
Abstract
Cannabis affects cognitive performance through the activation of the endocannabinoid system, and the molecular mechanisms involved in this process are poorly understood. Using the novel object-recognition memory test in mice, we found that the main psychoactive component of cannabis, delta9-tetrahydrocannabinol (THC), alters short-term object-recognition memory specifically involving protein kinase C (PKC)-dependent signaling. Indeed, the systemic or intra-hippocampal pre-treatment with the PKC inhibitors prevented the short-term, but not the long-term, memory impairment induced by THC. In contrast, systemic pre-treatment with mammalian target of rapamycin complex 1 inhibitors, known to block the amnesic-like effects of THC on long-term memory, did not modify such a short-term cognitive deficit. Immunoblot analysis revealed a transient increase in PKC signaling activity in the hippocampus after THC treatment. Thus, THC administration induced the phosphorylation of a specific Ser residue in the hydrophobic-motif at the C-terminal tail of several PKC isoforms. This significant immunoreactive band that paralleled cognitive performance did not match in size with the major PKC isoforms expressed in the hippocampus except for PKCθ. Moreover, THC transiently enhanced the phosphorylation of the postsynaptic calmodulin-binding protein neurogranin in a PKC dependent manner. These data demonstrate that THC alters short-term object-recognition memory through hippocampal PKC/neurogranin signaling.
Similar content being viewed by others
Introduction
Cannabis sativa preparations are widely consumed mainly for recreational purposes (Mounteney et al, 2016). These preparations are enriched in delta9-tetrahydrocannabinol (THC) (Gaoni and Mechoulam, 1964), the compound responsible for most central effects produced by cannabis (Hall and Degenhardt, 2009). THC produces cognitive impairment through the activation of type-1 cannabinoid (CB1) receptors in the brain, a behavioral effect extensively studied in human and animal models (Broyd, et al, 2016; Ranganathan and D'Souza, 2006; Zanettini et al, 2011). The hippocampus has a relevant role in those memory deficits produced by THC, since microinjection of CB1 receptor agonists in this brain region impairs memory (Barna et al, 2007), whereas the microinjection of CB1 receptor antagonists prevents the memory deficits produced by systemic administration of cannabinoids (Wise et al, 2009). THC systemic administration rapidly activates pathways involving MAPK (Derkinderen et al, 2003), Akt/GSK3 (Ozaita et al, 2007), and mammalian target of rapamycin complex 1 (mTORC1) (Puighermanal et al, 2009) in the hippocampus. The participation of these signaling events in the THC cognitive alterations has only been currently described for mTORC1, as specific inhibitors of this pathway manage to abolish the deficit of THC in long-term non-emotional memory (Puighermanal et al, 2009; Puighermanal et al, 2013). Whether other signaling pathways might be related to specific effects of THC over cognition remains to be elucidated.
Interestingly, the over-activation of the MAPK, Akt/GSK3, and mTORC1 signaling pathways by THC in the hippocampus is prevented by the previous blockade of N-methyl-D-aspartate (NMDA) receptors (Derkinderen et al, 2003; Puighermanal et al, 2009). Moreover, the blockade of these glutamate receptors prevents the long-term memory deficit induced by THC pointing to a necessary activation of these receptors (Puighermanal et al, 2009; Han et al, 2012). One of the key steps following NMDA receptor activation involves protein kinase C (PKC)/calmodulin (CaM) signaling (MacDonald et al, 2001). However, the effects of THC on this specific signaling pathway have not been explored to date.
PKC is a family of serine/threonine kinases modulated by phosphorylation in three conserved sites, the activation-loop, the turn-motif, and the hydrophobic-motif that control PKC catalytic activity, stability, and intracellular localization (Freeley et al, 2011). The PKC family consists of several isoforms: (i) conventional (α, βI, βII, and γ isoforms, activated by Ca2+ and diacylglycerol), (ii) novel (δ, ɛ, η, and θ isoforms, activated by diacylglycerol), and (iii) atypical (ζ and λ (in human ι) isoforms, sensitive to phospholipids (Newton, 2010; Freeley et al, 2011). Notably, CaM is a relevant downstream target of PKC signaling at synapses where it has a crucial role in synaptic function and plasticity owing to its ability to mediate Ca2+ signaling. CaM signaling fine-tuning involves neurogranin/RC3 in the postsynaptic sites, and neuromodulin/GAP-43 in the presynaptic sites, both of them forming complexes with CaM to maintain it in an inactive state (Xia and Storm, 2005; Díez-Guerra, 2010). PKC phosphorylation in specific residues of neurogranin and neuromodulin affect the interaction with CaM (Petersen and Gerges, 2015), leading to the liberation of CaM for downstream activation of CaM-dependent kinase signaling.
In this study, we identified the specific involvement of the hippocampal PKC signaling as a new molecular player in the deleterious effects of THC on short-term novel object-recognition memory. In this regard, we reveal a THC-induced PKC-dependent phosphorylation of hippocampal neurogranin, a relevant player for postsynaptic CaM function. Together these data point to differential mechanisms underlying the long-term and short-term consequences of cannabis abuse in cognition.
Materials and methods
Animals
Male CD-1 mice (Charles River, Lyon, France) weighting 25–30 g were used. Mice were housed in cages of four and maintained at a controlled temperature (21±1 °C) and humidity (55±10%). Food and water were available ad libitum. Lighting was maintained at 12 h cycles (on at 8 am and off at 8 pm). All the experiments were performed during the light phase of the dark/light cycle. Animals were habituated to the experimental room and handled for one week before starting the experiments. All animal procedures were conducted in accordance with the standard ethical guidelines (European Communities Directive 2010/63/EU) and approved by the local ethical committee (Comitè Ètic d'Experimentació Animal, CEEA-PRBB). All behavioral analysis was performed blind to the experimental conditions.
Drugs and Treatments
Delta9-THC was purchased from THC-Pharm-GmbH (Frankfurt, Germany); rapamycin and chelerythrine (CHE) were purchased from Calbiochem (San Diego, CA, USA); cremophor-EL, MK801, PEAQX, NPC-15437 and anisomycin were purchased from Sigma–Aldrich (Madrid, Spain); temsirolimus was purchased from LC Laboratories (Woburn, MA); RO 25-6981 was purchased from Tocris Bioscience (Bristol, UK) and rimonabant was kindly provided by Sanofi-Aventis (Sanofi-Aventis Recherche, Montpellier, France).
MK801, NPC-15437, RO 25-6981, and PEAQX were dissolved in 0.9% sodium chloride (saline). THC and rimonabant were diluted in 5% ethanol, 5% cremophor-EL, and 90% saline. CHE was dissolved in dimethyl sulfoxide (DMSO) as a stock, 14.3 mM (equivalent to 50 mg/kg), and diluted in saline to the doses indicated (0.01, 0.1, 1, 3 mg/kg). NPC-15437 was dissolved in saline for intra-hippocampal microinjections and used at 0.3, 1, 2.5, and 5 μg/0.5 μl, bilaterally. Rapamycin was dissolved in DMSO. All drugs were administered intraperitoneally in a volume of 10 ml/kg, except rapamycin (2 ml/kg). Rimonabant (1 mg/kg), temsirolimus (1 mg/kg), anisomycin (18 mg/kg), MK801 (0.1 mg/kg), NPC-15437 dihydrochloride hydrate (1 mg/kg), RO 25-6981 maleate (6 mg/kg), PEAQX tetrasodium hydrate (1.2 mg/kg), and CHE (0.01 mg/kg) were administered 20 min before THC administration. Rapamycin (1 mg/kg) was administered once daily for 5 d, and mice were tested 3 h after the last rapamycin injection.
For the surgery, ketamine hydrochloride (Imalgène; Merial Laboratorios S.A., Barcelona, Spain) and medetomidine hydrochloride (Domtor; Esteve, Barcelona, Spain) were mixed and dissolved in sterile 0.9% physiological saline and administered intraperitoneally in a volume of 10 ml/kg of body weight. Atipamezole hydrochloride (Revertor; Virbac, Barcelona, Spain) and Meloxicam (Metacam; Boehringer Ingelheim, Rhein, Germany) were dissolved in sterile 0.9% physiological saline and administered subcutaneously in an injection volume of 10 ml/kg of body weight. Gentamicine (Genta-Gobens; Laboratorios Normon, S.A., Madrid, Spain) was dissolved in sterile 0.9% physiological saline and administered intraperitoneally in an injection volume of 10 ml/kg of body weight.
Immunoblot Analysis
Brain samples were obtained 30 min or 1 h after THC or vehicle treatment. Hippocampal tissues were dissected from brains, frozen on dry ice and stored at −80 °C until used as previously reported (Puighermanal et al, 2009). The antibodies used for immunoblot were: anti-phospho-Ser PKC (1:2,000), anti-phospho-PKC(pan)(βII Ser660) (1:500) (Cell Signaling, Beverly, MA, USA); anti-phospho-neurogranin (Ser36)/phospho-neuromodulin (Ser41) (1:1,000), (Millipore, Darmstadt, Germany), anti-neurogranin (1:500), anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:15,000), PKC alpha (1:500), PKCβI (1:500), PKCβII (1:500), PKCδ (1:500), PKCɛ (1:500), PKCη (1:500) and PKCθ (1:500) (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The optical densities obtained with the phospho-specific antibody against pNeurogranin were normalized to the detection of total Neurogranin or GAPDH in the same sample and expressed as a percentage of control/vehicle treatment. The optical densities obtained for the rest of antibodies were normalized to the detection of GAPDH.
Object-Recognition Task
Object-recognition memory was assayed in the V-maze paradigm 3 h (short-term memory assay) or 24 h (long-term memory assay) after the training session as described previously (Busquets-Garcia et al, 2011). On day 1, mice were habituated to the empty maze for 9 min. On the second day, mice were introduced in the maze for 9 min, where two identical objects were presented. For the memory test, mice were placed again in the V-maze for a period of 9 min, where one of the familiar objects was replaced by a novel object, and the total time spent exploring each of the two objects (novel and familiar) was recorded. Object exploration was defined as the orientation of the nose towards the object at a distance less than 2 cm. A discrimination index (DI) was calculated as the difference between the time spent exploring either the novel (Tn) or familiar (Tf) object divided by the total time exploring both objects (Tn+Tf) (DI=(Tn−Tf)/(Tn+Tf)). A positive DI reflects memory retention for the familiar object as mice explore the novel object for longer than the familiar object. This DI was different between control groups depending on animal batches or experimental conditions (eg, local vs systemic injections). For this reason, experimental groups to be compared were run in parallel. Drugs were injected immediately after training (rimonabant, temsirolimus, anisomycin, MK801, RO 25-6981, PEAQX, NPC-15437, and CHE) or 20 min after training (THC). Rapamycin was administered during 5 days previous to the task, and the fifth administration was performed immediately after training following the procedure described previously (Puighermanal et al, 2009). Given the variability in the absolute DI values, all experimental groups to be compared were run in parallel in continuous sessions where experimental groups were intermingled.
Surgery
Mice were deeply anaesthetized by intraperitoneal injection of a mixture of ketamine (75 mg/kg) and medetomidine (1 mg/kg). After surgery, anesthesia was reversed by subcutaneous injection of the synthetic α2 adrenergic receptor antagonist, atipamezole (2.5 mg/kg) indicated for the reversal of the sedative and analgesic effects of medetomidine (α2 adrenergic receptor agonist). In addition, mice received an intraperitoneal injection of gentamicine (1 mg/kg) along with subcutaneous administration of the analgesic meloxicam (2 mg/kg). During surgery, mice were placed in a stereotaxic frame (David Kopf, Tujunga, CA) under sterile conditions, and the upper incisor bar was set 1.0 mm below the interaural line so that the skull was horizontal between bregma and lambda. An incision was made over the skull, thus the skull was surgically exposed, the periosteum removed, and burr holes were subsequently drilled with the size of the guide cannula. A bilateral 26-gauge guide cannula (Plastics One, Roanoke, VA) was implanted into the dorsal hippocampus to be used to guide a bilateral injection cannula (33-gauge internal cannula, Plastics One). The bilateral guide cannula was held in place using dental cement (Dentalon plus, Heraeus Kulzer GmbH, Hanau, Germany) and two stainless-steel screws. The placement was set at 1 mm above the target injection site and the guide cannula was sealed with a dummy of stainless-steel wire with 0.5 mm of projection to prevent obstruction. The target injection site coordinates were as follows: anteroposterior, −1.80 mm; mediolateral, ±1.00 mm; dorsoventral, 2.00 mm (Paxinos and Franklin, 2001). Animals were kept on a 37 °C heating pad during the surgery, and until recovery from anesthesia. The behavioral experiments started 4 days after surgery.
Intra-Hippocampal Microinjections
Immediately after training, mice received a bilateral intra-hippocampal injection of 0.50 μl of NPC (0.3, 1, 2.5, or 5 μg) or vehicle at a constant rate of 0.25 μl/min by using a microinfusion pump (Harvard Apparatus, Holliston, MA) during 2 min. The 33-gauge bilateral injection cannula was connected to polyethylene tubing (PE-20, Plastics One) attached to a 10 μl Hamilton microsyringe (Hamilton, Reno, NV). The injection cannula projected 1.00 mm below the ventral tip of the implanted guide cannula. The displacement of an air bubble inside the length of the polyethylene tubing that connected the syringe to the injection needle was used to monitor the microinjections. After infusion, the injection cannula was left in place for an additional period of 2 min to allow the fluid to diffuse and to prevent reflux, then it was slowly withdrawn. In order to minimize surgical interventions, memory assessment was performed in the minimum number of subjects (n=3–6) to reveal a significant effect of pharmacological treatments.
Verification of Cannula Position
After behavioral experiment, mice were killed by cervical dislocation, and the brains were immediately removed, frozen and stored at -80 °C until sectioning. Coronal sections (30 μm) were cut on a cryostat and mounted on slides. The slides were stained with cresyl violet (Supplementary Figure S4A), and the injection sites were verified under a light microscope by an experimenter blind to the treatment. Mice with cannula location outside of the hippocampus were excluded from the results.
Tissue Preparation for Immunofluorescence
Mice were deeply anesthetized by i.p. injection (0.2 ml/10 g of body weight) of a mixture of ketamine (100 mg/kg) and xylazine (20 mg/kg) prior to intracardiac perfusion with 4% PFA in 0.1 M Na2HPO4/ 0.1 M NaH2PO4 buffer (PB), pH 7.5, delivered with a peristaltic pump at 19 ml per min for 3 min. Subsequently, brains were extracted and post-fixed with 4% PFA for 24 h and transferred to PB 0.1 M at 4 °C. Coronal frozen sections (30 μm) of the dorsal hippocampus (coordinates relative to bregma, –1.22 mm to –1.82 mm) were obtained on a vibratome and stored in the freezer with cryoprotectant solution until used.
Immunofluorescence
Free-floating brain slices were rinsed in PB 0.1M. Brain slices were blocked with 3% normal goat serum, 0.3% Triton X-100 in PB (NGS-T-PB) at room temperature for 2 h, and incubated overnight in the same solution with the primary antibody to anti-phospho-PKC(pan)(βII Ser660) (1:500 and 1:300) (Cell Signaling), anti-neurogranin (1:500), anti-PKCθ (1:500) (Santa Cruz Biotechnology), anti-CB1 (1:500) (Frontier Institute Co.Ltd, Japan), anti-neuronal nuclei (NeuN) (1:500) (Millipore) and anti-microtubule-associated protein 2 (MAP2) (1:500) (Millipore) at 4 °C. The next day, after three rinses in PB 0.1 M, sections were incubated at room temperature with the secondary antibody to rabbit or mouse conjugated to Alexa 555 (Cy3), Alexa 488 (Cy2), or Alexa 647 (IR) (1:500, Jackson ImmunoResearch, West Grove, USA) in NGS-T-PB for 2 h. After incubation, brain sections were rinsed and mounted immediately after onto glass slides coated with gelatin. Mowiol was used as mounting medium.
Confocal Imaging
Double immunofluorescence (pPKC (S660)/CB1) and triple immunofluorescence (neurogranin, pPKC (S660) and PKCθ/NeuN/MAP2) confocal images were obtained by using a Leica TCS SP8 confocal microscope adapted to an inverted Leica DMi8 microscope. General hippocampus images were obtained with a 10 × 0.3 NA dry Leica Semi-apochromatic objective and specific CA1 images with a × 63 1.3 NA glycerin immersion Leica Plan Apochromatic CS2. Alexa 488 (Cy2), Alexa 555 (Cy3), and Alexa 647 (IR) were excited with a 488 nm line of argon laser, a 561 nm diode-pumped solid state laser and a 633 nm helium neon laser. An acousto-optical beam splitter was used detecting on windows 501–566 nm (Alexa 488), 566–647 nm (Alexa 555) and 638–783 nm (Alexa 647). All images were 1024 × 1024 pixels, 8 bits.
Statistical Analysis
Comparisons between two groups were performed by Student’s t-test. One-way or two-way analysis of variance (ANOVA) were performed where appropriate for multiple-group comparisons as indicated in the text. Post hoc comparisons were performed by Student–Newman–Keuls test only when significant main effect of ANOVA (one-way ANOVA) or significant interaction between factors (two-way ANOVA) was revealed. All results are expressed as mean±SEM and differences were considered significant at P<0.05. Statistica Software was used for these statistical comparisons.
Results
Different Mechanisms for Short-Term and Long-Term Object-Recognition Memory Impairment of THC
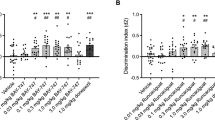
The effect of THC on short-term object-recognition memory was assessed 3 h after the training session. Acute THC administration 20 min after the training phase in the object-recognition test produced dose-dependent memory impairment (Figure 1a). We found that THC dose-dependently produced a decrease of the DI in comparison with the vehicle group (THC effect: F(1,30)=30.644, p<0.001). The dose of THC 3 mg/kg obliterated novel object discrimination (p<0.001), whereas no differences were found in the total exploration time during the memory test (Supplementary Figure S1A). The role of CB1 receptors in the effect of THC on short-term memory was confirmed by a pre-treatment with the specific CB1 receptor antagonist rimonabant (1 mg/kg) 20 min before THC administration. Thus, two-way ANOVA showed an interaction between THC treatment and rimonabant pre-treatment (p<0.01; rimonabant effect: F(1,28)=5.40, p<0.05; THC effect: F(1,28)=7.23, p<0.05; interaction: F(1,28)=6.79, p<0.05), and post hoc analysis revealed a significant effect of rimonabant (p<0.01) (Figure 1b), that did not affect exploratory behavior (Supplementary Figure S1B). Pre-treatment with a low dose of the NMDA receptor antagonist MK801 (0.1 mg/kg) prevented the amnesic-like effect of THC in object-recognition short-term memory (p<0.001) (MK801 effect: F(1,19)=15.416, p<0.001; THC effect: F(1,19)=15.130, p<0.001; interaction: F(1,19)=7.367, p<0.05) without affecting the total exploration time (Supplementary Figure S1C). Interestingly, the NR2A-preferring NMDA receptor antagonist PEAQX (1.2 mg/kg, i.p.) (Fox et al, 2006) and the NR2B-preferring NMDA receptor antagonist RO25-6981 (6 mg/kg, i.p.) (Fox et al, 2006) similarly prevented the THC-induced memory deficit (p<0.001) (PEAQX and RO25-6981 effect: F(1,49)=29.491, p<0.001; THC effect: F(1,49)=16.845, p<0.001; interaction: F(1,49)=9.537, p<0.001) (Figure 1d), further involving the activity of NMDA receptors in the memory deficits produced by THC administration.

Characteristics of the short-term object-recognition memory impairment produced by THC. (a) THC dose-dependently impaired short-term memory in the object recognition task in comparison with vehicle (Veh) groups. Different doses of THC were administered after the training phase and discrimination indexes were obtained 3 h after training (n=4–7 animals per group). Statistical significance was calculated by one-way ANOVA. The pre-treatment with rimonabant (1 mg/kg) (n=5–9 animals per group) (b), MK801 (0.1 mg/kg) (n=5–6 animals per group) (c), or the NR2A- and NR2B-preferring NMDA receptor antagonists PEAQX and RO 25-6981 maleate, respectively (n=4–9 animals per group) (d), block the amnesic-like effects of THC (3 mg/kg) in short-term memory. All drugs were injected immediately after the training session and 20 min prior to THC. (e–f) Discrimination index values obtained in the object recognition test of mice treated with THC (3 mg/kg) after pre-treatment with rapamycin, temsirolimus and anisomycin (n=7–9 animals per group). Data are expressed as mean±SEM. *P<0.05, ***P<0.001 (THC compared with vehicle) and ##P<0.01 (rimonabant+THC compared with vehicle+THC), ###P<0.001 (MK801+THC compared to saline+THC and PEAQX+THC, RO 25-6981 maleate+THC compared with vehicle+THC).
Notably, low doses of specific inhibitors of the mTORC1 pathway that readily prevent the long-term amnesic-like effect of THC, such as rapamycin (Puighermanal et al, 2009) or temsirolimus (Puighermanal et al, 2013), did not affect the short-term object-recognition memory deficit produced by THC (rapamycin and temsirolimus effect: F(1,35)=0.720, p=0.493; THC effect: F(1,35)=95.825, p<0.001; interaction: F(1,35)=2.867, p=0.07) (Figure 1e). In this regard, the protein synthesis inhibitor anisomycin, shown to prevent the deleterious effect of THC on long-term object-recognition memory (Puighermanal et al, 2009), did not affect those deficits produced by THC on short-term memory (anisomycin effect: F(1,17)=1.042, p=0.321; THC effect: F(1,17)=202.188, p<0.001; interaction: F(1,17)=0.585, p=0.367) Figure 1f). These data indicate that THC influences short-term object-recognition memory by disturbing different intracellular mechanisms from those involved in the deficits observed in long-term object recognition memory.
PKC Signaling Mediates the Effects of THC on Short-Term Object-Recognition Memory
We evaluated the activity of the PKC signaling pathway in the hippocampus after an acute administration of THC since this signaling pathway is closely related to the activation of NMDA receptors (MacDonald et al, 2001), found to be critical in the short-term deficit of THC. To this end, we used an antibody that recognizes proteins phosphorylated at the PKC substrate consensus: Ser residues surrounded by Arg or Lys at the −2 and +2 positions and a hydrophobic residue at the +1 position (anti-phospho-(Ser) PKC substrate). The immunoblot analysis of hippocampal samples revealed a significant enhancement in phosphorylated PKC substrates after THC administration, reaching a peak 1 h after THC administration (30 min: Student’s t-test, t=−0.199, p=0.846, df=10; 1 h: Student’s t-test, t=−3.852, p<0.01, df=10; 2 h: Student’s t-test, t=−0.972, p=0.353, df=10) (Figure 2a). Interestingly, MK801 pre-treatment, under those conditions that prevent the short-term memory deficits produced by THC, also blocked the enhancement in PKC signaling found 1 h after THC treatment (Figure 2b).

THC-effects on short-term object recognition memory are mediated by PKC signaling. (a) Time course for the phosphorylation of PKC substrates in hippocampal samples of mice treated with vehicle or THC (3 mg/kg). Hippocampus were extracted 30 min, 1 h, or 2 h after THC injection (n=5–6 animals per group). (b) Phosphorylation of PKC substrates in hippocampal samples of mice after pre-treatment with MK801 or saline 20 min before THC or vehicle administration. Samples were collected 30 min after drug injection (n=6–7 animals per group). Data are expressed as mean±SEM. **P<0.01 (THC compared with vehicle); ##P<0.01 (MK801+THC compared with saline+THC).
We assessed the functional relevance of the PKC signaling by using two specific PKC inhibitors, NPC-15437 (NPC, 1 mg/kg) and CHE (0.01 mg/kg), at doses that did not affect cognitive performance on their own (Figure 3). Noteworthy, doses of CHE above 0.01 mg/kg (0.1, 1 and 3 mg/kg) had a profound effect over short-term memory performance, without affecting long-term memory (Supplementary Figure S2). Both PKC inhibitors (NPC or CHE) prevented the short-term amnesic-like effect of THC (NPC pre-treatment effect: F(1,36)=8.314, p<0.01; THC effect: F(1,36)= 34.522, p<0.001; interaction: F(1,36)=15.999, p<0.001. CHE pre-treatment effect: F(1,28)=6.161, p<0.05; THC effect: F(1,28)=44.954, p<0.001; interaction: F(1,28)=17.754, p<0.001) (Figures 3a and b), without affecting the total exploration time during the test session (Supplementary Figure S3). Remarkably, both PKC inhibitors were ineffective preventing the object-recognition long-term memory deficits produced by THC (NPC-15437 and chelerythrine effect: F(1,30)=0.373, p=0.691; THC effect: F(1,30)=57.187, p<0.001; interaction: F(1,30)=1.582, p=0.222) (Figure 3c). Similarly, hippocampal pre-treatment with a sub-effective dose of NPC-15437 (1 μg/0.5 μl per side) that do not affect the total exploration time (Supplementary Figure S4B-C), was able to block the short-term memory deficits produced by THC (NPC pre-treatment effect: F(1,10)=15.032, p<0.01; THC effect: F(1,10)= 11.474, p<0.01; interaction: F(1,10)=9.795, p<0.05). (Figure 3d) but no effect was observed in long-term memory (NPC pre-treatment effect: F(1,14)=4.802, p<0.05; THC effect: F(1,14)= 38.5, p<0.001; interaction: F(1,14)=0.583, p=0.475) (Figure 3e). No differences in total exploration time were observed in any case (Supplementary Figure S4D-E). We found that at the biochemical level, NPC pre-treatment also reduced THC-enhanced PKC signaling in the hippocampus under those conditions that prevented the memory impairment of THC in short-term memory (NPC effect: F(1,16)=1.596, p=0.224; THC effect: F(1,16)=33.081, p<0.001; interaction: F(1,16)=7.904, p<0.05) (Figure 3f). These data support the specificity and relevance of PKC signaling, modulated by specific inhibitors, in the deleterious effects of THC on short-term but not long-term object-recognition memory.

THC memory impairment in short-term object recognition memory is prevented by PKC inhibitors. Discrimination index values obtained in the object recognition short-term memory test of mice treated with THC (3 mg/kg) after pre-treatment with the PKC inhibitors NPC-15437 (n=6–12 animals per group) (a) or chelerythrine (n=5–11 animals per group) (b). (c) None of these two drugs administered 20 min before THC prevented the cognitive impairment produced by THC on long-term object-recognition memory (n=6 animals per group). Discrimination index values obtained when intra-hippocampal pre-treatment with NPC-15437 (1 μg/0.5 μl per side) was performed in short-term (n=3–4 animals per group) (d) and long-term (n=3-6 animals per group) (e) memory showing a similar effect to systemic administration. (f) Phosphorylation of PKC substrates in hippocampal samples of mice after pre-treatment with NPC or vehicle 20 min before THC or vehicle administration (n=5–6 animals per group). Data are expressed as mean±SEM. **P<0.01, ***P<0.001 (THC compared with vehicle); # P<0.05 (NPC-15437+THC compared with saline+THC), ##P<0.01 (NPC-15437+THC compared with saline+THC); ###P<0.001 (NPC-15437+THC and chelerythrine+THC compared with saline+THC).
THC Administration Enhances the Phosphorylation of PKC Isoforms in the Hippocampus
We next assessed the phosphorylation status of a Ser residue in the hydrophobic motif, a motif highly conserved in most PKC isoforms that is required for PKC activation (Freeley et al, 2011). We used an antibody against phospho-PKC (pan) (βII Ser660). This antibody detects PKCα, βI, βII, δ, ɛ, η, and θ isoforms only when phosphorylated at the carboxy-terminal residue homologous to Ser660 of PKCβII. Immunoblot analysis of hippocampal samples revealed three main immunoreactive bands at ≈60 kDa (p60), ≈85 kDa (p85), and ≈95 kDa (p95) (Figure 4a). The immunoreactive band at 60 kDa was specifically enhanced 1 h after THC administration, whereas the most relevant bands at 85-95 kDa were not affected by THC treatment (Figures 4a and b). NPC pre-treatment under those conditions that prevented the short-term object-recognition memory deficit of THC also showed a non-significant trend to reduce the detection of p60 with anti-phospho-PKC (pan) (βII Ser660) (Veh_Veh vs Veh_THC: Student’s t-test, t=−2.676, p<0.05, df=8; Veh_THC vs NPC_THC: Student’s t-test, t=1.430, p=0.186, df=9) (Figure 4b). At the histological level, phospho-PKC (pan) (βII Ser660) immunoreactivity was detected mainly in the dendritic area of stratum radiatum of the hippocampus (Figure 4c) where it did not co-localize with CB1 receptor immunoreactivity (Figure 4d).

THC administration enhances the phosphorylation of PKC isoforms in the hippocampus (a) An antibody against phospho-PKC (pan) (βII Ser660) detecting PKC α, βI, βII, δ, ɛ, η, and θ isoforms only when phosphorylated at a carboxy-terminal residue homologous to Ser660 of PKC βII reveal a modulation at 60 kDa after THC administration (n=5–6 animals per group). (b) The modulation produced by THC observed at 60 kDa by using the antibody against phospho-PKC (pan) (βII Ser660) was partially reversed when animals were pre-treated with the PKC inhibitor, NPC-15437 (n=5–6 animals per group). (c) Phospho-PKC (pan) (βII Ser660) immunoreactivity was detected mainly in the dendritic area of stratum radiatum of the hippocampus (d) where it did not co-localize with CB1 receptor immunoreactivity. All samples were collected 1 h after drug administration. Data are expressed as mean±SEM. *P<0.05 (THC compared with vehicle). Scale bar 300 μm, 50 μm, and 20 μm.
To study the possible preferential involvement of specific PKC isoforms in this effect of THC, we immunodetected all the isoforms bearing a Ser residue at the hydrophobic domain, PKC α, βI, βII, δ, ɛ, η, and θ isoforms. Using specific antibodies against the carboxyl terminus of these isoforms we found that the relative electrophoretic mobility compared to that of the immunoreactive bands detected by anti-phospho-PKC (pan) (βII Ser660), the p60 band modulated by THC did not correspond to abundantly expressed conventional isoforms such as PKCα, βI, βII, or novel isoforms such as PKCδ, ɛ, and η, but to the PKCθ isoform (Supplementary Figure S5). Furthermore, PKCθ immunoreactivity was detected in the stratum radiatum of the hippocampus (Supplementary Figure S5B), supporting the potential modulation of this subunit under those conditions of THC administration.
THC Modulates the Post-Synaptic PKC Substrate Neurogranin
PKC signaling modulates neurogranin and neuromodulin, both CaM-binding proteins acting as CaM reservoirs at post-synaptic and pre-synaptic sites, respectively (Zhong et al, 2011). Neurogranin is heavily expressed in the hippocampus (Figure 5a). In the CA1 region, neurogranin was found in the cell bodies and apical dendrites of pyramidal cells, as detected by the co-localization with the dendritic marker MAP2 (Figure 5a). Neurogranin phosphorylation at Ser36 by PKC signaling is a key step to release CaM from neurogranin (Prichard et al, 1999; Kumar et al, 2013). THC administration under those amnesic conditions enhancing PKC signaling significantly increased the phosphorylation of neurogranin in the hippocampus (Veh vs THC: Student’s t-test, t=−2.345, p<0.05, df=9) (Figure 5b). The enhancement in phospho-neurogranin was sensitive to the pre-treatment with NPC (Veh/THC vs NPC/THC: Student’s t-test, t=2.565, p<0.05, df=9) (Figure 5b) and pre-treatment with CHE (Veh/THC vs CHE/THC: Student’s t-test, t=2.759, p<0.05, df=9) (Figure 5b). Instead, the pre-treatment with the mTORC1 inhibitor temsirolimus did not modify the increased phosphorylation of neurogranin produced by THC (Veh_Veh vs Veh_THC: Student’s t-test, t=−3.419, p<0.01, df=10; Veh_THC vs Temsi_THC: Student’s t-test, t=0.074, p=0.941, df=10) (Figure 5c). Insterestingly, neuromodulin phosphorylation was not modified in the same samples (Veh vs THC: Student’s t-test, t=−1.378, p=0.198, df=10) (Figure 5d). These data reveal the selective modulation by THC of PKC-dependent signaling in the THC-induced short-term memory impairment.

PKC-dependent neurogranin, but not neuromodulin, phosphorylation after THC administration. (a) Neurogranin was found in the cell bodies and apical dendrites of pyramidal cells in the CA1 region of the hippocampus, as detected by the co-localization with the dendritic marker MAP2. (b) THC produced a significant increase in the phosphorylation of neurogranin, an effect reverted by the pre-treatment with NPC-15437 and chelerythrine. (c) The pre-treatment with temsirolimus (1 mg/kg), an inhibitor of the mTORC1 pathway, did not normalize the increased phosphorylation of neurogranin. (d) No differences were found in the phosphorylation of hippocampal neuromodulin after THC administration. All samples were obtained 1 h after the drug administration (n=5–6 animals per group in all experiments). Data are expressed as mean±SEM. *P<0.05, **P<0.01 (THC compared with vehicle); #P<0.05 (NPC+THC and chelerythrine+THC compared with vehicle+THC). Scale bar 300 μm, 50 μm, and 20 μm.
Discussion
The present study reveals that specific signaling pathways activated by the acute administration of an amnesic dose of THC (3 mg/kg) are responsible for different cognitive alterations produced by this cannabinoid agonist. In this regard, the modulation of hippocampal PKC signaling has a significant relevance for the short-term, but not the long-term, object-recognition memory deficits produced by THC. Moreover, THC-induced phosphorylation of neurogranin, a key postsynaptic protein with crucial roles in calmodulin (CaM) -associated signaling was also associated to this short-term memory deficit.
CB1 receptors, the major cannabinoid receptor in the brain, are especially relevant at synaptic terminals, controlling synaptic homeostasis (Hillard, 2015; Wilson and Nicoll, 2002; Freund et al, 2003), and in astroglia, promoting glutamate release at both synaptic and extrasynaptic sites (Navarrete and Araque, 2010; Han et al, 2012; Navarrete et al, 2014). THC alters synaptic plasticity in the brain mainly by activating presynaptic CB1 receptors in glutamatergic and GABAergic terminals (Hoffman and Lupica, 2013). In the hippocampus, CB1 receptors are mainly expressed in GABAergic terminals over glutamatergic terminals (Kawamura et al, 2006). Moreover, THC may act as a partial agonist to inhibit glutamate-mediated synaptic responses (Hoffman et al, 2010), and as a full agonist to inhibit GABA release from inhibitory terminals (Laaris et al, 2010), whereas astrocytic CB1 receptors enhance glutamate release from this cell type (Han et al, 2012; Navarrete et al, 2014). As a result, glutamatergic tone in the hippocampus could be enhanced by acute THC administration, resulting in cognitive dysfunction. In agreement, NMDA receptor antagonist pre-treatment prevented the amnesic-like and the intracellular signaling effects induced by THC administration (Derkinderer et al, 2003; Puighermanal et al, 2009; Han et al, 2012). Nevertheless, certain differences were highlighted between the THC-induced short-term novel object-recognition memory deficits described in the present study and those described for the CB1 receptor agonist HU210 on spatial working memory (Han et al, 2012). In our study, both NR2A- and NR2B-preferring NMDA receptor antagonists exerted the same inhibitory effect over THC impairment in short-term object-recognition memory, whereas the same NR2A-NMDA antagonist did not modify the spatial working memory impairment produced by HU210 (Han et al, 2012). These observations are in agreement with the possible involvement of differential molecular mechanisms in the different aspects of cognition impaired by cannabinoid agonists.
The involvement of NMDA receptors on THC-induced memory deficits points to the possibility that THC may increase aberrantly the postsynaptic depolarization enhancing Akt/mTORC1 signaling (Puighermanal et al, 2009; Puighermanal et al, 2012; Puighermanal et al, 2013) and, as revealed in the present study, PKC signaling. Our data showed an enhancement in PKC phosphorylated substrates shortly after THC administration, which is blocked by the NMDA receptor antagonist MK801. MK801 also prevented the activation of the Akt/mTORC1 pathway that mediates the deficits in long-term object-recognition memory produced by THC (Puighermanal et al, 2009). Therefore, a similar NMDA receptor mechanism might be responsible for the increased PKC and mTORC1 activity after an amnesic dose of THC. The use of PKC and mTORC1 inhibitors, at concentrations that do not affect memory on their own, demonstrated the independent mechanisms underlying the short-term (PKC signaling) and long-term (mTORC1 signaling) object-recognition memory deficits produced by THC. In this regard, PKC signaling has been shown to regulate synaptic plasticity at CA1 synapses (Hsu et al, 2011). At this level, PKC signaling is implicated in facilitating long-term plasticity by reducing the threshold necessary for long-term potentiation induction (Miura et al, 2002).
The PKC family of proteins is composed by a considerable number of members grouped in three categories according to their structure and mechanism of activation (Newton, 2010), all expressed in the hippocampus (Lein et al, 2007). Our biochemical results demonstrate a significant increase in the phosphorylation levels of several substrates of PKC after acute THC treatment. In an attempt to identify those PKC isoforms involved, we investigated the phosphorylation of the PKC hydrophobic-motif which has been associated to enzymatic stimulation in some instances (Freeley et al, 2011). THC only modulated such residue in a PKC isoform weighting ~60 kDa. Such result allowed discarding most of PKC isoforms bearing a Ser in the hydrophobic-motif except PKCθ, since all other PKC isoforms have a molecular weight above 75 kDa. The degree of phosphorylation on this residue was enhanced by THC and reduced by PKC inhibitor pre-treatment in mice that received THC. Interestingly, Ser-to-Ala mutation of the hydrophobic-motif in PKCθ has been found to reduce its activity (Liu et al, 2002), pointing to the idea that the phosphorylation status of hydrophobic-motif in PKCθ is a relevant site to determine its activity status.
Among those relevant PKC substrates for synaptic function, neurogranin and neuromodulin are crucial CaM-binding proteins playing a buffering effect over CaM function. Neurogranin is a postsynaptic protein abundantly expressed in brain regions involved in cognitive functions (Díez-Guerra, 2010; Prichard et al, 1999). The phosphorylation of neurogranin at Ser36 by PKC signaling reduces the binding between neurogranin and CaM, allowing CaM to bind to calcium and activate CaM-dependent pathways such as calcium-calmodulin-dependent protein kinase II signaling cascades, which are relevant in learning and memory (Kumar et al, 2013; Gaertner et al, 2004). When Ser36 was mutated to Ala, the neurogranin mutant was unable to release CaM, which prevented synaptic potentiation (Zhong et al, 2009) revealing the importance of PKC-signaling on CaM-neurogranin-dependent synaptic plasticity (Zhong and Gerges, 2012). Indeed, previous studies have shown that the ratio of phosphorylated/unphosphorylated neurogranin could determine the threshold to obtain LTP/LTD in concert with calcium/CaM-dependent kinase II (Gerendasy and Sutcliffe, 1997; Pak et al, 2000; Díez-Guerra, 2010). Our data reveal that THC increases PKC-dependent phosphorylation of neurogranin. This effect could prevent the buffering effect of non-phosphorylated neurogranin over CaM leading to an anomalously long CaM signaling. Accordingly, neurogranin knockout mice showed impaired spatial learning and altered short- and long-term hippocampal plasticity (Pak et al, 2000). Notably, THC did not modify the phosphorylation of neuromodulin, a relevant presynaptic target of PKC that also plays a role as a CaM-binding protein at those sites (Biewenga et al, 1996).
In this study, the use of specific intracellular signaling inhibitors to selectively modify biochemical and behavioral effects of THC has allowed dissociating the molecular mechanisms involved in one of the most deleterious effects of cannabis abuse, the cognitive impairment. Indeed, two different signaling pathways, the PKC and the mTORC1 intracellular cascades, are modulated by THC and are specifically responsible for the short-term and the long-term memory deficits, respectively. Altogether, our study identifies the relevance of hippocampal PKC signaling in the specific deleterious effect of THC over short-term episodic memories.
Funding and disclosure
Predoctoral fellowship from the Spanish Ministry of Education (AB-G and EP). Partial support by FP7-PEOPLE-2013-IEF-623638 (AB-G). Partial support by FRAXA Research Foundation (MG-G). Predoctoral fellowship by FPI (MINEICO/FEDER, EU) (VS-M and LG-L). Research supported by BFU2015-68568 P (AO), (MINECO/FEDER, UE); Redes temáticas de investigación cooperativa en salud (RETICS) del Instituto de Salud Carlos III (#RD16/0017/0020)-Red de trastornos adictivos (RTA) (RM); Generalitat de Catalunya, AGAUR (#2014-SGR-1547) (RM); ICREA (Institució Catalana de Recerca i Estudis Avançats) Acadèmia (RM and AO). The authors declare no conflict of interest.
References
Barna I, Soproni K, Arszovszki A, Csabai K, Haller J (2007). WIN-55,212-2 chronically implanted into the CA3 region of the dorsal hippocampus impairs learning: a novel method for studying chronic, brain-area-specific effects of cannabinoids. Behav Pharmacol 18: 515–520.
Biewenga JE, Schrama LH, Gispen WH (1996). Presynaptic phosphoprotein B-50/GAP-43 in neuronal and synaptic plasticity. Acta Biochim Pol 43: 327–338.
Broyd SJ, van Hell HH, Beale C, Yücel M, Solowij N (2016). Acute and chronic effects of cannabinoids on human cognition-A systematic review. Biol Psychiatry 79: 557–567.
Busquets-Garcia A, Puighermanal E, Pastor A, de la Torre R, Maldonado R, Ozaita A (2011). Differential role of anandamide and 2-arachidonoylglycerol in memory and anxiety-like responses. Biol Psychiatry 70: 479–486.
Derkinderen P, Valjent E, Toutant M, Corvol JC, Enslen H, Ledent C et al (2003). Regulation of extracellular signal-regulated kinase by cannabinoids in hippocampus. J Neurosci 23: 2371–2382.
Díez-Guerra FJ (2010). Neurogranin, a link between calcium/calmodulin and protein kinase C signaling in synaptic plasticity. IUBMB Life 62: 597–606.
Fox CJ, Russell KI, Wang YT, Christie BR (2006). Contribution of NR2A and NR2B NMDA subunits to bidirectional synaptic plasticity in the hippocampus in vivo. Hippocampus 16: 907–915.
Freeley M, Kelleher D, Long A (2011). Regulation of Protein Kinase C function by phosphorylation on conserved and non-conserved sites. Cell Signal 23: 753–762.
Freund TF, Katona I, Piomelli D (2003). Role of endogenous cannabinoids in synaptic signaling. Physiol Rev 83: 1017–1066.
Gaertner TR, Putkey JA, Waxham MN (2004). RC3/neurogranin and Ca2+/calmodulin dependent protein kinase II produce opposing effects on the affinity of calmodulin for calcium. J Biol Chem 279: 39374–39382.
Gaoni Y, Mechoulam R (1964). Isolation, structure, and partial synthesis of an active constituent of hashish. J Am Chem Soc 86: 1646–1647.
Gerendasy DD, Sutcliffe JG (1997). RC3/neurogranin, a postsynaptic calpacitin for setting the response threshold to calcium influxes. Mol Neurobiol 15: 131–163.
Hall W, Degenhardt L (2009). Adverse health effects of non-medical cannabis use. Lancet 374: 1383–1391.
Han J, Kesner P, Metna-Laurent M, Duan T, Xu L, Georges F et al (2012). Acute cannabinoids impair working memory through astroglial CB1 receptor modulation of hippocampal LTD. Cell 148: 1039–1050.
Hillard CJ (2015). The endocannabinoid signaling system in the CNS: a primer. Int Rev Neurobiol 125: 1–47.
Hoffman AF, Laaris N, Kawamura M, Masino SA, Lupica CR (2010). Control of cannabinoid CB1 receptor function on glutamate axon terminals by endogenous adenosine acting at A1 receptors. J Neurosci 30: 545–555.
Hoffman AF, Lupica CR (2013). Synaptic targets of Δ9-tetrahydrocannabinol in the central nervous system. Cold Spring Harb Perspect Med 3 pii: a012237.
Hsu JC, Cheng SJ, Yang HW, Wang HJ, Chiu TH, Min MY et al (2011). Bidirectional synaptic plasticity induced by conditioned stimulations with different number of pulse at hippocampal CA1 synapses: roles of N-methyl-D-aspartate and metabotropic glutamate receptors. Synapse 65: 795–803.
Kawamura Y, Fukaya M, Maejima T, Yoshida T, Miura E, Watanabe M et al (2006). The CB1 cannabinoid receptor is the major cannabinoid receptor at excitatory presynaptic sites in the hippocampus and cerebellum. J Neurosci 26: 2991–3001.
Kumar V, Chichili VP, Zhong L, Tang X, Velazquez-Campoy A, Sheu FS et al (2013). Structural basis for the interaction of unstructured neuron specific substrates neuromodulin and neurogranin with Calmodulin. Sci Rep 3: 1392.
Laaris N, Good CH, Lupica CR (2010). Delta9-tetrahydrocannabinol is a full agonist at CB1 receptors on GABA neuron axon terminals in the hippocampus. Neuropharmacology 59: 121–127.
Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A et al (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445: 168–176.
Liu Y, Graham C, Li A, Fisher RJ, Shaw S (2002). Phosphorylation of the protein kinase C-theta activation loop and hydrophobic motif regulates its kinase activity, but only activation loop phosphorylation is critical to in vivo nuclear-factor-kappaB induction. Biochem J 361 (Pt 2): 255–265.
MacDonald JF, Kotecha SA, Lu WY, Jackson MF (2001). Convergence of PKC-dependent kinase signal cascades on NMDA receptors. Curr Drug Targets 2: 299–312.
Miura M, Watanabe M, Offermanns S, Simon MI, Kano M (2002). Group I metabotropic glutamate receptor signaling via Galpha q/Galpha 11 secures the induction of long-term potentiation in the hippocampal area CA1. J Neurosci 22: 8379–8390.
Mounteney J, Griffiths P, Sedefov R, Noor A, Vicente J, Simon R (2016). The drug situation in Europe: an overview of data available on illicit drugs and new psychoactive substances from European monitoring in 2015. Addiction 111: 34–48.
Navarrete M, Araque A (2010). Endocannabinoids potentiate synaptic transmission through stimulation of astrocytes. Neuron 68: 113–126.
Navarrete M, Díez A, Araque A (2014). Astrocytes in endocannabinoid signalling. Philos Trans R Soc Lond B Biol Sci 369: 20130599.
Newton AC (2010). Protein kinase C: poised to signal. Am J Physiol Endocrinol Metab 298: E395–E402.
Ozaita A, Puighermanal E, Maldonado R (2007). Regulation of PI3K/Akt/GSK-3 pathway by cannabinoids in the brain. J Neurochem 102: 1105–1114.
Pak JH, Huang FL, Li J, Balschun D, Reymann KG, Chiang C et al (2000). Involvement of neurogranin in the modulation of calcium/calmodulin-dependent protein kinase II, synaptic plasticity, and spatial learning: a study with knockout mice. Proc Natl Acad Sci USA 97: 11232–11237.
Paxinos G, Franklin KBJ (2001). The Mouse Brain in Stereotaxic Coordinates, 2nd edn. Academic Press: San Diego, CA..
Petersen A, Gerges NZ (2015). Neurogranin regulates CaM dynamics at dendritic spines. Sci Rep 5: 11135.
Prichard L, Deloulme JC, Storm DR (1999). Interactions between neurogranin and calmodulin in vivo. J Biol Chem 274: 7689–7694.
Puighermanal E, Marsicano G, Busquets-Garcia A, Lutz B, Maldonado R, Ozaita A (2009). Cannabinoid modulation of hippocampal long-term memory is mediated by mTOR signaling. Nat Neurosci 12: 1152–1158.
Puighermanal E, Busquets-Garcia A, Maldonado R, Ozaita A (2012). Cellular and intracellular mechanisms involved in the cognitive impairment of cannabinoids. Philos Trans R Soc Lond B Biol Sci 367: 3254–3263.
Puighermanal E, Busquets-Garcia A, Gomis-González M, Marsicano G, Maldonado R, Ozaita A (2013). Dissociation of the pharmacological effects of THC by mTOR blockade. Neuropsychopharmacology 38: 1334–1343.
Ranganathan M, D'Souza DC (2006). The acute effects of cannabinoids on memory in humans: a review. Psychopharmacology (Berl) 188: 425–444.
Wilson RI, Nicoll RA (2002). Endocannabinoid signaling in the brain. Science 296: 678–682.
Wise LE, Thorpe AJ, Lichtman AH, Hippocampal CB (2009). (1) receptors mediate the memory impairing effects of Delta(9)-tetrahydrocannabinol. Neuropsychopharmacology 34: 2072–2080.
Xia Z, Storm DR (2005). The role of calmodulin as a signal integrator for synaptic plasticity. Nat Rev Neurosci 6: 267–276.
Zanettini C, Panlilio LV, Alicki M, Goldberg SR, Haller J, Yasar S (2011). Effects of endocannabinoid system modulation on cognitive and emotional behavior. Front Behav Neurosci 5: 57.
Zhong L, Cherry T, Bies CE, Florence MA, Gerges NZ (2009). Neurogranin enhances synaptic strength through its interaction with calmodulin. EMBO J 28: 3027–3039.
Zhong L, Kaleka KS, Gerges NZ (2011). Neurogranin phosphorylation fine-tunes long-term potentiation. Eur J Neurosci 33: 244–250.
Zhong L, Gerges NZ (2012). Neurogranin targets calmodulin and lowers the threshold for the induction of long-term potentiation. PLoS One 7: e41275..
Acknowledgements
We are grateful to Cristina Fernández-Avilés, Dulce Real, and Francisco Porrón for expert technical assistance and the Laboratori de Neurofarmacologia-NeuroPhar for helpful discussion.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Supplementary information
Rights and permissions
About this article

Cite this article
Busquets-Garcia, A., Gomis-González, M., Salgado-Mendialdúa, V. et al. Hippocampal Protein Kinase C Signaling Mediates the Short-Term Memory Impairment Induced by Delta9-Tetrahydrocannabinol. Neuropsychopharmacol. 43, 1021–1031 (2018). https://doi.org/10.1038/npp.2017.175
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2017.175
This article is cited by
-
Protein Kinase C-Gamma Knockout Mice Show Impaired Hippocampal Short-Term Memory While Preserved Long-Term Memory
Molecular Neurobiology (2021)
-
Adenosine A2A-Cannabinoid CB1 Receptor Heteromers in the Hippocampus: Cannabidiol Blunts Δ9-Tetrahydrocannabinol-Induced Cognitive Impairment
Molecular Neurobiology (2019)
