhang, D. et al. Metabolism, pharmacokinetics, and protein covalent binding of radiolabeled MaxiPost (BMS-204352) in humans. Drug Metab. Dispos. 33, 83–93 (2005).
Tummino, P. J. & Copeland, R. A. Residence time of receptor-ligand complexes and its effect on biological function. Biochemistry 47, 5481–5492 (2008).
Copeland, R. A., Pompliano, D. L. & Meek, T. D. Drug-target residence time and its implications for lead optimization. Nature Rev. Drug Discov. 5, 730–739 (2006).
Copeland, R. A. Evaluation of enzyme inhibitors in drug discovery: A guide for medicinal chemists and pharmacologists. 1–265 (Wiley, Hoboken, New Jersey, 2005).
Lu, H. & Tonge, P. J. Drug-target residence time: critical information for lead optimization. Curr. Opin. Chem. Biol. 14, 467–474 (2010).
Rajashekhar, B., Fitzpatrick, P. F., Colombo, G. & Villafranca, J. J. Synthesis of several 2-substituted 3-(p-hydroxyphenyl)-1-propenes and their characterization as mechanism-based inhibitors of dopamine β-hydroxylase. J. Biol. Chem. 259, 6925–6930 (1984).
Wu, J. C. & Fritz, L. C. Irreversible caspase inhibitors: tools for studying apoptosis. Methods 17, 320–328 (1999).
Ahn, K. et al. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem. Biol. 16, 411–420 (2009).
Kuntz, I. D., Chen, K., Sharp, K. A. & Kollman, P. A. The maximal affinity of ligands. Proc. Natl Acad. Sci. USA 96, 9997–10002 (1999).
Hopkins, A. L., Groom, C. R. & Alex, A. Ligand efficiency: a useful metric for lead selection. Drug Discov. Today 9, 430–431 (2004).
Hajduk, P. J. Fragment-based drug design: how big is too big? J. Med. Chem. 49, 6972–6976 (2006).
Smith, A. J., Zhang, X., Leach, A. G. & Houk, K. N. Beyond picomolar affinities: quantitative aspects of noncovalent and covalent binding of drugs to proteins. J. Med. Chem. 52, 225–233 (2009). An insightful review of the potential for covalent drugs to achieve excellent potency towards drug targets.
Wissner, A. et al. Synthesis and structure–activity relationships of 6,7-disubstituted 4-anilinoquinoline-3-carbonitriles. The design of an orally active, irreversible inhibitor of the tyrosine kinase activity of the epidermal growth factor receptor (EGFR) and the human epidermal growth factor receptor-2 (HER-2). J. Med. Chem. 46, 49–63 (2003).
Jencks, W. P. Binding energy, specificity, and enzymic catalysis: the circe effect. Adv. Enzymol. Relat. Areas Mol. Biol. 43, 219–410 (1975).
Menger, F. M. On the source of intramolecular and enzymatic reactivity. Acc. Chem. Res. 18, 128–134 (1985).
Puente, X. S., Sanchez, L. M., Overall, C. M. & Lopez-Otin, C. Human and mouse proteases: a comparative genomic approach. Nature Rev. Genet. 4, 544–558 (2003).
Turk, B. Targeting proteases: successes, failures and future prospects. Nature Rev. Drug Discov. 5, 785–799 (2006).
Honigberg, L. A. et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl Acad. Sci. USA 107, 13075–13080 (2010).
Hagel, M. Selective irreversible inhibition of a protease by targeting a non-catalytic cysteine. Nature Chem. Biol. 7, 22–24 (2010). This was the first reported example of a TCI of a protease that targeted a non-catalytic residue.
Lewandowicz, A., Tyler, P. C., Evans, G. B., Furneaux, R. H. & Schramm, V. L. Achieving the ultimate physiological goal in transition state analogue inhibitors for purine nucleoside phosphorylase. J. Biol. Chem. 278, 31465–31468 (2003).
Sachs, G., Shin, J. M. & Howden, C. W. Review article: the clinical pharmacology of proton pump inhibitors. Aliment. Pharmacol. Ther. 23 (Suppl. 2), 2–8 (2006).
Kwak, E. L. et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc. Natl Acad. Sci. USA 102, 7665–7670 (2005).
Pao, W. et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2, e73 (2005).
Carter, T. A. et al. Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. Proc. Natl Acad. Sci. USA 102, 11011–11016 (2005).
Yun, C. H. et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl Acad. Sci. USA 105, 2070–2075 (2008).
Singh, J. et al. Structure-based design of a potent, selective, and irreversible inhibitor of the catalytic domain of the erbB receptor subfamily of protein tyrosine kinases. J. Med. Chem. 40, 1130–1135 (1997). The original outline of the TCI strategy targeted towards EGFR.
Fry, D. W. Inhibition of the epidermal growth factor receptor family of tyrosine kinases as an approach to cancer chemotherapy: progression from reversible to irreversible inhibitors. Pharmacol. Ther. 82, 207–218 (1999).
Wissner, A. & Mansour, T. S. The development of HKI-272 and related compounds for the treatment of cancer. Arch. Pharm. (Weinheim) 341, 465–477 (2008).
Cohen, M. S., Zhang, C., Shokat, K. M. & Taunton, J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science 308, 1318–1321 (2005).
Zhou, W. et al. A structure-guided approach to creating covalent FGFR inhibitors. Chem. Biol. 17, 285–295 (2010).
Schirmer, A., Kennedy, J., Murli, S., Reid, R. & Santi, D. V. Targeted covalent inactivation of protein kinases by resorcylic acid lactone polyketides. Proc. Natl Acad. Sci. USA 103, 4234–4239 (2006).
Choi, S., Connelly, S., Reixach, N., Wilson, I. A. & Kelly, J. W. Chemoselective small molecules that covalently modify one lysine in a non-enzyme protein in plasma. Nature Chem. Biol. 6, 133–139 (2010).
Fry, D. W. Site-directed irreversible inhibitors of the erbB family of receptor tyrosine kinases as novel chemotherapeutic agents for cancer. Anticancer Drug Des. 15, 3–16 (2000).
Fry, D. W. et al. Specific, irreversible inactivation of the epidermal growth factor receptor and erbB2, by a new class of tyrosine kinase inhibitor. Proc. Natl Acad. Sci. USA 95, 12022–12027 (1998). This was the first example of targeted covalent inhibition of a kinase with detailed in vitro and in vivo characterization.
Gan, J., Harper, T. W., Hsueh, M. M., Qu, Q. & Humphreys, W. G. Dansyl glutathione as a trapping agent for the quantitative estimation and identification of reactive metabolites. Chem. Res. Toxicol. 18, 896–903 (2005).
Torrance, C. J. et al. Combinatorial chemoprevention of intestinal neoplasia. Nature Med. 6, 1024–1028 (2000).
Chandrasekaran, A. et al. Reversible covalent binding of neratinib to human serum albumin in vitro. Drug Metab. Lett. 4, 220–227 (2010).
Wang, J. et al. Characterization of HKI-272 covalent binding to human serum albumin. Drug Metab. Dispos. 38, 1083–1093 (2010).
Johnson, D. S., Weerapana, E. & Cravatt, B. F. Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Future Med. Chem. 2, 949–964 (2010).
Yap, T. A. et al. Phase I trial of the irreversible EGFR and HER2 kinase inhibitor BIBW 2992 in patients with advanced solid tumors. J. Clin. Oncol. 28, 3965–3972 (2010).
Gonzales, A. J. et al. Antitumor activity and pharmacokinetic properties of PF-00299804, a second-generation irreversible pan-erbB receptor tyrosine kinase inhibitor. Mol. Cancer Ther. 7, 1880–1889 (2008).
Burstein, H. J. et al. Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer. J. Clin. Oncol. 28, 1301–1307 (2010). Promising early clinical data with the TCI neratinib.
Singh, J., Petter, R. C. & Kluge, A. F. Targeted covalent drugs of the kinase family. Curr. Opin. Chem. Biol. 14, 475–480 (2010).
Vogiatzi, P. & Claudio, P. P. Efficacy of abiraterone acetate in post-docetaxel castration-resistant prostate cancer. Expert. Rev. Anticancer Ther. 10, 1027–1030 (2010).
O'Connor, O. A. et al. A phase 1 dose escalation study of the safety and pharmacokinetics of the novel proteasome inhibitor carfilzomib (PR-171) in patients with hematologic malignancies. Clin. Cancer Res. 15, 7085–7091 (2009).
Ryan, C. J. et al. Phase I clinical trial of the CYP17 inhibitor abiraterone acetate demonstrating clinical activity in patients with castration-resistant prostate cancer who received prior ketoconazole therapy. J. Clin. Oncol. 28, 1481–1488 (2010).
O'Shaughnessy, J. et al. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N. Engl. J. Med. 364, 205–214 (2011).
Lin, C., Kwong, A. D. & Perni, R. B. Discovery and development of VX-950, a novel, covalent, and reversible inhibitor of hepatitis C virus NS34A serine protease. Infect. Disord. Drug Targets 6, 3–16 (2006).
Kwo, P. Y. et al. Efficacy of boceprevir, an NS3 protease inhibitor, in combination with peginterferon α2b and ribavirin in treatment-naive patients with genotype 1 hepatitis C infection (SPRINT-1): an open-label, randomised, multicentre phase 2 trial. Lancet 376, 705–716 (2010).
Gierse, J. K., Koboldt, C. M., Walker, M. C., Seibert, K. & Isakson, P. C. Kinetic basis for selective inhibition of cyclo-oxygenases. Biochem. J. 339, 607–614 (1999).
Acknowledgements
We would like to thank the Avila team for their help and encouragement in the preparation of this manuscript. We would also like to thank K. Houk, D. Fry and A. Wissner for their helpful comments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Juswinder Singh and Russell C. Petter are employees of Avila Therapeutics.
Thomas A. Baillie and Adrian Whitty are paid consultants of Avila Therapeutics.
Supplementary information
Supplementary information Table S1
(PDF 327 kb)
Related links
Related links
DATABASES
FURTHER INFORMATION
Glossary
- Idiosyncratic drug-related toxicity
-
(IDT). A rare adverse event that is observed after administration of certain drugs and is frequently immunogenic in origin.
- Targeted covalent inhibitor
-
An inhibitor bearing a bond-forming functional group of low reactivity that, following binding to the target protein, is positioned to react rapidly with a specific non-catalytic residue at the target site. For the purposes of this Review, it is assumed that covalent modification is essentially irreversible.
- Covalent inhibitor
-
An inhibitor that reacts with its target protein to form a covalent complex in which the protein has lost its function. Covalent inhibitors can be reversible or irreversible, depending on the rate of the reverse reaction. In this Review, we predominantly consider irreversible inhibitors, and so the terms 'covalent inhibitor' and 'irreversible inhibitor' are used interchangeably.
- Irreversible inhibitor
-
An inhibitor that possesses an off-rate that is slow relative to the rate of re-synthesis of the target protein in vivo, so that once the target protein is inhibited, it does not regain activity.
- Re-synthesis rate
-
The rate at which a cell and/or organism replaces a protein target with freshly synthesized functional protein. The re-synthesis rate defines the rate at which an irreversibly inhibited protein target will recover activity in vivo, once the inhibitor is no longer present.
Rights and permissions
About this article
Cite this article
Singh, J., Petter, R., Baillie, T. et al. The resurgence of covalent drugs. Nat Rev Drug Discov 10, 307–317 (2011). https://doi.org/10.1038/nrd3410
Issue Date:
DOI: https://doi.org/10.1038/nrd3410
This article is cited by
-
Z-REX: shepherding reactive electrophiles to specific proteins expressed tissue specifically or ubiquitously, and recording the resultant functional electrophile-induced redox responses in larval fish
Nature Protocols (2023)
-
Discovery of andrographolide hit analog as a potent cyclooxygenase-2 inhibitor through consensus MD-simulation, electrostatic potential energy simulation and ligand efficiency metrics
Scientific Reports (2023)
-
Discovering covalent inhibitors of protein–protein interactions from trillions of sulfur(VI) fluoride exchange-modified oligonucleotides
Nature Chemistry (2023)
-
A simple method for developing lysine targeted covalent protein reagents
Nature Communications (2023)
-
Species-specific lipophilicities of fluorinated diketones in complex equilibria systems and their potential as multifaceted reversible covalent warheads
Communications Chemistry (2023)
