
Key Points
-
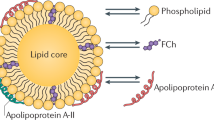
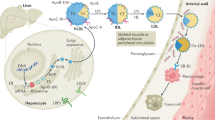
HDL protects against atherosclerosis through multiple mechanisms that include amelioration of endothelial dysfunction, removal of excess cholesterol from macrophages, and antioxidative, anti-inflammatory, and antiapoptotic effects
-
Under particular circumstances, HDL loses its atheroprotective properties, resulting in the formation of dysfunctional HDL particles
-
Dysfunctional HDL particles increase proinflammatory signalling and reduce the efflux of cholesterol from macrophages by the ATP-binding cassette transporter A1
-
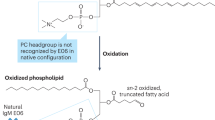
In prospective studies, myeloperoxidase-mediated oxidation of particular residues on apolipoprotein A-I creates a dysfunctional HDL particle that is associated with an increased incidence of cardiovascular events
Abstract
High-density lipoproteins (HDLs) protect against atherosclerosis by removing excess cholesterol from macrophages through the ATP-binding cassette transporter A1 (ABCA1) and ATP-binding cassette transporter G1 (ABCG1) pathways involved in reverse cholesterol transport. Factors that impair the availability of functional apolipoproteins or the activities of ABCA1 and ABCG1 could, therefore, strongly influence atherogenesis. HDL also inhibits lipid oxidation, restores endothelial function, exerts anti-inflammatory and antiapoptotic actions, and exerts anti-inflammatory actions in animal models. Such properties could contribute considerably to the capacity of HDL to inhibit atherosclerosis. Systemic and vascular inflammation has been proposed to convert HDL to a dysfunctional form that has impaired antiatherogenic effects. A loss of anti-inflammatory and antioxidative proteins, perhaps in combination with a gain of proinflammatory proteins, might be another important component in rendering HDL dysfunctional. The proinflammatory enzyme myeloperoxidase induces both oxidative modification and nitrosylation of specific residues on plasma and arterial apolipoprotein A-I to render HDL dysfunctional, which results in impaired ABCA1 macrophage transport, the activation of inflammatory pathways, and an increased risk of coronary artery disease. Understanding the features of dysfunctional HDL or apolipoprotein A-I in clinical practice might lead to new diagnostic and therapeutic approaches to atherosclerosis.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others
References
Rosenson, R. S. et al. Translation of high-density lipoprotein function into clinical practice: current prospects and future challenges. Circulation 128, 1256–1267 (2013).
Rosenson, R. S. et al. HDL measures, particle heterogeneity, proposed nomenclature, and relation to atherosclerotic cardiovascular events. Clin. Chem. 57, 392–410 (2011).
Rosenson, R. S. et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation 125, 1905–1919 (2012).
Du, X. et al. HDL particle size is a critical determinant of ABCA1-mediated macrophage cellular cholesterol export. Circ. Res. 116, 1133–1142 (2015).
Riwanto, M. et al. Altered activation of endothelial anti- and proapoptotic pathways by high-density lipoprotein from patients with coronary artery disease: role of high-density lipoprotein-proteome remodeling. Circulation 127, 891–904 (2013).
Huang, Y. et al. An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat. Med. 20, 193–203 (2014).
Rosenson, R. S., Brewer, H. B. & Rader, D. J. Lipoproteins as biomarkers and therapeutic targets in the setting of acute coronary syndrome. Circ. Res. 114, 1880–1889 (2014).
Camont, L. et al. Small, dense high-density lipoprotein-3 particles are enriched in negatively charged phospholipids: relevance to cellular cholesterol efflux, antioxidative, antithrombotic, anti-inflammatory, and antiapoptotic functionalities. Arterioscler. Thromb. Vasc. Biol. 33, 2715–2723 (2013).
Patel, S. et al. Acute hypertriglyceridaemia in humans increases the triglyceride content and decreases the anti-inflammatory capacity of high density lipoproteins. Atherosclerosis 204, 424–428 (2009).
Rohatgi, A. et al. HDL cholesterol efflux capacity and incident cardiovascular events. N. Engl. J. Med. 371, 2383–2393 (2014).
Saleheen, D. et al. Association of HDL cholesterol efflux capacity with incident coronary heart disease events: a prospective case–control study. Lancet Diabetes Endocrinol. 3, 507–513 (2015).
DiDonato, J. A. et al. Site-specific nitration of apolipoprotein A-I at tyrosine 166 is both abundant within human atherosclerotic plaque and dysfunctional. J. Biol. Chem. 289, 10276–10292 (2014).
Shao, B. et al. Humans with atherosclerosis have impaired ABCA1 cholesterol efflux and enhanced high-density lipoprotein oxidation by myeloperoxidase. Circ. Res. 114, 1733–1742 (2014).
Van Lenten, B. J. et al. Anti-inflammatory HDL becomes pro-inflammatory during the acute phase response. Loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures. J. Clin. Invest. 96, 2758–2767 (1995).
Navab, M. et al. Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: steps 2 and 3. J. Lipid Res. 41, 1495–1508 (2000).
Ansell, B. J. et al. Inflammatory/antiinflammatory properties of high-density lipoprotein distinguish patients from control subjects better than high-density lipoprotein cholesterol levels and are favorably affected by simvastatin treatment. Circulation 108, 2751–2756 (2003).
Berard, A. M. et al. High plasma HDL concentrations associated with enhanced atherosclerosis in transgenic mice overexpressing lecithin-cholesteryl acyltransferase. Nat. Med. 3, 744–749 (1997).
Vaisman, B. L. et al. Overexpression of human lecithin cholesterol acyltransferase leads to hyperalphalipoproteinemia in transgenic mice. J. Biol. Chem. 270, 12269–12275 (1995).
Foger, B. et al. Cholesteryl ester transfer protein corrects dysfunctional high density lipoproteins and reduces aortic atherosclerosis in lecithin cholesterol acyltransferase transgenic mice. J. Biol. Chem. 274, 36912–36920 (1999).
Dugi, K. A. et al. Adenovirus-mediated expression of hepatic lipase in LCAT transgenic mice. J. Lipid Res. 38, 1822–1832 (1997).
Persegol, L., Verges, B., Foissac, M., Gambert, P. & Duvillard, L. Inability of HDL from type 2 diabetic patients to counteract the inhibitory effect of oxidised LDL on endothelium-dependent vasorelaxation. Diabetologia 49, 1380–1386 (2006).
Persegol, L. et al. HDL particles from type 1 diabetic patients are unable to reverse the inhibitory effect of oxidised LDL on endothelium-dependent vasorelaxation. Diabetologia 50, 2384–2387 (2007).
Sorrentino, S. A. et al. Endothelial-vasoprotective effects of high-density lipoprotein are impaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy. Circulation 121, 110–122 (2010).
Besler, C. et al. Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J. Clin. Invest. 121, 2693–2708 (2011).
Speer, T. et al. Abnormal high-density lipoprotein induces endothelial dysfunction via activation of Toll-like receptor-2. Immunity 38, 754–768 (2013).
Kontush, A. & Chapman, M. J. Antiatherogenic function of HDL particle subpopulations: focus on antioxidative activities. Curr. Opin. Lipidol. 21, 312–318 (2010).
Hansel, B. et al. Metabolic syndrome is associated with elevated oxidative stress and dysfunctional dense high-density lipoprotein particles displaying impaired antioxidative activity. J. Clin. Endocrinol. Metab. 89, 4963–4971 (2004).
Nobecourt, E. et al. Defective antioxidative activity of small dense HDL3 particles in type 2 diabetes: relationship to elevated oxidative stress and hyperglycaemia. Diabetologia 48, 529–538 (2005).
Kontush, A., de Faria, E. C., Chantepie, S. & Chapman, M. J. Antioxidative activity of HDL particle subspecies is impaired in hyperalphalipoproteinemia: relevance of enzymatic and physicochemical properties. Arterioscler. Thromb. Vasc. Biol. 24, 526–533 (2004).
Kontush, A., de Faria, E. C., Chantepie, S. & Chapman, M. J. A normotriglyceridemic, low HDL-cholesterol phenotype is characterised by elevated oxidative stress and HDL particles with attenuated antioxidative activity. Atherosclerosis 182, 277–285 (2005).
Banka, C. L. et al. Serum amyloid A (SAA): influence on HDL-mediated cellular cholesterol efflux. J. Lipid Res. 36, 1058–1065 (1995).
Cavallero, E. et al. Abnormal reverse cholesterol transport in controlled type II diabetic patients. Studies on fasting and postprandial LpA-I particles. Arterioscler. Thromb. Vasc. Biol. 15, 2130–2135 (1995).
Brites, F. D. et al. Alterations in the main steps of reverse cholesterol transport in male patients with primary hypertriglyceridemia and low HDL-cholesterol levels. Atherosclerosis 152, 181–192 (2000).
Pennathur, S. et al. Human atherosclerotic intima and blood of patients with established coronary artery disease contain high density lipoprotein damaged by reactive nitrogen species. J. Biol. Chem. 279, 42977–42983 (2004).
Teslovich, T. M. et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466, 707–713 (2010).
Voight, B. F. et al. Plasma HDL cholesterol and risk of myocardial infarction: a Mendelian randomisation study. Lancet 380, 572–580 (2012).
Global Lipids Genetics, C. et al. Discovery and refinement of loci associated with lipid levels. Nat. Genet. 45, 1274–1283 (2013).
Do, R. et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat. Genet. 45, 1345–1352 (2013).
Rosenson, R. S., Davidson, M. H., Hirsh, B. J., Kathiresan, S. & Gaudet, D. Genetics and causality of triglyceride-rich lipoproteins in atherosclerotic cardiovascular disease. J. Am. Coll. Cardiol. 64, 2525–2540 (2014).
Holmes, M. V. et al. Mendelian randomization of blood lipids for coronary heart disease. Eur. Heart J. 36, 539–550 (2015).
Harrison, S. C., Holmes, M. V. & Humphries, S. E. Mendelian randomisation, lipids, and cardiovascular disease. Lancet 380, 543–545 (2012).
The CARDIoGRAMplusC4D Consortium. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat. Genet. 45, 25–33 (2013).
Westerterp, M. et al. ATP-binding cassette transporters, atherosclerosis, and inflammation. Circ. Res. 114, 157–170 (2014).
Pirillo, A., Uboldi, P., Bolego, C., Kuhn, H. & Catapano, A. L. The 15-lipoxygenase-modified high density lipoproteins 3 fail to inhibit the TNF-alpha-induced inflammatory response in human endothelial cells. J. Immunol. 181, 2821–2830 (2008).
Cabana, V. G., Lukens, J. R., Rice, K. S., Hawkins, T. J. & Getz, G. S. HDL content and composition in acute phase response in three species: triglyceride enrichment of HDL a factor in its decrease. J. Lipid Res. 37, 2662–2674 (1996).
Fisher, E. A., Feig, J. E., Hewing, B., Hazen, S. L. & Smith, J. D. High-density lipoprotein function, dysfunction, and reverse cholesterol transport. Arterioscler. Thromb. Vasc. Biol. 32, 2813–2820 (2012).
Alwaili, K. et al. The HDL proteome in acute coronary syndromes shifts to an inflammatory profile. Biochim. Biophys. Acta 1821, 405–415 (2012).
Rached, F. et al. Defective functionality of small, dense HDL3 subpopulations in ST segment elevation myocardial infarction: relevance of enrichment in lysophosphatidylcholine, phosphatidic acid and serum amyloid A. Biochim. Biophys. Acta 1851, 1254–1261 (2015).
Kawakami, A. et al. Apolipoprotein CIII in apolipoprotein B lipoproteins enhances the adhesion of human monocytic cells to endothelial cells. Circulation 113, 691–700 (2006).
Jensen, M. K., Rimm, E. B., Furtado, J. D. & Sacks, F. M. Apolipoprotein C-III as a potential modulator of the association between HDL-cholesterol and incident coronary heart disease. J. Am. Heart Assoc. 1, e000232 (2012).
Daugherty, A., Dunn, J. L., Rateri, D. L. & Heinecke, J. W. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J. Clin. Invest. 94, 437–444 (1994).
Zheng, L. et al. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J. Clin. Invest. 114, 529–541 (2004).
Shao, B., Oda, M. N., Oram, J. F. & Heinecke, J. W. Myeloperoxidase: an oxidative pathway for generating dysfunctional high-density lipoprotein. Chem. Res. Toxicol. 23, 447–454 (2010).
Heinecke, J. W. Pathways for oxidation of low density lipoprotein by myeloperoxidase: tyrosyl radical, reactive aldehydes, hypochlorous acid and molecular chlorine. Biofactors 6, 145–155 (1997).
Undurti, A. et al. Modification of high density lipoprotein by myeloperoxidase generates a pro-inflammatory particle. J. Biol. Chem. 284, 30825–30835 (2009).
Takeshita, J. et al. Myeloperoxidase generates 5-chlorouracil in human atherosclerotic tissue: a potential pathway for somatic mutagenesis by macrophages. J. Biol. Chem. 281, 3096–3104 (2006).
Baldus, S. et al. Endothelial transcytosis of myeloperoxidase confers specificity to vascular ECM proteins as targets of tyrosine nitration. J. Clin. Invest. 108, 1759–1770 (2001).
Shao, B. et al. Tyrosine 192 in apolipoprotein A-I is the major site of nitration and chlorination by myeloperoxidase, but only chlorination markedly impairs ABCA1-dependent cholesterol transport. J. Biol. Chem. 280, 5983–5993 (2005).
Bergt, C., Fu, X., Huq, N. P., Kao, J. & Heinecke, J. W. Lysine residues direct the chlorination of tyrosines in YXXK motifs of apolipoprotein A-I when hypochlorous acid oxidizes high density lipoprotein. J. Biol. Chem. 279, 7856–7866 (2004).
Bergt, C. et al. The myeloperoxidase product hypochlorous acid oxidizes HDL in the human artery wall and impairs ABCA1-dependent cholesterol transport. Proc. Natl Acad. Sci. USA 101, 13032–13037 (2004).
Hewing, B. et al. Effects of native and myeloperoxidase-modified apolipoprotein A-I on reverse cholesterol transport and atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 34, 779–789 (2014).
Shao, B., Tang, C., Heinecke, J. W. & Oram, J. F. Oxidation of apolipoprotein A-I by myeloperoxidase impairs the initial interactions with ABCA1 required for signaling and cholesterol export. J. Lipid Res. 51, 1849–1858 (2010).
Shao, B., Cavigiolio, G., Brot, N., Oda, M. N. & Heinecke, J. W. Methionine oxidation impairs reverse cholesterol transport by apolipoprotein A-I. Proc. Natl Acad. Sci. USA 105, 12224–12229 (2008).
Peng, D. Q. et al. Apolipoprotein A-I tryptophan substitution leads to resistance to myeloperoxidase-mediated loss of function. Arterioscler. Thromb. Vasc. Biol. 28, 2063–2070 (2008).
Van Lenten, B. J. et al. High-density lipoprotein loses its anti-inflammatory properties during acute influenza A infection. Circulation 103, 2283–2288 (2001).
Coetzee, G. A. et al. Serum amyloid A-containing human high density lipoprotein 3. Density, size, and apolipoprotein composition. J. Biol. Chem. 261, 9644–9651 (1986).
Wroblewski, J. M. et al. Nascent HDL formation by hepatocytes is reduced by the concerted action of serum amyloid A and endothelial lipase. J. Lipid Res. 52, 2255–2261 (2011).
Han, C. Y. et al. Reciprocal and coordinate regulation of serum amyloid A versus apolipoprotein A-I and paraoxonase-1 by inflammation in murine hepatocytes. Arterioscler. Thromb. Vasc. Biol. 26, 1806–1813 (2006).
Vaisar, T. et al. Inflammatory remodeling of the HDL proteome impairs cholesterol efflux capacity. J. Lipid Res. 56, 1519–1530 (2015).
Zhang, C. et al. Studies on protective effects of human paraoxonases 1 and 3 on atherosclerosis in apolipoprotein E knockout mice. Gene Ther. 17, 626–633 (2010).
Birjmohun, R. S. et al. Both paraoxonase-1 genotype and activity do not predict the risk of future coronary artery disease; the EPIC-Norfolk Prospective Population Study. PLoS ONE 4, e6809 (2009).
Marsillach, J. et al. Paraoxonase-3 is depleted from the high-density lipoproteins of autoimmune disease patients with subclinical atherosclerosis. J. Proteome Res. 14, 2046–2054 (2015).
Marathe, G. K., Zimmerman, G. A. & McIntyre, T. M. Platelet-activating factor acetylhydrolase, and not paraoxonase-1, is the oxidized phospholipid hydrolase of high density lipoprotein particles. J. Biol. Chem. 278, 3937–3947 (2003).
Kriska, T., Marathe, G. K., Schmidt, J. C., McIntyre, T. M. & Girotti, A. W. Phospholipase action of platelet-activating factor acetylhydrolase, but not paraoxonase-1, on long fatty acyl chain phospholipid hydroperoxides. J. Biol. Chem. 282, 100–108 (2007).
Rosenson, R. S. & Gelb, M. H. Secretory phospholipase A2: a multifaceted family of proatherogenic enzymes. Curr. Cardiol. Rep. 11, 445–451 (2009).
de Beer, F. C. et al. HDL modification by secretory phospholipase A2 promotes scavenger receptor class B type I interaction and accelerates HDL catabolism. J. Lipid Res. 41, 1849–1857 (2000).
de Beer, F. C. et al. Secretory non-pancreatic phospholipase A2: influence on lipoprotein metabolism. J. Lipid Res. 38, 2232–2239 (1997).
Tietge, U. J. et al. Overexpression of secretory phospholipase A2 causes rapid catabolism and altered tissue uptake of high density lipoprotein cholesteryl ester and apolipoprotein A-I. J. Biol. Chem. 275, 10077–10084 (2000).
McGillicuddy, F. C. et al. Inflammation impairs reverse cholesterol transport in vivo. Circulation 119, 1135–1145 (2009).
Kar, S. et al. Oxidized phospholipid content destabilizes the structure of reconstituted high density lipoprotein particles and changes their function. Biochim. Biophys. Acta 1821, 1200–1210 (2012).
Khera, A. V. et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 364, 127–135 (2011).
Li, X. M. et al. Paradoxical association of enhanced cholesterol efflux with increased incident cardiovascular risks. Arterioscler. Thromb. Vasc. Biol. 33, 1696–1705 (2013).
Patel, P. J., Khera, A. V., Wilensky, R. L. & Rader, D. J. Anti-oxidative and cholesterol efflux capacities of high-density lipoprotein are reduced in ischaemic cardiomyopathy. Eur. J. Heart Fail. 15, 1215–1219 (2013).
Miyazaki, O., Ogihara, J., Fukamachi, I. & Kasumi, T. Evidence for the presence of lipid-free monomolecular apolipoprotein A-1 in plasma. J. Lipid Res. 55, 214–225 (2014).
Spieker, L. E. et al. High-density lipoprotein restores endothelial function in hypercholesterolemic men. Circulation 105, 1399–1402 (2002).
Bisoendial, R. J. et al. Restoration of endothelial function by increasing high-density lipoprotein in subjects with isolated low high-density lipoprotein. Circulation 107, 2944–2948 (2003).
Yuhanna, I. S. et al. High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat. Med. 7, 853–857 (2001).
Zhao, Y., Sparks, D. L. & Marcel, Y. L. Specific phospholipid association with apolipoprotein A-I stimulates cholesterol efflux from human fibroblasts. Studies with reconstituted sonicated lipoproteins. J. Biol. Chem. 271, 25145–25151 (1996).
Carvalho, L. S. et al. HDL levels and oxidizability during myocardial infarction are associated with reduced endothelial-mediated vasodilation and nitric oxide bioavailability. Atherosclerosis 237, 840–846 (2014).
McMillen, T. S., Heinecke, J. W. & LeBoeuf, R. C. Expression of human myeloperoxidase by macrophages promotes atherosclerosis in mice. Circulation 111, 2798–2804 (2005).
Kaysen, G. A. Disorders in high-density metabolism with insulin resistance and chronic kidney disease. J. Ren. Nutr. 17, 4–8 (2007).
Morgantini, C. et al. Anti-inflammatory and antioxidant properties of HDLs are impaired in type 2 diabetes. Diabetes 60, 2617–2623 (2011).
Kontush, A. & Chapman, M. J. Why is HDL functionally deficient in type 2 diabetes? Curr. Diab. Rep. 8, 51–59 (2008).
Choudhury, R. P. & Leyva, F. C-Reactive protein, serum amyloid A protein, and coronary events. Circulation 100, e65–e66 (1999).
Bagdade, J. D., Buchanan, W. E., Kuusi, T. & Taskinen, M. R. Persistent abnormalities in lipoprotein composition in noninsulin-dependent diabetes after intensive insulin therapy. Arteriosclerosis 10, 232–239 (1990).
de Souza, J. A. et al. Metabolic syndrome features small, apolipoprotein A-I-poor, triglyceride-rich HDL3 particles with defective anti-apoptotic activity. Atherosclerosis 197, 84–94 (2008).
Zerrad-Saadi, A. et al. HDL3-mediated inactivation of LDL-associated phospholipid hydroperoxides is determined by the redox status of apolipoprotein A-I and HDL particle surface lipid rigidity: relevance to inflammation and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 29, 2169–2175 (2009).
Roberts, C. K., Ng, C., Hama, S., Eliseo, A. J. & Barnard, R. J. Effect of a short-term diet and exercise intervention on inflammatory/anti-inflammatory properties of HDL in overweight/obese men with cardiovascular risk factors. J. Appl. Physiol. 101, 1727–1732 (2006).
Hoofnagle, A. N. et al. Low clusterin levels in high-density lipoprotein associate with insulin resistance, obesity, and dyslipoproteinemia. Arterioscler. Thromb. Vasc. Biol. 30, 2528–2534 (2010).
Li, S. et al. Reduction of cold ischemia-reperfusion injury by graft-expressing clusterin in heart transplantation. J. Heart Lung Transplant. 30, 819–826 (2011).
Beauchamp, A. et al. Associations among smoking status, lifestyle and lipoprotein subclasses. J. Clin. Lipidol. 4, 522–530 (2010).
Park, K. H., Shin, D. G. & Cho, K. H. Dysfunctional lipoproteins from young smokers exacerbate cellular senescence and atherogenesis with smaller particle size and severe oxidation and glycation. Toxicol. Sci. 140, 16–25 (2014).
He, B. M., Zhao, S. P. & Peng, Z. Y. Effects of cigarette smoking on HDL quantity and function: implications for atherosclerosis. J. Cell. Biochem. 114, 2431–2436 (2013).
Song, W. et al. The implication of cigarette smoking and cessation on macrophage cholesterol efflux in coronary artery disease patients. J. Lipid Res. 56, 682–691 (2015).
Lüscher, T. F., Landmesser, U., von Eckardstein, A. & Fogelman, A. M. High-density lipoprotein: vascular protective effects, dysfunction, and potential as therapeutic target. Circ. Res. 114, 171–182 (2014).
Nicholls, S. J. et al. Consumption of saturated fat impairs the anti-inflammatory properties of high-density lipoproteins and endothelial function. J. Am. Coll. Cardiol. 48, 715–720 (2006).
Aron-Wisnewsky, J. et al. Effect of bariatric surgery-induced weight loss on SR-BI-, ABCG1-, and ABCA1-mediated cellular cholesterol efflux in obese women. J. Clin. Endocrinol. Metab. 96, 1151–1159 (2011).
Miyamoto-Sasaki, M. et al. Pitavastatin increases HDL particles functionally preserved with cholesterol efflux capacity and antioxidative actions in dyslipidemic patients. J. Atheroscler. Thromb. 20, 708–716 (2013).
Guerin, M. et al. Dose-dependent action of atorvastatin in type IIB hyperlipidemia: preferential and progressive reduction of atherogenic apoB-containing lipoprotein subclasses (VLDL-2, IDL, small dense LDL) and stimulation of cellular cholesterol efflux. Atherosclerosis 163, 287–296 (2002).
Niesor, E. J. et al. Statin-induced decrease in ATP-binding cassette transporter A1 expression via microRNA33 induction may counteract cholesterol efflux to high-density lipoprotein. Cardiovasc. Drugs Ther. 29, 7–14 (2015).
Yvan-Charvet, L. et al. Cholesterol efflux potential and antiinflammatory properties of high-density lipoprotein after treatment with niacin or anacetrapib. Arterioscler. Thromb. Vasc. Biol. 30, 1430–1438 (2010).
Khera, A. V., Patel, P. J., Reilly, M. P. & Rader, D. J. The addition of niacin to statin therapy improves high-density lipoprotein cholesterol levels but not metrics of functionality. J. Am. Coll. Cardiol. 62, 1909–1910 (2013).
The AIM-HIGH Investigators. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 365, 2255–2267 (2011).
The HPS2-THRIVE Collaborative Group. Effects of extended-release niacin with laropiprant in high-risk patients. N. Engl. J. Med. 371, 203–212 (2014).
Bays, H., Giezek, H., McKenney, J. M., O'Neill, E. A. & Tershakovec, A. M. Extended-release niacin/laropiprant effects on lipoprotein subfractions in patients with type 2 diabetes mellitus. Metab. Syndr. Relat. Disord. 10, 260–266 (2012).
Airan-Javia, S. L. et al. Atheroprotective lipoprotein effects of a niacin-simvastatin combination compared to low- and high-dose simvastatin monotherapy. Am. Heart J. 157, 687. e1–e8 (2009).
Barter, P. J. et al. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 357, 2109–2122 (2007).
Ballantyne, C. M. et al. Effect of dalcetrapib plus pravastatin on lipoprotein metabolism and high-density lipoprotein composition and function in dyslipidemic patients: results of a phase IIb dose-ranging study. Am. Heart J. 163, 515–521.e3 (2012).
Schwartz, G. G. et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 367, 2089–2099 (2012).
Nicholls, S. J. et al. Effects of the CETP inhibitor evacetrapib administered as monotherapy or in combination with statins on HDL and LDL cholesterol: a randomized controlled trial. JAMA 306, 2099–2109 (2011).
Tardif, J. C. et al. Pharmacogenomic determinants of the cardiovascular effects of dalcetrapib. Circ. Cardiovasc. Genet. 8, 372–383 (2015).
Ray, K. K. et al. The effect of cholesteryl ester transfer protein inhibition on lipids, lipoproteins, and markers of HDL function after an acute coronary syndrome: the dal-ACUTE randomized trial. Eur. Heart J. 35, 1792–1800 (2014).
Cannon, C. P. et al. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N. Engl. J. Med. 363, 2406–2415 (2010).
US National Library of Medicine. ClinicalTrials.gov [online]. (2015).
US National Library of Medicine. ClinicalTrials.gov [online], (2015).
Catalano, G. et al. Torcetrapib differentially modulates the biological activities of HDL2 and HDL3 particles in the reverse cholesterol transport pathway. Arterioscler. Thromb. Vasc. Biol. 29, 268–275 (2009).
Castro-Perez, J. et al. Anacetrapib promotes reverse cholesterol transport and bulk cholesterol excretion in Syrian golden hamsters. J. Lipid Res. 52, 1965–1973 (2011).
Nahrendorf, M. et al. Nanoparticle PET-CT imaging of macrophages in inflammatory atherosclerosis. Circulation 117, 379–387 (2008).
Nahrendorf, M. et al. Activatable magnetic resonance imaging agent reports myeloperoxidase activity in healing infarcts and noninvasively detects the antiinflammatory effects of atorvastatin on ischemia-reperfusion injury. Circulation 117, 1153–1160 (2008).
Ronald, J. A. et al. Enzyme-sensitive magnetic resonance imaging targeting myeloperoxidase identifies active inflammation in experimental rabbit atherosclerotic plaques. Circulation 120, 592–599 (2009).
Kelesidis, T. et al. Effects of lipid-probe interactions in biochemical fluorometric methods that assess HDL redox activity. Lipids Health Dis. 11, 87 (2012).
Kelesidis, T. et al. A high throughput biochemical fluorometric method for measuring lipid peroxidation in HDL. PLoS ONE 9, e111716 (2014).
Author information
Authors and Affiliations
Contributions
All the authors researched data for the article, substantially contributed to discussion of content, wrote the manuscript, and reviewed/edited it before submission.
Corresponding author
Ethics declarations
Competing interests
R.S.R. has served as a member of advisory boards for Amgen, AstraZeneca, Eli Lilly, Genzyme, GlaxoSmithKline, Novartis, Regeneron, and Sanofi; received honoraria from Kowa, travel support from LipoScience, and royalties from UpToDate, Inc.; and participates in clinical trials sponsored by Amgen, AstraZeneca, and Sanofi. H.B.B. has served as a member of advisory boards for Amgen, AstraZeneca, CSL, Eli Lilly, Merck, Pifzer, and Roche; received honoraria from Amgen, AstraZeneca, CSL, Eli Lilly, Merck, Pfizer, and Roche; received travel support from Amgen, AstraZeneca, Eli Lilly, Merck, and Roche; participates in clinical trials sponsored by Eli Lilly and Roche; is a patent holder for HDL Therapeutics; and receives royalties from AstraZeneca. B.J.A. is a member of an advisory board for Amgen; receives honoraria from Kowa; and is a shareholder in Amgen and Bruin Pharma. P.B. is a member of advisory boards for AstraZeneca, CSL, Merck, Novartis, Pfizer, and Roche; has received honoraria form Abbott, AstraZeneca, Merck, Novartis, Pfizer, and Roche; and participates in clinical trials sponsored by AstraZeneca, Merck, Pfizer, and Roche. J.C. receives research funding from CSL, Kowa, and Pfizer; is a member of advisory boards for Amgen, CSL, Danone, Merck, and Sanofi-Regeneron; has received honoraria from Amgen, Danone, Merck, Sanofi-Regeneron, and Unilever; participates in a clinical trial sponsored by AstraZeneca; and is a patent holder on the use of negatively charged phospholipids to optimize the biological function of recombinant HDL. J.W.H. is a member of advisory boards for Amgen, Bristol Myers Squibb, GlaxoSmithKline, Insilicos, and Merck; and is a patent holder for the use of oxidation markers to predict the risk of cardiovascular disease. A.K. participates in a clinical trial sponsored by CSL; and is a patent holder for the use of negatively charged phospholipids to optimize the biological function of recombinant HDL. A.R.T. is a member of advisory boards for Amgen, Arisaph, CSL, Eli Lilly, and Pfizer. N.R.W. declares no competing interests.
Rights and permissions
About this article

Cite this article
Rosenson, R., Brewer, H., Ansell, B. et al. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat Rev Cardiol 13, 48–60 (2016). https://doi.org/10.1038/nrcardio.2015.124
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrcardio.2015.124
This article is cited by
-
Sex-based association between high-density lipoprotein cholesterol and adverse outcomes after coronary artery bypass grafting
BMC Cardiovascular Disorders (2024)
-
High-density lipoprotein regulates angiogenesis by affecting autophagy via miRNA-181a-5p
Science China Life Sciences (2024)
-
Adipokine chemerin overexpression in trophoblasts leads to dyslipidemia in pregnant mice: implications for preeclampsia
Lipids in Health and Disease (2023)
-
Impact of gallstone disease on the risk of stroke and coronary artery disease: evidence from prospective observational studies and genetic analyses
BMC Medicine (2023)
-
Hypomethylation of ABCG1 in peripheral blood as a potential marker for the detection of coronary heart disease
Clinical Epigenetics (2023)
