
Abstract
Drugs of abuse cause persistent alterations in synaptic plasticity that may underlie addiction behaviors. Evidence suggests glial cells have an essential and underappreciated role in the development and maintenance of drug abuse by influencing neuronal and synaptic functions in multifaceted ways. Microglia and astrocytes perform critical functions in synapse formation and refinement in the developing brain, and there is growing evidence that disruptions in glial function may be implicated in numerous neurological disorders throughout the lifespan. Linking evidence of function in health and under pathological conditions, this review will outline the glial and neuroimmune mechanisms that may contribute to drug-abuse liability, exploring evidence from opioids, alcohol, and psychostimulants. Drugs of abuse can activate microglia and astrocytes through signaling at innate immune receptors, which in turn influence neuronal function not only through secretion of soluble factors (eg, cytokines and chemokines) but also potentially through direct remodeling of the synapses. In sum, this review will argue that neural–glial interactions represent an important avenue for advancing our understanding of substance abuse disorders.
Similar content being viewed by others
INTRODUCTION
Drug abuse remains an intractable and costly disorder (Degenhardt et al, 2013). For instance, the recent crisis of opioid abuse, driven in part by increased use of opioid-based prescription pain medication, has resulted in a dramatic rise of overdose-associated deaths in the past decade (Calcaterra et al, 2013; Case and Deaton, 2015). Drug dependence exacts an enormous cost beyond the loss of life, both in terms of individual suffering as well as the economic burden of treatment and incarceration for the use of illegal substances. To treat this epidemic, we must advance our understanding of the biological and environmental factors that contribute to the pathophysiology of drug abuse.
The vast majority of preclinical and clinical research on addiction has focused on uncovering neuronal adaptations that follow exposure to drugs of abuse. Despite decades of progress toward understanding the neurobiological consequences of acute and repeated drug exposure, the underlying mechanisms of addiction behavior—including drug-seeking, drug-taking, and relapse behavior following periods of abstinence—remain unsolved. Although drug-induced alterations in neuronal function remain an essential component for theories of drug addiction (Koob and Volkow, 2010), accumulating evidence suggests they are not the only cells impacted by drugs of abuse. Glial cells, including microglia and astrocytes, are also influenced by exposure to abused drugs, and their responses likely contribute to the behavioral outcomes associated with substance abuse (Coller and Hutchinson, 2012; Miguel-Hidalgo, 2009). Once viewed as passive support elements for neurons, it is now clear that both microglia and astrocytes can actively regulate many aspects of neuronal function, including neurotransmitter release, gene regulation, electrophysiology, dendritic morphology, synaptic connectivity, and cell viability (Araque et al, 2014; Eroglu and Barres, 2010; Kettenmann et al, 2013; Salter and Beggs, 2014). As immunocompetent sentinels of the central nervous system (CNS), they are also critical participants in the development and protection of the CNS, including neural development, cell migration, programmed cell death, and regulation of synapse maturation and elimination (Bilbo et al, 2012; Chung et al, 2015a; Hanisch and Kettenmann, 2007; Schafer et al, 2013).
In the following review, we argue that glia participate in the molecular and synaptic changes that occur in neurons following drug exposure, and these neural–glial interactions consequently influence behavioral responses to these drugs. We deliberately narrow our focus of ‘glia’ to microglia and astrocytes, given the wealth of existing literature on these cell types and the critical neuroimmune signals they produce. Nonetheless, there are other glial and non-neuronal cells in the brain—including oligodendrocytes, endothelial cells, vascular cells, and other cells of innate and adaptive immunity—and their potential contributions to addiction are undoubtedly important as well, but are beyond the scope of this review.
First, a brief overview of the neurobiology of drugs of abuse is presented, focusing on dopaminergic circuitry. Second, several mechanisms regulating glial physiology are reviewed, highlighting pathways of innate immune signaling. Third, we discuss recent evidence that microglia and astrocytes directly monitor synaptic activity in the healthy CNS and modify synaptic structures. Fourth, we explore how opioids, alcohol, and psychostimulants can influence glia and neuroimmune signaling, tracing evidence from signaling to behavior. Finally, future research and clinical implications will be discussed.
NEUROCIRCUITRY INVOLVED IN REWARD AND REINFORCEMENT OF ABUSED SUBSTANCES
Patterned neural activity between brain regions coordinates the manifestation of motivated behavior toward natural stimuli that promote survival and reproductive needs, such as food, water, or a potential mate. Although the specifics of these needs vary considerably within and across species, they share a common characteristic of eliciting purposive behaviors to seek and obtain a particular goal (Sternson, 2013). In a similar manner, drugs of abuse exhibit remarkable diversity, differing in terms of chemical structure, affinity for various target receptors within the brain and periphery, solubility across the blood–brain barrier, metabolism and eventual clearance from the organism. Despite these structural and functional differences, drugs of abuse (like natural rewards) share a common characteristic of being reinforcing; humans and animals will work to acquire and consume these substances, rapidly associating environmental and interoceptive cues that predict their availability and increasing the frequency of behaviors required to obtain them (Hyman et al, 2006). Repeated exposure to drugs of abuse can produce the condition of drug addiction, characterized by compulsive drug-seeking and drug-taking behavior that continues despite harmful consequences (Deroche-Gamonet et al, 2004; Koob and Volkow, 2010). A full review of the mechanisms of drug addiction is beyond the scope of this review, but the basic evidence for a neuronal basis of drug reinforcement is provided.
Most investigations into the pathogenesis of drug addiction and the physiological consequences from acute and repeated drug exposure have focused on the function and adaptations of dopamine-producing neurons in the ventral midbrain. Two anatomical divisions—the ventral tegmental area (VTA) and the substantia nigra pars compacta (SNc)—provide dopaminergic innervation throughout the forebrain, including the prefrontal cortex (PFC), amygdala, hippocampus, and striatum (Tritsch and Sabatini, 2012). Dopaminergic projections from the SNc largely terminate in the dorsal striatum (the nigrostriatal pathway), while neurons originating from the VTA send projections to the nucleus accumbens (NAc), a component of the ventral striatum, as well as the PFC (the mesolimbic and mesocortical pathways, respectively) (Björklund and Dunnett, 2007; Oades and Halliday, 1987). Initial insight into the functional consequence of dopaminergic transmission in the context of reward was strongly influenced by experiments of intracranial self-stimulation (Olds and Milner, 1954). Rats with electrodes implanted in certain locations, including fibers of passage for the mesocorticolimbic pathway, would exert extraordinary effort in order to receive electrical stimulation (Crow, 1972; Olds, 1958). In addition, fast-scan cyclic voltammetry, which can resolve fluctuations in extracellular dopamine levels at the subsecond scale, have largely confirmed that drugs of abuse increase the amplitude of phasic dopamine release (Phillips et al, 2003; Vander Weele et al, 2014). Taken together, these results suggest that dopaminergic transmission in response to drug administration may be a common neurobiological substrate that may account for the overwhelmingly reinforcing properties of various abused substances (Lüscher and Malenka, 2011).
Although the dynamics of mesocorticolimbic dopamine release are important for certain aspects of reinforcement learning, it is clear that dopamine is not always necessary for the development of reward-like behavior (Salamone and Correa, 2012). For instance, transgenic mice incapable of producing dopamine still exhibit preference for sweetened sucrose solution over water (Cannon and Palmiter, 2003) and develop conditioned place preference (CPP) for a morphine-paired chamber (Hnasko et al, 2005). There are some caveats to the validity of this transgenic method (eg, the mice must be treated daily with L-DOPA to survive and given caffeine to overcome hypoactivity), but these experiments and others indicate that non-dopaminergic molecular mechanisms may also mediate reinforcement learning.
There are multiple, non-mutually exclusive explanations for why researchers have faced challenges in unraveling the neurocircuitry involved in addiction. For one, the composition of cells within the midbrain reward system are more complex than originally presumed (Lammel et al, 2014; Lammel et al, 2012). Neurons within the VTA are not identical in the synaptic input they receive, nor do they project to identical targets (Beier et al, 2015). In addition, VTA neurons may release more than just dopamine, often co-releasing glutamate and GABA simultaneously (Hnasko et al, 2010; Root et al, 2014; Tritsch and Sabatini, 2012), which complicates the characterization of post-synaptic effects. An additional hindrance in translating addiction research involves the necessity of comparing results across experiments that can vary dramatically by a multitude of factors, including the drug concentration, method of delivery (eg, experimenter administered vs self-administration), time-course of drug access (eg, hours of access per day, number of days of access), and the consequent neuroadaptations that occur from repeated drug exposure. To understand how reward and reinforcement is encoded in the brain, we need to understand the pathways and the participants—that is, understanding not only the detailed neuronal circuitry but also characterizing which cell types can contribute to a given behavioral outcome. To this end, there is a growing appreciation that glia and non-neuronal cell types can be key participants in a wide array of cognitive and behavioral functions. The following sections serve as an entry point to discussing mechanisms that impact glial-neuroimmune signaling and therefore the neurocircuitry underlying abuse liability and reward behavior.
GLIAL PHYSIOLOGY: MECHANISMS REGULATING FUNCTION
Microglia
Microglia have long been considered the resident immune cells of the brain and spinal cord, sharing many functional characteristics with circulating and tissue-specific macrophages of myeloid origin (del Río-Hortega, 1932; Thomas, 1992). They are distributed throughout all brain regions but in varying densities, with the highest concentration of cells appearing in the substantia nigra, basal ganglia, and hippocampus (Lawson et al, 1990). Each cell has numerous ramified processes that extend throughout the surrounding neuropil, but their processes rarely overlap with those of a neighboring microglial cell (Kierdorf and Prinz, 2013). This spatial organization within the brain demonstrates how microglia are active surveyors of their tissue microenvironment, sensing the location of other microglia and receiving regulatory signals from surrounding neurons that influence their location. The surface membrane of microglia is replete with a tremendous diversity of receptors for mounting stimulus-specific responses against a variety of potential threats to tissue homeostasis (for review see Hanisch et al, 2007; Lucin and Wyss-Coray, 2009; Ransohoff and Perry, 2009; Saijo and Glass, 2011). This includes an extensive catalog of exogenous factors, such as infectious microbes (eg, bacterial, viral, or fungal pathogens), toxins, and other foreign substances (ie, xenobiotics), as well as noxious endogenous compounds released from dead or dying cells that often result from conditions such as traumatic brain injury, ischemia, or neurodegeneration (Xanthos and Sandkühler, 2014). Upon detection of these threats, microglia mount specialized responses that can include phagocytosis, lysosomal degradation, and secretion of cytokines, chemotactic cytokines (ie, chemokines), or growth factors that can effect neuronal function, recruit additional immune cells, aid in tissue repair, or induce apoptosis (Kierdorf and Prinz, 2013). In addition to these signaling principles that are initiated by the presence of exogenous factors that are not usually seen in the CNS or endogenous factors not normally seen at such concentrations (such as intracellular release of heat shock proteins), there are also a number of endogenous membrane-bound and secreted factors originating from neurons that influence microglial activity (Hanisch et al, 2007). These so called ‘on’ and ‘off’ signals that regulate microglial responses are extremely varied and beyond the scope of this review (see Biber et al, 2007). We focus in the sections that follow on the signaling pathways associated with Toll-like receptor (TLR) complexes, as these are the most extensively characterized receptors for host defense, as well as in the response to drugs of abuse (Gay et al, 2014; Trotta et al, 2014). See Box 1 for an in-depth discussion of TLRs and their unique properties of recognition and signaling.

Astrocytes
Astrocytes have traditionally been characterized as supportive cells in the brain for their roles in maintaining neuron homeostasis and survival. The mammalian nervous system requires astrocytes; genetic elimination of astrocyte precursors is lethal to pups within 2 weeks of birth, which coincides with the period of exuberant synaptic outgrowth in the rodent cerebral cortex (Li et al, 2012a). Similar to microglia, in the healthy brain astrocytes extend a network of cellular processes throughout the CNS parenchyma that ensheath synapses and establish connections with blood vessels. A single astrocyte can make contact with a large number of synapses emerging from a single neuron as well as across a population of neurons, ranging from hundreds of synaptic contacts per astrocyte in the cortex to tens of thousands of synapses in the hippocampus (Bushong et al, 2002; Halassa et al, 2007). As a result, astrocytes are well-positioned to integrate signals from a great number of synapses at once, which may have implications for higher-order information processing.
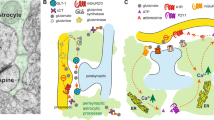
The role of astrocytes at the synaptic cleft are diverse and involve reuptake of neurotransmitters, buffering of extracellular ion concentrations, and release of many diffusible signaling molecules (see Araque et al, 2014; Fields et al, 2015). In particular, astrocytes have been shown to be essential for glutamate regulation between neurons. Inhibition of astrocyte proliferation leads to profound sensitivity to glutamate toxicity in vitro (Rosenberg and Aizenman, 1989), while in vivo disruption of astrocyte potassium channel activity results in impaired synaptic glutamate clearance with devastating effects to survival (Djukic et al, 2007). Astrocytes express glutamate transporters, including GLAST and GLT-1, which are critical for clearing synaptic glutamate. This was confirmed by intracerebroventricular delivery of antisense oligonucleotides against GLAST or GLT-1, which results in excessive glutamate release, striatal neurotoxicity, and progressive motor deficits (Rothstein et al, 1996) (see Figure 1 and Table 1).

Glia modify synaptic plasticity via secreted factors, synapse formation, and synaptic pruning. (a) Synaptic elements from neurons are engulfed by microglia, which can alter synaptic circuitry. (b) Secreted factors from microglia and neurons include chemokines and cytokines. This allows for communication between different cell types (eg, neuronal expressed IL-34 can bind to microglial CSF1R). Blue represents neuronal origin, while green represents microglial origin. (c) Synapse formation via astrocytes (in purple) is positively and negatively regulated by hevin (yellow spheres) and SPARC (blue spheres), respectively. Astrocytes can also modulate neuronal activity through their effects on glutamate receptor trafficking and synaptic glutamate reuptake via GLT-1 and GLAST. (d) Synaptic pruning can occur in the CNS through a complement-mediated mechanism. C1q and C3 expressed on synapses can induce microglial engulfment and phagocytosis of synaptic elements. Within figure citations: 1, Trotta et al, 2014; 2, Lee and Kim, 2007; 3, Li et al, 2010a;4, Ginhoux et al, 2010; 5, Lin et al, 2008; 6, Chung et al, 2015a; 7, Clarke and Barres, 2013; 8, Schafer et al, 2012; 9, Stevens et al, 2007.
Although the immunoregulatory properties of astrocytes are perhaps less appreciated than microglia, astrocytes can also participate in inflammatory signaling within the CNS as well as regulating immune cell trafficking at blood vessels (see Tian et al, 2012). In response to injury or other insults, astrocytes can secrete a variety of neuroimmune signaling molecules, including IL-1α, IL-6, TNFα, and IFN-γ (Lau and Yu, 2001). Release of immune-related molecules from astrocytes can signal to and regulate glial cell function as well as modulate synaptic function. In particular, release of the cytokine TNFα from astrocytes has been shown to increase AMPA glutamate receptor trafficking and modulate synaptic scaling of hippocampal neurons in culture (Beattie et al, 2002; Stellwagen and Malenka, 2006). In total, astrocytes and microglia are both essential contributors to maintaining tissue homeostasis in the CNS through a variety of signaling mechanisms.
GLIAL REGULATION OF NEURONAL FUNCTION AND STRUCTURE
Microglia
Resident tissue macrophages, including microglia, have the capacity to defend the organism against threats to tissue homeostasis. Under healthy conditions, it was traditionally assumed that microglia remained in a resting or quiescent state before rapidly mounting stimulus-specific responses to various signals of injury or danger, termed activation (Kreutzberg, 1996). Activation was most evident by alterations in cell morphology, in which resting microglia retract their highly ramified processes and adopt an amoeboid shape distinguished by high motility (Stence et al, 2001). However, the notion of microglia ‘resting’ in the healthy CNS was upended by a series of reports that devised an elegant preparation to visualize mammalian microglia in vivo (Davalos et al, 2005; Nimmerjahn et al, 2005). Microglia were fluorescently labeled in transgenic mice expressing enhanced GFP at the locus of the Cx3cr1 gene (Jung et al, 2000) and visualized by thinning the skull and performing two-photon microscopy. This preparation revealed that the soma of cortical microglia remain relatively stationary, but in contrast, their ramified processes are highly motile, retracting and extending protrusions throughout the parenchyma and surveying the entire brain every few hours (Nimmerjahn et al, 2005). This surveillance allows microglia to dynamically respond to signs of injury, which rapidly extend their processes toward sites injured by focal laser ablation (Davalos et al, 2005). Similar results were also obtained from zebrafish embryos; like their mammalian counterparts, microglial cells expressing GFP under the promotion of the apolipoprotein-E locus display highly mobile, extensively branched processes, which phagocytose apoptotic neurons for lysosomal degradation (Peri and Nüsslein-Volhard, 2008). Taken together, these results indicate that microglia are not simply ‘reactive’ immune cells that mobilize following infection or injury, but are active sentinels that are constantly surveying the healthy CNS.
On the basis of these observations, researchers have sought a potential explanation for why an organism might expend considerable energy to continuously scan the neuropil under homeostatic conditions. As a result, several groups have argued that microglia might have an essential role in monitoring synaptic activity (Bilbo and Schwarz, 2012; Graeber, 2010; Kettenmann et al, 2013; Schafer and Stevens, 2013). In support of this hypothesis, ultrastructural analysis in mice has revealed that microglial processes make direct contact not only with axon terminals and dendritic spines but also astrocytes and their perisynaptic processes that ensheath dendritic branches (Tremblay et al, 2010). Similarly, simultaneous in vivo two-photon imaging of fluorescently labeled neurons and microglia confirmed that microglial processes make frequent but brief contact with dendritic spines (Wake et al, 2009). These results put microglia in a prime position to monitor fluctuations in secreted elements at synapses, but it was unclear if their function could be altered by changes in neuronal activity. Remarkably, reducing basal neural activity in the visual cortex by either binocular occlusion or intraocular injection of tetrodotoxin (TTX) reduces the frequency that processes make contact with neurons (Wake et al, 2009). Similar results were obtained when animals were reared in complete darkness during a critical period of visual cortex development (Tremblay et al, 2010). In contrast, increasing neuronal activity in zebrafish by either glutamate uncaging or repetitive visual stimulation results in increased contact by microglial processes, which preferentially ensheath neurons with high spontaneous calcium transients through a mechanism that likely involves calcium-induced release of ATP from neurons (Li et al, 2012b). Contact by microglial processes was also associated with a subsequent decrease in spontaneous spiking frequency (Li et al, 2012b). Therefore, microglia appear to monitor synaptic function and alter their physiological behavior in response to changes in neuronal activity, which in turn can exert influence on the activity properties of contacted neurons. Likewise, microglial surveillance of synapses is likely an evolutionarily conserved process, as similar properties are observed from zebrafish to mice. On the basis of their macrophage-related lineage, many wondered if microglia have a role in phagocytosing structural rudiments of the CNS, including synapses.
In seeking answers to these questions, it has become increasing clear that microglia can be active participants in sculpting neural circuits. The initial insight for synaptic stripping originated from the facial nerve injury model, in which unilateral incision of the facial nerve would reliably produce microglial activation on the transected side. Light and electron microscopy revealed that microglial processes make direct contact with dendritic shafts and perikarya of damaged neurons and remove synaptic terminals from these branches, suggesting that under conditions of sterile injury, microglia can remove synaptic structures from axotomized neurons (Blinzinger and Kreutzberg, 1968). A similar outcome was described in the sensorimotor cortex following immune activation; focal targeting of cortical microglia by stereotaxic delivery of heat-killed bacteria and subsequent peripheral bacterial challenge resulted in increased contact of Iba1+ and CD68+ microglia processes with neurons and decreased the number of presynaptic terminals compared with saline injections (Trapp et al, 2007). Increased elimination of dendritic spines is also observed under other conditions of injury that are characterized by microglial activation, including ischemia (Wake et al, 2009) and craniotomy (Xu et al, 2007). These results are consistent with the notion that microglia are sensitive to changes in the neuronal milieu and that certain molecular signals can promote pruning of synapses. However, it was previously unclear what molecular signals may promote synapse elimination and, more importantly, if this process was restricted to states of severe injury, or if it may have broader implications for shaping neuronal connectivity.
These questions were methodically addressed in the context of retinogeniculate system development. Axons from retinal ganglion cells (RGCs) form exuberant synaptic connections in the dorsal lateral geniculate nucleus (dLGN) of the thalamus early in development. These imprecise and overlapping projections are then selectively eliminated in an activity-dependent fashion, resulting in remarkably precise eye-specific segregation of synaptic input (Luo and O'Leary, 2005; Schafer et al, 2013a). During the postnatal period when synaptic refinement is still ongoing (P5), microglial phagocytosis of RGC inputs is high, compared with time periods after eye-segregation is established (P9 or P30) (Schafer et al, 2012). This synaptic pruning is exquisitely dependent upon neuronal activity. When neuronal activity in one eye is inhibited by intraocular injection of TTX, microglia preferentially engulf RGC input from the TTX-treated eye; conversely, when neuronal activity in one eye is augmented by forskolin (an adenylyl cyclase activator that potentiates neuronal activity), microglia preferentially engulf RGC input from the saline-treated eye (Schafer et al, 2012). Thus, microglia participate in synaptic refinement during development by engulfing the synapse with relatively ‘weak’ synaptic strength.
Subsequent synaptic refinement experiments determined a somewhat surprising role for classical complement proteins of the immune system in this process; specifically, C1q and C3 localize to synapses intended for elimination. Microglia that express the complement receptor for C3 (also known as CD11b/CD18) are activated for phagocytosis of these ‘opsonized’ or labeled synapses. Mice with transgenic knockout of either the ligand C3 or its receptor CR3 both show a significant decrease in RGC synaptic engulfment compared with wild type, and this disruption in synaptic pruning results in greater overlap between ipsilateral and contralateral RGC input to the dLGN, a deficit which persists well into adulthood (Schafer et al, 2012). These results represent some of the most compelling evidence that microglia directly alter neural circuitry by actively phagocytosing structural elements from neurons, a process driven by changes in neuronal activity.
Astrocytes
The participation of astrocytes in the development, maturation, and refinement of neuronal synapses has been well-established for over two decades. Early work with in vitro preparations of neuronal cultures noted that astrocytes were required for proper axon outgrowth, spatial arrangement, and neuron cell survival (Banker, 1980; Noble et al, 1984). The ability to identify astrocyte-specific factors regulating neuron function was achieved with the development of a purified culture of neurons (ie, RGCs), whose survival can be maintained in the absence of glia with the addition of several growth factors (Meyer-Franke et al, 1995). When cultured in isolation, these RGC neurons extend their processes but produce very few functional synapses. Interestingly, when cultured with a feeder layer of astrocytes for several days, these neurons produce numerous functional synapses, based on characterization of their electrophysiological properties, immunostaining for synaptic markers, and evaluation by electron microscopy (Pfrieger and Barres, 1997; Ullian et al, 2001). Culturing RGCs with astrocyte conditioned medium (ACM) results in similar synaptic development, implying that secreted signals from astrocytes promote synapse formation (Christopherson et al, 2005).
To this end, a great number of synaptogenic molecules from astrocytes have been identified. In the context of early brain development, a critical role for thrombospondins (TSPs) has been characterized. TSPs are extracellular matrix proteins secreted by astrocytes that appear to be important for synapse formation. Purified TSP1 or TSP2 delivered to RGCs replicates the development of synapses seen following ACM treatment (Christopherson et al, 2005). Consistent with these in vitro data, genetic knockout of TSP1 or TSP2 causes a dramatic reduction in synaptic puncta in the mouse cerebral cortex (Christopherson et al, 2005). Subsequent experiments identified the calcium channel subunit α2-δ1 as the synaptic receptor responsible for the synaptogenic action of TSPs (Eroglu et al, 2009). Although the synapses formed by TSPs are functionally ‘silent’ by lacking AMPA receptors, astrocytes can also promote GluA1-containing AMPA receptor expression to produce postsynaptically ‘active’ synapses through release of glypicans 4 and 6 (Allen et al, 2012).
In addition to TSPs, the secreted matricellular protein hevin (also known as secreted protein acidic and rich in cysteine (SPARC)-like 1, or SPARCL1) is important for RGC synapse formation in vitro and for establishing synaptic connectivity to the superior colliculus in vivo (Kucukdereli et al, 2011). Interestingly, the protein SPARC is also released by astrocytes and functions to inhibit the synaptogenic action of hevin (Kucukdereli et al, 2011). The positive and negative actions of these molecules on synapse formation demonstrate that astrocytes can carefully tune the formation of synaptic elements in a bidirectional manner.
There are many other molecules released by astrocytes that have been shown to be synaptogenic, and importantly, their expression is dynamically regulated at different developmental time points (for an extensive review see Chung et al, 2015a; Clarke and Barres, 2013). Interactions between astrocytes and microglia may determine synapse elimination as well. The complement protein C1q that opsonizes synapses for elimination is expressed by purified RGCs, and its transcription is regulated by astrocytic release of the cytokine transforming growth factor (TGF)-β (Bialas and Stevens, 2013). On the basis of these results and others, it has been proposed that astrocytic control of synaptic transmission and plasticity may have an important role in the development and maintenance of substance abuse (Haydon et al, 2009).
These collective data suggest that under pathological conditions in the CNS or in conditions of altered glial activation, there may also be inappropriate remodeling of synaptic structures and persistent alterations in neuronal activity (Chung et al, 2015b; Robel and Sontheimer, 2016). On the basis of the ability of microglia and astrocytes to monitor, effect, create and eliminate synapses (see Figure 1), one could hypothesize that synaptic remodeling may occur under the conditions of drug exposure and abuse and could contribute to the persistent behavioral effects characteristic of substance abuse disorders (Coller and Hutchinson, 2012; Miguel-Hidalgo, 2009).
OPIOIDS, GLIA, AND NEUROIMMUNE SIGNALING
Drugs of abuse could influence glia directly through interactions at surface receptors sensitive to these natural or synthetic compounds, or indirectly through their effects on neighboring neurons surveyed by microglia or astrocytes, which include but are not limited to changes in neurotransmitter release or synthesis, protein trafficking, spontaneous or evoked patterns of action potentials, and release of cytokines, chemokines, or other immune signaling molecules. In turn, glial responses to drugs of abuse could influence neuronal function directly through physical remodeling of structural components (Brown and Neher, 2014; Corty and Freeman, 2013), as described in the previous section, or indirectly through a variety of released compounds (see Block et al, 2007). This chemical communication from glia can be both anti-inflammatory and neuroprotective (eg, IL-10, TGF-β, and BDNF) or pro-inflammatory and neurotoxic (eg, IL-1β, TNFα, and NADPH oxidase) (see Figure 1). In particular, the expanding literature implicating glia as critical effectors of synaptic maturation and pruning can be extended to predict that glia may participate in regulating synaptic plasticity in the face of drug exposure. The following section will explore the physiological effects that opioids exert on glial biology, in particular changes to neuroimmune signaling, as well as the evidence that glial activity may influence behavioral responses to these drugs. The phrase ‘neuroimmune signaling’ is used deliberately to emphasize that, while microglia and astrocytes are the principal cells of immune regulation in the CNS, they signal to and receive signals from all cells of the CNS, including neurons, microglia, astrocytes, oligodendrocytes, non-parenchymal CNS macrophages, and endothelial cells (see Hutchinson and Watkins, 2014).
Derivatives of the opium poppy (Papaver somniferum), particularly morphine, have been used for thousands of years due to their potent analgesic and rewarding effects. Opioids have varying degrees of selectivity for the classical heterotrimeric Gi/o-coupled opioid receptors—μ, δ, and κ receptors—but the reinforcing effects are largely attributed to their signaling at μ-opioid receptors (Matthes et al, 1996), which is due in part to suppression of local GABAergic inhibition of VTA dopamine neurons (Di Chiara and Imperato, 1988; Johnson and North, 1992; Shoaib et al, 1995). Apart from their effects on neurons, there is some evidence that microglia can be influenced through classical opioid receptor signaling. Cell cultures of neonatal cortical microglia from rats show immunoreactivity for all three receptor classes and dynamically regulate their expression in response to morphine exposure (Turchan-Cholewo et al, 2008), while others have detected μ- and κ-receptors on cultured human microglia (Chao et al, 1996, 1997). Treatment with morphine or the selective μ-opioid receptor agonist DAMGO inhibits chemotaxis in human cultures (Chao et al, 1997), although increases in morphine-induced chemotaxis were observed in rodent cultures in a manner that requires P2X4 purinergic receptors (Horvath and DeLeo, 2009; Takayama and Ueda, 2005). These differences could be species-specific or due to disparities in the assays used to quantify chemotaxis. Alternatively, a compound used by Horvath and DeLeo (2009) as a μ-opioid receptor antagonist (CTAP) has also been described as an antagonist for TLR4 (Grace et al, 2014b), making it difficult to interpret these results. In contrast to cultured cells, fluorescence-activated cell sorting (FACS) of purified microglia and astrocytes from the NAc revealed near undetectable relative gene expression for μ-, δ-, and κ-opioid receptors on either cell type (Schwarz et al, 2013). These results could implicate heterogeneous expression of opioid-sensitive glia across regions and developmental time points, but also suggests that the role of opioid receptors on glia awaits further description.
Beyond the classical opioid receptors, a fascinating area of research receiving considerable interest has originated from the observations of overlapping cellular responses between opioids and typical activators of the innate immune system (Jacobsen et al, 2014). Similar to an exposure of bacterial lipopolysaccharide (LPS), it was discovered that repeated injections of morphine increases microglial CD11b and astrocytic GFAP densitometry in the rat brain (Hutchinson et al, 2009). Importantly, these measures of glial activation are not found across the entire brain, but are rather restricted within certain brain regions, including the VTA, dentate gyrus, periaqueductal gray, dorsal caudate, and other regions that have previously been associated with opioid dependence (Hutchinson et al, 2009). In a similar manner, an acute injection of morphine rapidly increases mRNA expression of both microglial CD11b and astrocytic GFAP in the NAc, but not the hippocampus (Schwarz et al, 2011). Concomitant systemic treatment with a pharmacological modulator of glial activation such as ibudilast, which functions as a non-selective phosphodiesterase inhibitor but has broad effects on glial function (Gibson et al, 2006), is effective at preventing CD11b and GFAP induction by morphine (Hutchinson et al, 2009), a result replicated by targeted intra-NAc infusions of ibudilast (Schwarz et al, 2011). Minocycline, a highly lipid-soluble tetracycline antibiotic that can also modulate glial activity, is capable of preventing LPS-induced upregulation of microglial Iba1 but has no effect on astrocytic GFAP upregulation following LPS (Yoon et al, 2012). In vitro assessment of cultured rat microglia confirms that minocycline can inhibit microglial activation to morphine, measured by a dose-dependent reduction in morphine-induced cyclooxygenase-1 production following minocycline treatment (Hutchinson et al, 2008). These results indicate that opioids may stimulate microglia and astrocytes into an activated state through a shared mechanism of LPS detection, although their activation appears to be confined to discrete locations within the brain. Systemic or region-specific injections of glial modulators prevent morphine-induced elevations of CD11b or GFAP, although the precise mechanisms whereby ibudilast or minocycline suppress glial activity remain unclear. It is important to consider that both ibudilast and minocycline are known to affect the functional properties of other cells, so the mechanism of action for these pharmacological agents may not be exclusive to glia (González et al, 2007).
In addition to changes in surface markers indicative of glial activation, opioid administration increases a number of neuroimmune signaling molecules and receptors, including cytokines (IL-1β, TNFα), chemokines (CCL2), and related intracellular signaling pathways affecting gene transcription (extracellular signal-related kinase (ERK)/mitogen-activated protein kinase (MAPK), NF-κB, activator protein-1 (AP-1)) (reviewed in Coller and Hutchinson, 2012), which are all reminiscent of the downstream inflammatory profile induced by innate immune PRR signaling (see Rivest, 2009). Consequently, opioid-induced neuroimmune signaling appears to be largely pro-inflammatory, although transcription of the anti-inflammatory cytokine IL-10 is also induced by acute morphine (Schwarz et al, 2011), an effect that parallels LPS injection (de Waal Malefyt et al, 1991). For instance, acute treatment of morphine rapidly increases gene expression of interferon-γ (IFN-γ) and several classes of chemokines within the NAc, an effect which is suppressed by pre-treatment with ibudilast (Schwarz et al, 2011). This effect was mediated in part through significant induction of anti-inflammatory IL-10 gene expression in response to ibudilast treatment (Schwarz et al, 2011).
Because of the striking similarities in the neuroimmune signaling response between endotoxin (ie, LPS) and opioid drugs, investigators predicted there would be a commonality in how these disparate xenobiotics initiate comparable outcomes. This prediction has led to the characterization of TLR4 as a novel, non-classical opioid-binding site that encompasses mechanisms of innate immunity to alter glial function (see Box 1). Opioid-induced activation through non-classical binding has experimental precedent in the context of neuropathic pain (Watkins et al, 2009); opioid-induced hyperalgesia was reported in opioid receptor triple knockout mice, suggesting alternative sites of action for altering nociception (Juni et al, 2007). In silico docking simulations predicted that opioids would bind to the LPS-binding pocket of MD-2, the co-receptor for TLR4 (Hutchinson et al, 2010), which was later confirmed in vitro (Wang et al, 2012a). In an extensive series of experiments, morphine was determined to induce conformational changes at the MD-2 structure to induce TLR4 oligomerization, resulting in pro-inflammatory interleukin production that was blocked by pharmacological or RNA interference at the TLR4/MD-2 complex (Wang et al, 2012a). Likewise, (+)-naloxone and (+)-naltrexone, which are stereoisomers of traditional antagonists to opioid receptors, have been characterized as selective antagonists to TLR4 signaling with no appreciable function at opioid receptors (Hutchinson et al, 2011; Wang et al, 2016). These initial insights have expanded the notion of TLRs as not just sensors for pathogen- or damage-associated molecular patterns, but are perhaps sensitive to a wider range of xenobiotic or ‘non-self’-associated molecular patterns.
Considering the previous evidence that opioids likely contribute to glial activation through TLR4-dependent signaling, one could predict that consequences of this signaling pathway would influence responding to opioids. Consistent with this prediction, (+)-naloxone (a TLR4 antagonist) administered immediately before morphine injections prevents the establishment of morphine CPP in rats, while transgenic mice deficient in either TLR4 or MyD88 both failed to demonstrate CPP for oxycodone (Hutchinson et al, 2012). An explanation for this effect may partly derive from alterations in dopaminergic transmission; similar to the result obtained using ibudilast, (+)-naloxone suppresses morphine-induced dopamine release into the NAc as measured by in vivo microdialysis (Hutchinson et al, 2012). Opioid-induced TLR-mediated signaling appears to depend on microglial p38 MAPK (Zhang et al, 2012), a pathway associated with cAMP-responsive element-binding protein (CREB) -mediated transcription of pro-inflammatory cytokines and chemokines (Gay et al, 2014). Thus, in addition to opioid receptor-mediated effects on neurons, TLR signaling can strongly influence conditioning to the motivational properties of different classes of abused opioid drugs.
Experiments designed to investigate if microglia or astrocytes contribute to self-administration of opioids are desperately needed, as this behavioral paradigm has strong validity for modeling voluntary acquisition of abused substances. In rats, self-administration of remifentanil, an ultra-short-acting synthetic opioid, is dose-dependently reduced by (+)-naloxone treatment (Hutchinson et al, 2012). In a similar fashion, chronic antagonism of TLR4 with (+)-naltrexone delivered via osmotic minipumps during withdrawal reduces cue-induced heroine self-administration (Theberge et al, 2013). Importantly, a single dose of (+)-naltrexone administered before extinction testing was ineffective at reducing heroin intake, suggesting that TLR4 antagonism may not acutely diminish the reinforcing properties of heroin after repeated exposure but rather influence synaptic remodeling during the withdrawal period.
Importantly, mechanisms of drug metabolism can also influence neuroimmune activation. For instance, the morphine metabolite morphine-3-glucuronide exhibits strong affinity for the TLR4 receptor, while morphine-6-glucuronide exhibits no TLR4 activation (Lewis et al, 2010; Wang et al, 2012a). Alternative synthetic opioids would produce different metabolites, which could be an important factor when considering similarities and differences in behavioral outcomes to these opioid drugs. On the basis of the differences in pharmacodynamics and cellular metabolism, glial activation and the resultant neuroimmune signaling will likely vary across different classes of abused substances, and may even vary within opioids.
Although a comprehensive explanation of the underlying neuronal adaptations to opioids remains an essential and ongoing research endeavor, these results affirm that neuroimmune signaling contributes to the pathogenesis of opioid abuse and offers a potential entry point for future development of therapeutics.
ALCOHOL, GLIA, AND NEUROIMMUNE SIGNALING
Alcohol or ethanol (hereafter referred to as alcohol) has been extensively characterized for its effects on neuronal physiology, neurotransmission, and synaptic plasticity (Weiss and Porrino, 2002). The pharmacological effects of alcohol in the brain are diverse and include alterations in lipid cell membrane permeability, GABA release, GABAA and NMDA receptor functionality, and adaptations in other neurotransmitter systems (see Valenzuela, 1997). The positive reinforcing properties of alcohol suggest that dopaminergic neurotransmission is altered during repeated drug exposure, and indeed, alcohol administration leads to changes in VTA dopamine neuron activity using in vitro and in vivo models (Brodie et al, 1999; Weiss et al, 1993; Yim and Gonzales, 2000). Functional alterations in related circuits, including the central amygdala, frontal cortex, NAc, and bed nucleus of the stria terminalis (BNST) have been vigorously examined, and several molecular mechanisms have been implicated in the transition from acute to chronic alcohol consumption, including corticotropin-releasing factor (CRF), neuropeptide Y (NPY), and endogenous opioids (Mitchell et al, 2012; Pleil et al, 2015; Roberto et al, 2010).
Although alcohol-induced effects on glia have not been as extensively characterized as its effects on neurons, accumulating evidence suggests that both microglia and astrocytes are impacted by acute and chronic alcohol. Early in vitro work with astrocyte progenitors cultured from fetal rat brains found that acute alcohol resulted in reduced GFAP expression in addition to stunted astrocyte cell proliferation (Guerri et al, 1990; Renau-Piqueras et al, 1989). In vivo, rats given free access to alcohol show increased activation of astrocytes (measured by GFAP) in discrete subregions of the hippocampus following shorter exposure periods (4–12 weeks), but decreased astrocyte activation was observed following extended drug access (36 weeks) (Franke, 1995). Likewise, analysis of post-mortem human tissue reveals reduced GFAP immunoreactivity in dorsolateral prefrontal cortex (Miguel-Hidalgo et al, 2002) and a dramatic loss of astrocytes in the hippocampus of alcoholics (Korbo, 1999). Microglia also show classical signs of activation in response to alcohol. Chronic alcohol treatment results in increased CD11b densitometry in the frontal cortex of female mice (Alfonso-Loeches et al, 2010). Likewise, increased immunoreactivity for microglial Iba1 is observed in the cingulate cortex of alcohol-dependent humans (He and Crews, 2008). These results offer indirect evidence that alcohol can influence cell viability and activation state in microglia and astrocytes, although it is important to note that these glial responses do not appear to be uniform throughout the brain but rather differ based on brain region assessed, the concentration and frequency of drug exposure, and the developmental period during exposure. Further studies are required to delineate the mechanisms explaining these selective glial responses to alcohol.
Alcohol is a potent neurotoxic substance to the tissues of the CNS and its administration is associated with a cascade of various neuroimmune signaling molecules. Similar to LPS treatment, application of alcohol to cultured microglia results in marked elevations of soluble IL-1β, IL-6, CCL3, CXCL2, and TNFα (Boyadjieva and Sarkar, 2010). Treating developing hypothalamic neurons in culture with alcohol-activated microglia-conditioned media results in apoptotic death of neurons—an effect that is blocked by pre-treating conditioned media with blocking antibodies against TNFα—suggesting that improper TNFα release in response to alcohol could damage neighboring cells (Boyadjieva and Sarkar, 2010). Cytokine signaling within discrete brain regions can also have functional consequences on neurotransmission. Bath treatment of IL-1β alters action potential-independent vesicular GABA release in a subset of mouse central amygdala neurons, an effect that is reversed by co-administration of alcohol (Bajo et al, 2015). Because both neurons and astrocytes express IL-1 receptors, it cannot be determined if the electrophysiological consequences are driven by glial dependent or independent mechanisms, a caveat that is worth considering for many of the studies examined here. Regardless, inhibition of IL-1 signaling in the basolateral amygdala via local infusions of IL-1 receptor antagonist reduced acquisition of alcohol, suggesting that cytokine expression in the amygdala could modulate the reinforcing effects of alcohol (Marshall et al, 2016).
The regulation of oxidative stress, a critical function of glia for maintaining homeostasis, is another important pathway of interaction between neurons and glia in response to alcohol. Cultured neonatal hamster microglia exposed to alcohol for 24–48 h show increased levels of superoxide anions (Colton et al, 1998). Consequently, shifts in oxidative stress can impact neuronal cell viability. An in vitro application of microglia-conditioned media after 24 h alcohol incubation increased alcohol-induced apoptosis of developing hypothalamic neurons and produced elevations in intracellular production of O2−, reactive oxygen species (ROS), and nitrite while reducing several antioxidative substances (Boyadjieva and Sarkar, 2013). Likewise, increased expression of NADPH oxidase is found in cortical tissue from both alcohol-treated mice and alcohol-dependent humans (Qin and Crews, 2012). Increased catalytic production of superoxides can potentially lead to neurodegeneration either directly through oxidation of proteins and cell membranes, or indirectly through amplification of NF-κB-mediated transcription of TNFα or other pro-inflammatory cytokines (Crews and Vetreno, 2016).
Genetic analyses have an important role in identifying many immune-related genes associated with alcohol abuse in both rodents and humans (Kimpel et al, 2007; Liu et al, 2006; Mulligan et al, 2006). Validating several candidate genes from these studies (including Cd14, Il1ra, and Il6) through single-gene null mutations in mice confirmed that knockout of each gene resulted in decreased alcohol consumption on a two-bottle choice test (Blednov et al, 2012). In humans, polymorphisms with genes encoding for IL-1β, IL-1RN, IL-10, and other immune-related genes have been associated with alcohol dependence, the functional impact of these allelic variants generally supporting a link between increased pro-inflammatory signaling and increased susceptibility to alcoholism (Marcos et al, 2008; Pastor et al, 2005). Consistent with this notion, when whole-brain transcriptome analysis was conducted across multiple strains of rats, the functional relationship among candidate genes most strongly associated with alcohol consumption included gene networks related to neuron-glia communication and neuroimmune signaling (Saba et al, 2015). As a whole, these investigations implicate immune gene regulation as a fundamental signaling network in the context of alcohol abuse.
Similar to opioids, there is considerable evidence that innate immune signaling pathways, including TLRs, are implicated in the neuroimmune response to alcohol (for a detailed review see Crews and Vetreno, 2016; Crews et al, 2015). In alcohol-preferring rats, self-administration of alcohol is associated with increased TLR4 protein expression in the VTA, but not ventral pallidum (June et al, 2015). Several members of the receptor complex—TLR4, MD-2, and CD14—are critical for alcohol-induced activation of astrocytes in culture, as small interfering RNA (siRNA) to knockdown expression of any of these three targets prevented nuclear translocation of the NF-κB-p65 subunit in response to alcohol (Alfonso-Loeches et al, 2010). As predicted from these results, TLR4-deficient mice, unlike their wild-type counterparts, show no evidence of alcohol-induced effects on NF-κB-p65 activation, pro-inflammatory cytokine production, or caspase-3 cleavage, a marker for apoptosis (Alfonso-Loeches et al, 2010). Null mutations in CD14 can broadly disrupt many of the LPS- or alcohol-induced changes in the electrophysiological properties of GABA neurons in mouse central amygdala, supporting the notion that TLR signaling contributes to alcohol-induced changes in neuronal activity (Bajo et al, 2014).
Disruptions to innate immune signaling appears to contribute to the behavioral consequences of alcohol. Acute alcohol-induced sedation was significantly attenuated in mice genetically deficient for either TLR4 or MyD88, and was nearly absent in mice lacking TLR2 (Corrigan et al, 2015; Wu et al, 2012). The authors also observed interesting interactions between alcohol and morphine, such that the loss of righting reflex that is potentiated by alcohol and morphine co-administration in wild-type mice is absent in Tlr2−/− and Myd88−/− mice, but not Tlr4−/− mice (Corrigan et al, 2015). These complex findings highlight how innate immune signaling pathways frequently exhibit synergistic or compensatory interactions, and the relative contribution of different TLRs to drug-induced behavioral effects are still a matter of ongoing investigation, especially in the case of polydrug abuse. Acute administration of both morphine and alcohol also results in increased Iba1+ microglia and NF-κB-p65 expression in layer 5 of motor cortex, but these drug-induced changes in glial activation and transcription factor phosphorylation are not observed in mice lacking TLR2 or TLR4 (Corrigan et al, 2015). Direct manipulation of TLR4 expression can also influence drinking behavior. Viral mediated delivery of siRNA to knockdown expression of TLR4 in the central amygdala was found to inhibit binge drinking in rats (Liu et al, 2011). In a similar fashion, delivery of siRNA against either TLR4 or CCL2 into the VTA produced a reduction in voluntary alcohol consumption in rats, while consumption of sucrose was unaffected (June et al, 2015). Although these results suggest a potential role for both TLR4 and TLR2, there are still many questions that remain regarding their relative contributions to neuroimmune signaling cascades induced by alcohol. Future studies should endeavor to disentangle the cell-type-specific contributions of these innate immune receptors.
There are also age-dependent effects of alcohol on neuronal and glial function, which can have significant health implications. For example, prenatal exposure to alcohol can result in fetal alcohol spectrum disorders, resulting in a broad spectrum of cognitive and behavioral abnormalities. By virtue of their critical role during brain development, researchers have hypothesized that improper glial activation during critical periods of development could lead to excessive cytokine and chemokine signaling, reduced cell survival, and possible aberrant sculpting of synapses (Bilbo and Schwarz, 2012; Wilhelm and Guizzetti, 2015). Indeed, acute alcohol exposure during the third trimester-equivalent time period in mice has been found to rapidly induce surface markers of microglial activation along with increased production of IL-1β and TNFα (Ahlers et al, 2015). This pro-inflammatory activity coincides with cortical neuronal apoptotic cell death, which likely drives the acute microglial response (Ahlers et al, 2015). This pattern may be reversed with chronic exposure, as repeated alcohol dosing in rats reduces the number and size of Iba1+ microglia in the dentate gyrus and CA1 subregions of the hippocampus (Boschen et al, 2016). However, it is unclear if these results are due to cell death, decreased proliferation, or cell migration.
Beyond early development, adolescence is also a developmental period marked by unique sensitivity to alcohol that can often result in long-lasting consequences to neuronal function. Properties of circuit function such as long-term potentiation, spontaneous interneuron firing, and alcohol-induced dopamine release in the NAc is impacted by adolescent alcohol exposure (Shnitko et al, 2016; Swartzwelder et al, 1995; Yan et al, 2010). Although the effects to neurons have been well characterized, much less is known about glial responses to alcohol during adolescence. Adolescent binge alcohol has been found to increase microglial activation (ie, Iba1 histology) in the hippocampus, consistent with exposure during adulthood (McClain et al, 2011). However, there is some evidence that alcohol may uniquely affect astrocytes during this developmental period. Intermittent alcohol exposure during adolescence is associated with changes in TSP expression within the hippocampus (Risher et al, 2015b). As previously mentioned, TSPs are glycoproteins secreted in part by astrocytes which mediate cell and matrix adhesion and are critical regulators of synaptogenesis during CNS development (Christopherson et al, 2005; Eroglu et al, 2009). Accordingly, the authors found that intermittent alcohol treatment results in exaggerated LTP in CA1 neurons and altered dendritic spine morphology in a manner indicative of spine immaturity (Risher et al, 2015a). As a result, one could conjecture that TSPs represent one of the soluble signals secreted by astrocytes to regulate excitatory synaptic restructuring in response to alcohol. Additional work is required to determine if astrocytes are required for this effect and if it is indeed restricted to certain brain regions during adolescence.
The evidence gained from this diverse set of methodologies converges on the hypothesis that innate immune signaling and pro-inflammatory neuroimmune gene regulation contributes to the biological and behavioral outcomes of alcohol abuse. Although microglia and astrocytes are the principle cells contributing to neuroimmune signaling within the CNS, definitive cell-type-specific characterization, particularly for in vivo experiments, is often lacking. Genetic manipulations restricted to defined cell types (such as manipulating genes of interest under the promotion of Cx3cr1 for microglia or Gfap for astrocytes) or in an inducible manner (such as using tamoxifen-inducible Cre) offer a promise for refined perturbations of molecular and cellular targets.
PSYCHOSTIMULANTS, GLIA, AND NEUROIMMUNE SIGNALING
Similar to opioids and alcohol, psychostimulants are remarkably reinforcing substances with a high potential for abuse (see Taylor et al, 2013 for review). The most commonly abused psychostimulants are cocaine and amphetamines, which includes the structurally related compounds of D-amphetamine, methamphetamine, and 3,4-methylenedioxymethamphetamine (MDMA or ecstasy). The pharmacological effects of psychostimulants are wide-ranging but are primarily characterized by their effects on elevating extracellular concentrations of monoamine neurotransmitters, including dopamine, norepinephrine, and serotonin (Ritz et al, 1990). Like other drugs of abuse, acute administration of psychostimulants results in increased dopamine release in the NAc, which is thought to occur by antagonism or reversal of dopamine transporter molecules (Volkow et al, 1997). The effects on presynaptic dopamine release are hypothesized to be an essential component to the highly motivational properties of these substances. However, mice lacking dopamine transporter will still self-administer cocaine, suggesting alternative or compensatory mechanisms for reinforcement (Rocha et al, 1998). On the basis of the extant literature of psychostimulants and their effects on glia, the following section will focus on cocaine, amphetamine, and methamphetamine.
There are several lines of evidence demonstrating that psychostimulants can alter the activation state of microglia and astrocytes in rodents. Cocaine and methamphetamine administration results in increased Iba1 densitometry across many regions, including striatum (Liao et al, 2016). Methamphetamine in particular can result in degeneration of dopaminergic terminals in the striatum, a process that is associated with elevated microglial activity (Thomas et al, 2004). Repeated amphetamine causes increased GFAP expression on astrocytes in the caudate-putamen but not the NAc or prefrontal cortex (Armstrong et al, 2004). Following withdrawal from prolonged cocaine self-administration GFAP expression is reduced in the NAc, an effect similar to alcohol (Scofield et al, 2016). In addition, post-mortem analysis of human cocaine abusers found evidence for increased microglial activation, particularly in the SNc (Little et al, 2009). Observing these changes in antigen expression demonstrates that the administration of some psychostimulants affect glia as well as neurons.
In terms of functional consequences, psychostimulants can profoundly alter the release of inflammatory mediators, although the responses are often dose-dependent and demonstrate drug- and brain region-specificity (see Coller and Hutchinson, 2012). In the brain, acute cocaine results in increased IL-1β mRNA in the NAc and cortex (Cearley et al, 2011), while methamphetamine leads to increased IL-6 and TNFα in hippocampus and frontal cortex (Gonçalves et al, 2008). Similarly, acute injections of cocaine results in elevated IL-1β mRNA in the VTA, an effect that is blocked with (+)-naloxone treatment (Northcutt et al, 2015). These data are supported by in vitro preparations, as cultured microglia exposed to cocaine upregulate transcription of TNFα, IL-6, and CCL2 (Liao et al, 2016) as well as IL-1β (Northcutt et al, 2015). Cocaine is cytotoxic to neurons and induces autophagy through a nitric oxide-mediated signaling pathway, which likely evokes an inflammatory or neuroprotective response from nearby astrocytes and surveillant microglia (Guha et al, 2016). Isolated microglia also show cocaine-induced cytotoxicity, causing their own release of ROS (Costa et al, 2013; Liao et al, 2016). Although the contribution of astrocytes cannot be ruled out, these results support the assumption that microglia produce inflammatory mediators in response to some psychostimulants, which may not necessarily be a secondary response to changes in neuronal activity or survival.
On the basis of the involvement of TLR signaling for opioids and alcohol, in addition to the antagonistic effects of (+)-naloxone treatment on cocaine-induced cytokine production, it was predicted that TLR activation would be associated with psychostimulant abuse. Indeed, recent data have implicated TLR2 and the TLR4/MD-2 signaling complex as critical pathways for the physiological and behavioral effects of cocaine. In a series of in vitro and in vivo experiments, Liao et al (2016) demonstrate that cocaine activates mouse microglia resulting in pro-inflammatory cytokine release, as well as upregulation of TLR2 mRNA and protein. Blocking transcription of TLR2 or inhibiting downstream oxidative stress with an NADPH oxidase inhibitor prevented cocaine-induced cytokine production. Beyond TLR2, Northcutt et al (2015) present compelling evidence that cocaine directly interacts with TLR4/MD-2. These data were supported by structural modeling, which found that cocaine docks to MD-2 at the same site as (+)-naloxone or (+)-naltrexone. More importantly, the authors found that pharmacological or genetic disruption of TLR4 signaling significantly decreased self-administration of intravenous cocaine for both mice and rats (Northcutt et al, 2015). Thus, these experiments lend support to the notion that innate immune receptors in the CNS have the capacity to recognize xenobiotics and the resulting inflammatory response may contribute to the reinforcing properties of cocaine.
It is currently unclear if other psychostimulants have direct activity with TLRs. Not all drugs may participate in TLR signaling; for instance, chronic antagonism of TLR4 with (+)-naltrexone during withdrawal does not modify cue-induced drug seeking for methamphetamine (Theberge et al, 2013). These results suggest that methamphetamine may act autonomously from TLR4, or alternatively, these results may be specific to the conditions of the behavioral paradigm (ie, incubation of drug-seeking). It is important to keep in mind that different classes of psychostimulants may have unique or divergent mechanisms for effecting glial cells. These mechanisms could involve direct receptor signaling on glia (eg, through TLRs) or as a consequence of their neurotoxicity on neurons or other cells. In addition, the extent to which glial cells or their immune signaling mechanisms influence behavioral responding for other psychostimulants, such as MDMA or the amphetamine-like synthetic cathinones (known as ‘bath salts’), still await characterization.
A recent report suggests that astrocytes within the NAc may have a direct role in the manifestation of cue-induced cocaine seeking in rats. To manipulate intracellular calcium signaling in a discrete population of astrocytes, the authors utilized viral mediated transfection to express the designer receptor exclusively activated by a designer drug (DREADD) hM3D (a designer Gq-protein coupled receptor) under the control of the GFAP promoter (Scofield et al, 2015). This method allows for pharmacological control of astrocyte intracellular signaling. Electrochemical recordings demonstrated that astroglial Gq-DREADD activation results in a reliable increase of glutamate release in the NAc core, and this manipulation is associated with an inhibition of cue-induced cocaine seeking (Scofield et al, 2015). Subsequent experiments suggested that the protective effect of astroglial stimulation was related to synaptic signaling for metabotropic glutamate receptors mGluR2/3 (Scofield et al, 2015). These data suggest that astrocytes can directly influence drug-seeking behavior for cocaine, and the methodologies employed emphasize the opportunities for glial cell-type-specific manipulations in the context of addiction.
In humans, there is conflicting data regarding the state of microglial activation after continued psychostimulant drug abuse. Minocycline treatment can reduce some of the subjective reinforcing effects of D-amphetamine, but it is difficult to determine if this response is glial mediated or not (Sofuoglu et al, 2011). Using positron emission tomography (PET) and the radioligand [11C](R)-PK11195 to measure microglial activity, the brains of abstinent methamphetamine abusers showed evidence of reactive microgliosis in the midbrain, striatum, thalamus, and the orbitofrontal and insular cortices (Sekine et al, 2008). In general, this measure of activation was negatively correlated with the duration of abstinence (Sekine et al, 2008). In contrast, in a group of abstinent cocaine abusers, a separate quantification of microgliosis via PET imaging revealed no difference in microglial activation between abusers and controls across cortical and subcortical brain regions (Narendran et al, 2014). The inconsistencies in these observations could be due to simple differences in radioligand specificity or pharmacological differences in the psychostimulant abused, but could also be explained by differences in how the experimenters dealt with normalization of nonspecific ligand binding (Narendran et al, 2014). These discrepancies not only highlight some of the challenges associated with translating the preclinical work to human models but also underscore the necessity for more research to interrogate the role of glia and immune signaling in the living human brain.
FUTURE RESEARCH DIRECTIONS AND CLINICAL IMPLICATIONS
Unfortunately, nature seems unaware of our intellectual need for convenience and unity, and very often takes delight in complication and diversity. —Ramón y Cajal (1906).
Neuroscientists have made remarkable progress in their efforts to characterize the neurophysiological consequences of drug exposure and associate these changes with the pathological behavioral phenotypes that manifest as substance abuse and addiction. With some notable exceptions highlighted in this review, the preponderance of the extant literature on the biological mechanisms of addiction has focused on the neurotransmitters, neuronal receptors, electrophysiological properties, intracellular signaling pathways, genetic and epigenetic factors that are specific to neurons and their associated circuits. Given the clear evidence that neuronal and synaptic changes occur in response to the drugs of abuse, the primacy of neurons as the fundamental cells responsible for behavior, and the long-standing assumption that glial cells serve largely supportive roles, this focus on neurons is an entirely logical preoccupation. Indeed, neurons should and will continue to remain at the center of the investigations and theories of addiction. However, we argue that neuroscientists, glial biologists, and addiction researchers should consider a focus on the role that neural–glial interactions have in the development and maintenance of substance abuse. Gaining basic insight into the foundation of psychiatric illness requires a well-validated animal model, which has often been an issue of concern when modeling other neuropsychiatric disorders (eg, depression, anxiety, schizophrenia, attention deficit/hyperactivity disorder, etc.); either the etiology of the disorder is unknown, or the behavioral outcome does not faithfully recapitulate the features of the human disease (Nestler and Hyman, 2010). In contrast, animal models of drug addiction are perhaps the best validated models for psychiatric disease (Fernando and Robbins, 2011): the precipitating factor is known (ie, exposure to drug), and both rodent and non-human primate procedures for drug self-administration can reproduce many of the behavioral features of addiction, including drug-seeking, drug-taking, relapse, and reinstatement following abstinence. Despite the face validity of these models, it is still unclear what accounts for the individual differences in addiction-like behavior, as only a relative minority of individuals who consume addictive substances transition to compulsive, dysregulated drug use (Warner et al, 1995). This heterogeneity in the behavioral outcome following drug exposure is influenced by the complex interactions of numerous genetic and non-genetic risk factors, including chronic stress, previous experience with other classes of drugs, comorbid psychiatric conditions, the type of drug abused, the route of administration, and the frequency of drug exposure. In addition, variations in early-life environmental conditions represent an important area for future research. For instance, rats that receive enriched maternal care show decreased preference for morphine as well as increased expression of anti-inflammatory IL-10 in the NAc, which may be persistently altered due to decreased Il-10 gene methylation in microglia (Schwarz et al, 2011). Given the unique developmental ontogeny of microglia (see Box 2) and their critical role in early brain development, it could be hypothesized that microglial-specific epigenetic modifications of inflammatory signaling could cause long-lasting changes in risk or resilience to opioid reinforcement.
On the basis of the ability of microglia and astrocytes to effect the formation, maturation, and elimination of synapses, one could predict that synaptic remodeling may occur under the conditions of drug abuse and could contribute to the persistent behavioral effects characteristic of the disorder. The mechanisms dictating microglial and astrocytic synapse refinement during normal development are still being refined and thus have yet to be applied under the conditions of acute or repeated drug exposure. Nevertheless, the review has examined evidence from a neuroimmune perspective that different classes of abused drugs can impact glial morphology and function, alter neuroimmune signaling, and influence drug-related behavior. The concept of glial mediated synaptic plasticity may represent a missing link connecting evidence of neuroimmune signaling to the persistent behaviors that result from pathological drug abuse. While acknowledging the interesting results reviewed thus far, it is still possible that neuroimmune signaling or glial mediated synaptic plasticity are not a fundamental component in the pathophysiology of addiction and represent an epiphenomenon of other, more essential processes. In this regard, drug abuse functions as an ideal behavioral model to further interrogate the causal or secondary role of glial cells in the development of a complex neuropsychiatric disorder.
There are a few considerations that are worth discussing. First, there is a need for greater specificity in the empirical characterization of neural–glial interactions in response to experimental manipulation. Despite its widespread use, the term ‘activation’ with respect to glial function does not signify a single type of response. Although the classification has its utility, it is clear that these cells do not exist in a binary state between ‘inactive’ and ‘active.’ For instance, microglia change their shape and function depending on the type and concentration of signals in its local environment, both through direct contact of membrane-bound adhesion molecules, and at a distance through secreted molecules. Historically, the morphology of microglia was used to infer its functional state—whether it was ramified and quiescent, or amoeboid and phagocytic—but morphology alone is often insufficient in infer the functional properties of the cell. In a similar fashion, the responses of microglia to differing insults may not be uniformly pro-inflammatory or anti-inflammatory. Along with increasing precision to measure and perturb genetic- and pathway-specific neural ensembles, there is a growing appreciation that these cellular subpopulations must be carefully and exhaustively characterized if their function is ever to be fully understood. Advancements in RNA sequencing (RNA-seq) should permit a higher fidelity of classifying cell-type-specific transcriptional dynamics during homeostasis and in response to drugs of abuse (Ginhoux et al, 2016). Efforts to develop searchable RNA-seq transcriptome libraries of the healthy mouse cerebral cortex have already highlighted novel cell-specific gene expression and alternative splicing in neurons, astrocytes, microglia, oligodendrocytes, and vascular cells (Zhang et al, 2014). Massively parallel single-cell sequencing should also help in identifying if there is heterogeneous transcriptional regulation in subpopulations of glial cells, which may unravel brain region- and drug-type-specific effects that are currently unexplained (Zeisel et al, 2015). To this end, FACS purification of CD11b+ microglia and GLT1+ astrocytes in the NAc following acute morphine found microglial-specific upregulation of chemokine ligands CCL4 and CCL17, their receptor CCR4, and anti-inflammatory IL-10 (Schwarz et al, 2013). The authors also found a concomitant downregulation of fractalkine ligand CX3CL1 in neurons, suggesting that opioid exposure rapidly changes neuron-glia crosstalk as well as autocrine regulation or glia-to-glia chemotaxis. Therefore, utilizing high-resolution cell sorting and sequencing of neurons and glia may ultimately reveal epigenetic and/or post-transcriptional modifications that may account for the developmental sensitivity and ‘priming’ of microglia by immunological or environmental factors.
As mentioned above, another topic of considerable interest relates to brain region-specific glial responses to acute or repeated drug exposure. The mechanisms that drive regional selectivity in glial activation, despite the penetrance of drugs of abuse throughout CNS parenchyma, are currently unknown. Although speculative, an enticing possibility may be related to the heterogeneity of tissue milieu, in particular variances in neuronal innervation patterns that dictate neurotransmitter release within a given region. Within the NAc, for example, simultaneous triggering of microglial or astrocytic TLR4/MD-2 by opioids that coincides with dopamine receptor binding from transient opioid- or cue-induced elevations in phasic dopamine release may produce a unique microglial phenotype within this region. This example is still a reductionist account of the staggering degree of complex molecular cascades initiated by opioid administration, such as known changes to GABAergic signaling in the VTA (Johnson and North, 1992; Laviolette et al, 2004; Taylor et al, 2016), all of which may have equally important roles in dictating the outcome of glial responses to drugs of abuse.
Beyond regional differences in network connectivity, others have suggested that microglia and astrocytes themselves are a heterogeneous population with specialized responses across the brain, some of which may be dictated by neuron–glial interactions (Bayraktar et al, 2015; Hanisch, 2013). For instance, astrocyte expression of TSP1 and TSP2 is found primarily in rodent cortex early in development (Christopherson et al, 2005), while TSP4 is expressed in astrocytes of the subventricular zone (Benner et al, 2013). Astrocyte-released glypicans 4 and 6 are also found primarily in the hippocampus and cerebellum, respectively (Allen et al, 2012). For microglia, signaling between the neuronal chemokine fractalkine (CX3CL1) and the microglial receptor CX3CR1 represents a critical pathway for bidirectional communication. CX3CL1 is expressed on subsets of neurons in the CNS, particularly within the striatum, hippocampus, and cortical layer II (Kim et al, 2011), although the mechanism regulating this heterogeneous expression is unknown. Disruptions to CX3CR1 can increase neurotoxicity, delay synapse maturation in the hippocampus, and result in social interaction deficits (Cardona et al, 2006; Paolicelli et al, 2011; Zhan et al, 2014). The maintenance and survival of microglia also depends on signaling at colony stimulating factor 1 receptor (CSF1R). The cytokine IL-34, which is primarily produced by neurons in the CNS, was identified as a separate ligand for CSF1R with a unique spatial distribution in the CNS; mice deficient in IL-34 have reduced numbers of microglia compared with wild-type mice, but only localized to brain regions where IL-34 would normally be expressed, including the cerebral cortex, basal ganglia, and hippocampus (Greter et al, 2012; Lin et al, 2008; Wang et al, 2012b). Moreover, microglia are curiously found in higher densities in regions that either produce or receive dopamine (Lawson et al, 1990), suggesting some unknown chemotactic or proliferation signal. Notably, a recent genome-wide transcriptional profile of rodent microglia uncovered remarkable phenotypic diversity across different regions in the healthy adult brain, identifying steady-state differences in immune vigilance and energy metabolism (Grabert et al, 2016). This regional heterogeneity could be programmed early in development and influence the migration and seeding of the embryonic brain, or the differences may emerge after exposure to local differences in the neurochemical environment, such as neurotransmitter innervation and blood–brain barrier permeability. Collectively, these results imply that glia may have regionally localized responses to drugs of abuse—for example, one could speculate that microglia or astrocytes in the NAc could mount an exaggerated pro-inflammatory response to cocaine compared with microglia in the cerebellum. As a result, neuroimmune responses or synaptic remodeling could be restricted within particular neural circuits associated with drug reinforcement.
With this consideration in mind, it is perhaps less perplexing how drug-induced neuroimmune signaling may produce a different physiological outcome compared with an alternative insult; that is, how a similar signaling pathway could be involved in sickness behavior and reinforcement for abused substances. Both LPS and opioids (as an example) may signal through TLR4, but there are differences between these molecules in terms of how and where they interact with TLR4. LPS does not easily diffuse across the blood–brain barrier under normal conditions (Banks and Robinson, 2010), but appears to nevertheless activate TLR4 receptors through diffusible signals originating from TLR4-dependent activity from cells within the circumventricular organs or resident macrophages in the choroid plexus, perivascular space, and endothelium (Chen et al, 2012; Rivest, 2009). In contrast, opioids can easily penetrate CNS tissue and signal directly at neurons and glia, suggesting independent patterns of cell recruitment (Hutchinson and Watkins, 2014). Drug-specific recruitment of multiple innate immune signaling pathways may also explain their unique neuroimmune and behavioral outcomes; opioids, for example, signal at TLR4 but there is also evidence that they may participate in signaling through TLR2 and TLR9 (He et al, 2011; Li et al, 2010b), although there is currently less evidence for these alternative pathways. Moreover, the affinity of the opioids morphine and fentanyl at the TLR4/MD-2 complex is considerably less than LPS in reporter HEK 293 cells (Stevens et al, 2013). In this case, opioid-induced production of cytokines and chemokines may not mimic the broadly neuroinflammatory response to LPS, but rather function in a neuromodulatory role (Jacobsen et al, 2014; Rostène et al, 2007).
Finally, while there is exciting evidence that suppression of the pro-inflammatory glial response to drugs of abuse is successful at reducing drug reward, efficaciously translating these results to the clinic will require a more nuanced approach. Therapeutics targeting TLRs certainly offers a promising avenue for investigation, given their involvement across many classes of drugs. For a discussion on the translational potential of TLR4 antagonism in the context of drug abuse, see Bachtell et al (2015). Alternatively, methods for manipulating anti-inflammatory cytokines such as IL-10 may yield some promise for treating drug-induced inflammation. IL-10 is a powerful suppressor of TLR-induced cytokine production (Curtale et al, 2013). In addition, IL-10 treatment has proven successful at treating animal models of neuropathic pain (Dengler et al, 2014; Kwilasz et al, 2015). However, the outcomes have been mixed for other preclinical models. In differing mouse models of Alzheimer’s disease, viral mediated overexpression of IL-10 worsens amyloid-β plaque deposition, while IL-10 deficiency promotes amyloid-β plaque clearance and improved outcomes on cognitive tasks (Chakrabarty et al, 2015; Guillot-Sestier et al, 2015). In contrast, intracerebroventricular infusions of IL-10 receptor blocking antibodies exacerbates disease onset in the SOD1G93A model of ALS, while targeted intrathecal overexpression of IL-10 in CD11b-expressing microglial cells greatly improves disease progression (Gravel et al, 2016). Although the disease models and methods of IL-10 manipulation vary significantly between these studies, they highlight the need for further studies to clarify how neuroimmune signaling is tuned in the context of disease as well as homeostasis.
CONCLUSION
Microglia and astrocytes have received considerable attention recently due to their contributions to a wide array of homeostatic and non-inflammatory processes, as well as during states of heightened inflammation and injury (Brown and Neher, 2014; Ginhoux and Jung, 2014; Prinz and Priller, 2014; Salter and Beggs, 2014; Walsh et al, 2014). Glial and neuroimmune mechanisms have been extensively characterized in the context of pathological pain (Grace et al, 2014a), while researchers attempting to decode the complex etiology of neurodegenerative disorders such as Alzheimer’s disease have also argued for a more holistic framework, where the interactions and compensatory responses between neurons, glia, and vascular cells all contribute to the progression of the disease (De Strooper and Karran, 2016). Given their role in immune surveillance and debris clearance, it is perhaps unsurprising that microglial activation in particular has been implicated in neurodegenerative diseases beyond Alzheimer’s disease, including multiple sclerosis and Parkinson’s disease (Chung et al, 2015b; Heneka et al, 2015; Hong et al, 2016). As evidence accumulates positioning glia as critical for synapse maturity and elimination, many have speculated that glia and immune signaling are associated with many neuropsychiatric disorders, from stress-related conditions such as depression (Hodes et al, 2015; Miller and Raison, 2015), to neurodevelopmental disorders such as autism and schizophrenia (Gupta et al, 2014; Sekar et al, 2016; Werling et al, 2016). Here, we propose that microglia and astrocytes have an important yet largely unexplored role in the development and maintenance of drug addiction. Advancements in our ability to control the activity state of these glial cells could allow us to further dissect the contribution of neurons and glia in regulating behavior in health and disease. This renewed focus on glia represents a necessary step in the continued elaboration of the deeply complex network of cells involved in motivation and addiction.
ABBREVIATIONS
ACM, astrocyte conditioned medium; ALS, amyotrophic lateral sclerosis; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; BDNF, brain-derived neurotrophic factor; CNS, central nervous system; CPP, conditioned place preference; CRF, corticotropin-releasing factor; CTAP, d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2; DAMGO, [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin; DREADD, designer receptor exclusively activated by a designer drug; FACS, fluorescence-activated cell sorting; GABA, γ-aminobutyric acid; GFAP, glial fibrillary acidic protein; GFP, green fluorescent protein; GLAST, glutamate-aspartate transporter; GLT-1, glutamate transporter 1; Glu, glutamate; l-DOPA, l-3,4-dihydroxyphenylalanine; MDMA, 3,4-methylenedioxymethamphetamine; NMDA, N-methyl-d-aspartate receptor; NPY, neuropeptide Y; PET, positron emission tomography; siRNA, small interfering RNA; SPARC, secreted protein acidic and rich in cysteine; TSP, thrombospondin; TTX, tetrodotoxin. See Table 1 for a complete list of abbreviations related to neuroimmune signaling.
FUNDING AND DISCLOSURE
The authors declare no conflict of interest.
References
Ahlers KE, Karacay B, Fuller L, Bonthius DJ, Dailey ME (2015). Transient activation of microglia following acute alcohol exposure in developing mouse neocortex is primarily driven by BAX-dependent neurodegeneration. Glia 63: 1694–1713.
Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FMV (2007). Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 10: 1538–1543.
Alfonso-Loeches S, Pascual-Lucas M, Blanco AM, Sanchez-Vera I, Guerri C (2010). Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. J Neurosci 30: 8285–8295.
Allen NJ, Bennett ML, Foo LC, Wang GX, Chakraborty C, Smith SJ et al (2012). Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 486: 410–414.
Alliot F, Godin I, Pessac B (1999). Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res 117: 145–152.
Araque A, Carmignoto G, Haydon PG, Oliet SH, Robitaille R, Volterra A (2014). Gliotransmitters travel in time and space. Neuron 81: 728–739.
Armstrong V, Reichel CM, Doti JF, Crawford CA, McDougall SA (2004). Repeated amphetamine treatment causes a persistent elevation of glial fibrillary acidic protein in the caudate-putamen. Eur J Pharmacol 488: 111–115.
Bachtell R, Hutchinson MR, Wang X, Rice KC, Maier SF, Watkins LR (2015). Targeting the toll of drug abuse: the translational potential of Toll-like receptor 4. CNS Neurol Disord Drug Targets 14: 692–699.
Bajo M, Herman MA, Varodayan FP, Oleata CS, Madamba SG, Harris RA et al (2015). Role of the IL-1 receptor antagonist in ethanol-induced regulation of GABAergic transmission in the central amygdala. Brain Behav Immun 45: 189–197.
Bajo M, Madamba SG, Roberto M, Blednov YA, Sagi VN, Roberts E et al (2014). Innate immune factors modulate ethanol interaction with GABAergic transmission in mouse central amygdala. Brain Behav Immun 40: 191–202.
Banker GA (1980). Trophic interactions between astroglial cells and hippocampal neurons in culture. Science 209: 809–810.
Banks WA, Robinson SM (2010). Minimal penetration of lipopolysaccharide across the murine blood-brain barrier. Brain Behav Immun 24: 102–109.
Bayraktar OA, Fuentealba LC, Alvarez-Buylla A, Rowitch DH (2015). Astrocyte development and heterogeneity. Cold Spring Harb Perspect Biol 7: a020362.
Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M et al (2002). Control of synaptic strength by glial TNFalpha. Science 295: 2282–2285.
Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA et al (2006). Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 103: 16021–16026.
Beier KT, Steinberg EE, DeLoach KE, Xie S, Miyamichi K, Schwarz L et al (2015). Circuit architecture of VTA dopamine neurons revealed by systematic input-output mapping. Cell 162: 622–634.
Benner EJ, Luciano D, Jo R, Abdi K, Paez-Gonzalez P, Sheng H et al (2013). Protective astrogenesis from the SVZ niche after injury is controlled by Notch modulator Thbs4. Nature 497: 369–373.
Bialas AR, Stevens B (2013). TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci 16: 1773–1782.
Biber K, Neumann H, Inoue K, Boddeke HWGM (2007). Neuronal 'On' and 'Off' signals control microglia. Trends Neurosci 30: 596–602.
Bilbo SD, Schwarz JM (2012). The immune system and developmental programming of brain and behavior. Front Neuroendocrinol 33: 267–286.
Bilbo SD, Smith SH, Schwarz JM (2012). A lifespan approach to neuroinflammatory and cognitive disorders: a critical role for glia. J Neuroimmune Pharmacol 7: 24–41.
Björklund A, Dunnett SB (2007). Dopamine neuron systems in the brain: an update. Trends Neurosci 30: 194–202.
Blednov YA, Ponomarev I, Geil C, Bergeson S, Koob GF, Harris RA (2012). Neuroimmune regulation of alcohol consumption: behavioral validation of genes obtained from genomic studies. Addict Biol 17: 108–120.
Blinzinger K, Kreutzberg G (1968). Displacement of synaptic terminals from regenerating motoneurons by microglial cells. Z Zellforsch Mikrosk Anat 85: 145–157.
Block ML, Zecca L, Hong JS (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8: 57–69.
Boschen KE, Ruggiero MJ, Klintsova AY (2016). Neonatal binge alcohol exposure increases microglial activation in the developing rat hippocampus. Neuroscience 324: 355–366.
Boyadjieva NI, Sarkar DK (2010). Role of microglia in ethanol's apoptotic action on hypothalamic neuronal cells in primary cultures. Alcohol Clin Exp Res 34: 1835–1842.
Boyadjieva NI, Sarkar DK (2013). Microglia play a role in ethanol-induced oxidative stress and apoptosis in developing hypothalamic neurons. Alcohol Clin Exp Res 37: 252–262.
Brodie MS, Pesold C, Appel SB (1999). Ethanol directly excites dopaminergic ventral tegmental area reward neurons. Alcohol Clin Exp Res 23: 1848–1852.
Brown GC, Neher JJ (2014). Microglial phagocytosis of live neurons. Nat Rev Neurosci 15: 209–216.
Bushong EA, Martone ME, Jones YZ, Ellisman MH (2002). Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci 22: 183–192.
Calcaterra S, Glanz J, Binswanger IA (2013). National trends in pharmaceutical opioid related overdose deaths compared to other substance related overdose deaths: 1999-2009. Drug Alcohol Depend 131: 263–270.
Cannon CM, Palmiter RD (2003). Reward without dopamine. J Neurosci 23: 10827–10831.
Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM et al (2006). Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci 9: 917–924.
Case A, Deaton A (2015). Rising morbidity and mortality in midlife among white non-Hispanic Americans in the 21st century. Proc Natl Acad Sci USA 112: 15078–15083.
Cearley CN, Blindheim K, Sorg BA, Krueger JM, Churchill L (2011). Acute cocaine increases interleukin-1beta mRNA and immunoreactive cells in the cortex and nucleus accumbens. Neurochem Res 36: 686–692.
Chakrabarty P, Li A, Ceballos-Diaz C, Eddy JA, Funk CC, Moore B et al (2015). IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron 85: 519–533.
Chao CC, Gekker G, Hu S, Sheng WS, Shark KB, Bu DF et al (1996). Kappa opioid receptors in human microglia downregulate human immunodeficiency virus 1 expression. Proc Natl Acad Sci USA 93: 8051–8056.
Chao CC, Hu S, Shark KB, Sheng WS, Gekker G, Peterson PK (1997). Activation of mu opioid receptors inhibits microglial cell chemotaxis. J Pharmacol Exp Ther 281: 998–1004.
Chen Z, Jalabi W, Shpargel KB, Farabaugh KT, Dutta R, Yin X et al (2012). Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J Neurosci 32: 11706–11715.
Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A et al (2005). Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120: 421–433.
Chung WS, Allen NJ, Eroglu C (2015a). Astrocytes control synapse formation, function, and elimination. Cold Spring Harb Perspect Biol 7: a020370.
Chung WS, Welsh CA, Barres BA, Stevens B (2015b). Do glia drive synaptic and cognitive impairment in disease? Nat Neurosci 18: 1539–1545.
Clarke LE, Barres BA (2013). Emerging roles of astrocytes in neural circuit development. Nat Rev Neurosci 14: 311–321.
Coller JK, Hutchinson MR (2012). Implications of central immune signaling caused by drugs of abuse: mechanisms, mediators and new therapeutic approaches for prediction and treatment of drug dependence. Pharmacol Ther 134: 219–245.
Colton CA, Snell-Callanan J, Chernyshev ON (1998). Ethanol induced changes in superoxide anion and nitric oxide in cultured microglia. Alcohol Clin Exp Res 22: 710–716.
Corrigan F, Wu Y, Tuke J, Coller JK, Rice KC, Diener KR et al (2015). Alcohol-induced sedation and synergistic interactions between alcohol and morphine: a key mechanistic role for Toll-like receptors and MyD88-dependent signaling. Brain Behav Immun 45: 245–252.
Corty MM, Freeman MR (2013). Architects in neural circuit design: glia control neuron numbers and connectivity. J Cell Biol 203: 395–405.
Costa BM, Yao H, Yang L, Buch S (2013). Role of endoplasmic reticulum (ER) stress in cocaine-induced microglial cell death. J Neuroimmune Pharmacol 8: 705–714.
Crews FT, Sarkar DK, Qin L, Zou J, Boyadjieva N, Vetreno RP (2015). Neuroimmune function and the consequences of alcohol exposure. Alcohol Res 37: 331–351.
Crews FT, Vetreno RP (2016). Mechanisms of neuroimmune gene induction in alcoholism. Psychopharmacology (Berl) 233: 1543–1557.
Crow TJ (1972). Catecholamine-containing neurones and electrical self-stimulation. 1. A review of some data. Psychol Med 2: 414–421.
Curtale G, Mirolo M, Renzi TA, Rossato M, Bazzoni F, Locati M (2013). Negative regulation of Toll-like receptor 4 signaling by IL-10-dependent microRNA-146b. Proc Natl Acad Sci USA 110: 11499–11504.
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S et al (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8: 752–758.
De Strooper B, Karran E (2016). The cellular phase of Alzheimer's disease. Cell 164: 603–615.
de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE (1991). Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med 174: 1209–1220.
Degenhardt L, Whiteford HA, Ferrari AJ, Baxter AJ, Charlson FJ, Hall WD et al (2013). Global burden of disease attributable to illicit drug use and dependence: findings from the Global Burden of Disease Study 2010. Lancet 382: 1564–1574.
del Río-Hortega P (1932). Microglia. In: Penfield W (ed) Cytology and Cellular Pathology of the Nervous System. Paul B. Hoeber: New York, NY, Vol 2 pp 482–534.
Dengler EC, Alberti LA, Bowman BN, Kerwin AA, Wilkerson JL, Moezzi DR et al (2014). Improvement of spinal non-viral IL-10 gene delivery by D-mannose as a transgene adjuvant to control chronic neuropathic pain. J Neuroinflammation 11: 92.
Deroche-Gamonet V, Belin D, Piazza PV (2004). Evidence for addiction-like behavior in the rat. Science 305: 1014–1017.
Di Chiara G, Imperato A (1988). Opposite effects of mu and kappa opiate agonists on dopamine release in the nucleus accumbens and in the dorsal caudate of freely moving rats. J Pharmacol Exp Ther 244: 1067–1080.
Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD (2007). Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci 27: 11354–11365.
Eroglu C, Allen NJ, Susman MW, O'Rourke NA, Park CY, Özkan E et al (2009). Gabapentin receptor α2δ-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139: 380–392.
Eroglu C, Barres BA (2010). Regulation of synaptic connectivity by glia. Nature 468: 223–231.
Fernando ABP, Robbins TW (2011). Animal models of neuropsychiatric disorders. Annu Rev Clin Psychol 7: 39–61.
Fields RD, Woo DH, Basser PJ (2015). Glial regulation of the neuronal connectome through local and long-distant communication. Neuron 86: 374–386.
Franke H (1995). Influence of chronic alcohol treatment on the GFAP-immunoreactivity in astrocytes of the hippocampus in rats. Acta Histochem 97: 263–271.
Gay NJ, Symmons MF, Gangloff M, Bryant CE (2014). Assembly and localization of Toll-like receptor signalling complexes. Nat Rev Immunol 14: 546–558.
Geissmann F, Gordon S, Hume DA, Mowat AM, Randolph GJ (2010). Unravelling mononuclear phagocyte heterogeneity. Nat Rev Immunol 10: 453–460.
Gibson LC, Hastings SF, McPhee I, Clayton RA, Darroch CE, Mackenzie A et al (2006). The inhibitory profile of Ibudilast against the human phosphodiesterase enzyme family. Eur J Pharmacol 538: 39–42.
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S et al (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330: 841–845.
Ginhoux F, Jung S (2014). Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol 14: 392–404.
Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK (2016). New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol 17: 34–40.
Gonçalves J, Martins T, Ferreira R, Milhazes N, Borges F, Ribeiro CF et al (2008). Methamphetamine-induced early increase of IL-6 and TNF-α mRNA expression in the mouse brain. Ann N Y Acad Sci 1139: 103–111.
González JC, Egea J, Del Carmen Godino M, Fernandez-Gomez FJ, Sánchez-Prieto J, Gandía L et al (2007). Neuroprotectant minocycline depresses glutamatergic neurotransmission and Ca(2+) signalling in hippocampal neurons. Eur J Neurosci 26: 2481–2495.
Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP et al (2016). Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci 19: 504–516.
Grace PM, Hutchinson MR, Maier SF, Watkins LR (2014a). Pathological pain and the neuroimmune interface. Nat Rev Immunol 14: 217–231.
Grace PM, Ramos KM, Rodgers KM, Wang X, Hutchinson MR, Lewis MT et al (2014b). Activation of adult rat CNS endothelial cells by opioid-induced toll-like receptor 4 (TLR4) signaling induces proinflammatory, biochemical, morphological, and behavioral sequelae. Neuroscience 280: 299–317.
Graeber MB (2010). Changing face of microglia. Science 330: 783–788.
Gravel M, Beland LC, Soucy G, Abdelhamid E, Rahimian R, Gravel C et al (2016). IL-10 controls early microglial phenotypes and disease onset in ALS caused by misfolded superoxide dismutase 1. J Neurosci 36: 1031–1048.
Greter M, Lelios I, Pelczar P, Hoeffel G, Price J, Leboeuf M et al (2012). Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity 37: 1050–1060.
Guerri C, Saez R, Sancho-Tello M, Martin de Aquilera E, Renau-Piqueras J (1990). Ethanol alters astrocyte development: a study of critical periods using primary cultures. Neurochem Res 15: 559–565.
Guha P, Harraz MM, Snyder SH (2016). Cocaine elicits autophagic cytotoxicity via a nitric oxide-GAPDH signaling cascade. Proc Natl Acad Sci USA 113: 1417–1422.
Guillot-Sestier MV, Doty KR, Gate D, Rodriguez J Jr, Leung BP, Rezai-Zadeh K et al (2015). Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron 85: 534–548.
Gupta S, Ellis SE, Ashar FN, Moes A, Bader JS, Zhan J et al (2014). Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat Commun 5: 5748.
Halassa MM, Fellin T, Takano H, Dong JH, Haydon PG (2007). Synaptic islands defined by the territory of a single astrocyte. J Neurosci 27: 6473–6477.
Hanisch UK (2013). Functional diversity of microglia—how heterogeneous are they to begin with? Front Cell Neurosci 7: 65.
Hanisch UK, Kettenmann H (2007). Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 10: 1387–1394.
Haydon PG, Blendy J, Moss SJ, Rob Jackson F (2009). Astrocytic control of synaptic transmission and plasticity: a target for drugs of abuse? Neuropharmacology 56 Suppl 1: 83–90.
He J, Crews FT (2008). Increased MCP-1 and microglia in various regions of the human alcoholic brain. Exp Neurol 210: 349–358.
He L, Li H, Chen L, Miao J, Jiang Y, Zhang Y et al (2011). Toll-like receptor 9 is required for opioid-induced microglia apoptosis. PLoS One 6: e18190.
Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL et al (2015). Neuroinflammation in Alzheimer's disease. Lancet Neurol 14: 388–405.
Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK et al (2013). The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 16: 1896–1905.
Hnasko TS, Chuhma N, Zhang H, Goh GY, Sulzer D, Palmiter RD et al (2010). Vesicular glutamate transport promotes dopamine storage and glutamate corelease in vivo. Neuron 65: 643–656.
Hnasko TS, Sotak BN, Palmiter RD (2005). Morphine reward in dopamine-deficient mice. Nature 438: 854–857.
Hodes GE, Kana V, Menard C, Merad M, Russo SJ (2015). Neuroimmune mechanisms of depression. Nat Neurosci 18: 1386–1393.
Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S et al (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352: 712–716.
Horvath RJ, DeLeo JA (2009). Morphine enhances microglial migration through modulation of P2X4 receptor signaling. J Neurosci 29: 998–1005.
Hutchinson MR, Lewis SS, Coats BD, Skyba DA, Crysdale NY, Berkelhammer DL et al (2009). Reduction of opioid withdrawal and potentiation of acute opioid analgesia by systemic AV411 (ibudilast). Brain Behav Immun 23: 240–250.
Hutchinson MR, Northcutt AL, Chao LW, Kearney JJ, Zhang Y, Berkelhammer DL et al (2008). Minocycline suppresses morphine-induced respiratory depression, suppresses morphine-induced reward, and enhances systemic morphine-induced analgesia. Brain Behav Immun 22: 1248–1256.
Hutchinson MR, Northcutt AL, Hiranita T, Wang X, Lewis SS, Thomas J et al (2012). Opioid activation of toll-like receptor 4 contributes to drug reinforcement. J Neurosci 32: 11187–11200.
Hutchinson MR, Shavit Y, Grace PM, Rice KC, Maier SF, Watkins LR (2011). Exploring the neuroimmunopharmacology of opioids: an integrative review of mechanisms of central immune signaling and their implications for opioid analgesia. Pharmacol Rev 63: 772–810.
Hutchinson MR, Watkins LR (2014). Why is neuroimmunopharmacology crucial for the future of addiction research? Neuropharmacology 76 Pt B: 218–227.
Hutchinson MR, Zhang Y, Shridhar M, Evans JH, Buchanan MM, Zhao TX et al (2010). Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav Immun 24: 83–95.
Hyman SE, Malenka RC, Nestler EJ (2006). Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci 29: 565–598.
Ivashkiv LB (2008). A signal-switch hypothesis for cross-regulation of cytokine and TLR signalling pathways. Nat Rev Immunol 8: 816–822.
Jack CS, Arbour N, Manusow J, Montgrain V, Blain M, McCrea E et al (2005). TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol 175: 4320–4330.
Jacobsen JH, Watkins LR, Hutchinson MR (2014). Discovery of a novel site of opioid action at the innate immune pattern-recognition receptor TLR4 and its role in addiction. Int Rev Neurobiol 118: 129–163.
Johnson SW, North RA (1992). Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci 12: 483–488.
Jones ME, Lebonville CL, Barrus D, Lysle DT (2015). The role of brain interleukin-1 in stress-enhanced fear learning. Neuropsychopharmacology 40: 1289–1296.
June HL, Liu J, Warnock KT, Bell KA, Balan I, Bollino D et al (2015). CRF-amplified neuronal TLR4/MCP-1 signaling regulates alcohol self-administration. Neuropsychopharmacology 40: 1549–1559.
Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A et al (2000). Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 20: 4106–4114.
Juni A, Klein G, Pintar JE, Kest B (2007). Nociception increases during opioid infusion in opioid receptor triple knock-out mice. Neuroscience 147: 439–444.
Kawai T, Akira S (2006). TLR signaling. Cell Death Differ 13: 816–825.
Kawai T, Takeuchi O, Fujita T, Inoue J, Muhlradt PF, Sato S et al (2001). Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol 167: 5887–5894.
Kettenmann H, Kirchhoff F, Verkhratsky A (2013). Microglia: new roles for the synaptic stripper. Neuron 77: 10–18.
Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG et al (2013). Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci 16: 273–280.
Kierdorf K, Prinz M (2013). Factors regulating microglia activation. Front Cell Neurosci 7: 44.
Kilic U, Kilic E, Matter CM, Bassetti CL, Hermann DM (2008). TLR-4 deficiency protects against focal cerebral ischemia and axotomy-induced neurodegeneration. Neurobiol Dis 31: 33–40.
Kim KW, Vallon-Eberhard A, Zigmond E, Farache J, Shezen E, Shakhar G et al (2011). In vivo structure/function and expression analysis of the CX3C chemokine fractalkine. Blood 118: e156–e167.
Kimpel MW, Strother WN, McClintick JN, Carr LG, Liang T, Edenberg HJ et al (2007). Functional gene expression differences between inbred alcohol-preferring and -non-preferring rats in five brain regions. Alcohol 41: 95–132.
Klein I, Cornejo JC, Polakos NK, John B, Wuensch SA, Topham DJ et al (2007). Kupffer cell heterogeneity: functional properties of bone marrow derived and sessile hepatic macrophages. Blood 110: 4077–4085.
Koob GF, Volkow ND (2010). Neurocircuitry of addiction. Neuropsychopharmacology 35: 217–238.
Korbo L (1999). Glial cell loss in the hippocampus of alcoholics. Alcohol Clin Exp Res 23: 164–168.
Kreutzberg GW (1996). Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19: 312–318.
Kucukdereli H, Allen NJ, Lee AT, Feng A, Ozlu MI, Conatser LM et al (2011). Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc Natl Acad Sci USA 108: E440–E449.
Kwilasz AJ, Grace PM, Serbedzija P, Maier SF, Watkins LR (2015). The therapeutic potential of interleukin-10 in neuroimmune diseases. Neuropharmacology 96: 55–69.
Lammel S, Lim BK, Malenka RC (2014). Reward and aversion in a heterogeneous midbrain dopamine system. Neuropharmacology 76 Pt B: 351–359.
Lammel S, Lim BK, Ran C, Huang KW, Betley MJ, Tye KM et al (2012). Input-specific control of reward and aversion in the ventral tegmental area. Nature 491: 212–217.
Lau LT, Yu AC (2001). Astrocytes produce and release interleukin-1, interleukin-6, tumor necrosis factor alpha and interferon-gamma following traumatic and metabolic injury. J Neurotrauma 18: 351–359.
Laviolette SR, Gallegos RA, Henriksen SJ, van der Kooy D (2004). Opiate state controls bi-directional reward signaling via GABAA receptors in the ventral tegmental area. Nat Neurosci 7: 160–169.
Lawson LJ, Perry VH, Dri P, Gordon S (1990). Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39: 151–170.
Lee MS, Kim YJ (2007). Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu Rev Biochem 76: 447–480.
Lewis SS, Hutchinson MR, Rezvani N, Loram LC, Zhang Y, Maier SF et al (2010). Evidence that intrathecal morphine-3-glucuronide may cause pain enhancement via toll-like receptor 4/MD-2 and interleukin-1beta. Neuroscience 165: 569–583.
Li X, Jiang S, Tapping RI (2010a). Toll-like receptor signaling in cell proliferation and survival. Cytokine 49: 1–9.
Li X, Newbern JM, Wu Y, Morgan-Smith M, Zhong J, Charron J et al (2012a). MEK is a key regulator of gliogenesis in the developing brain. Neuron 75: 1035–1050.
Li Y, Du XF, Liu CS, Wen ZL, Du JL (2012b). Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Dev Cell 23: 1189–1202.
Li Y, Li H, Zhang Y, Sun X, Hanley GA, LeSage G et al (2010b). Toll-like receptor 2 is required for opioids-induced neuronal apoptosis. Biochem Biophys Res Commun 391: 426–430.
Liao K, Guo M, Niu F, Yang L, Callen SE, Buch S (2016). Cocaine-mediated induction of microglial activation involves the ER stress-TLR2 axis. J Neuroinflammation 13: 33.
Lin H, Lee E, Hestir K, Leo C, Huang M, Bosch E et al (2008). Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science 320: 807–811.
Little KY, Ramssen E, Welchko R, Volberg V, Roland CJ, Cassin B (2009). Decreased brain dopamine cell numbers in human cocaine users. Psychiatry Res 168: 173–180.
Liu J, Lewohl JM, Harris RA, Iyer VR, Dodd PR, Randall PK et al (2006). Patterns of gene expression in the frontal cortex discriminate alcoholic from nonalcoholic individuals. Neuropsychopharmacology 31: 1574–1582.
Liu J, Yang AR, Kelly T, Puche A, Esoga C, June HL Jr et al (2011). Binge alcohol drinking is associated with GABAA α2-regulated Toll-like receptor 4 (TLR4) expression in the central amygdala. Proc Natl Acad Sci USA 108: 4465–4470.
Lucin KM, Wyss-Coray T (2009). Immune activation in brain aging and neurodegeneration: too much or too little? Neuron 64: 110–122.
Luo L, O'Leary DDM (2005). Axon retraction and degeneration in development and disease. Annu Rev Neurosci 28: 127–156.
Lüscher C, Malenka RC (2011). Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron 69: 650–663.
Marcos M, Pastor I, González-Sarmiento R, Laso FJ (2008). Interleukin-10 gene polymorphism is associated with alcoholism but not with alcoholic liver disease. Alcohol Alcohol 43: 523–528.
Marshall SA, Casachahua JD, Rinker JA, Blose AK, Lysle DT, Thiele TE (2016). IL-1 receptor signaling in the basolateral amygdala modulates binge-like ethanol consumption in male C57BL/6J mice. Brain Behav Immun 51: 258–267.
Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I et al (1996). Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature 383: 819–823.
McClain JA, Morris SA, Deeny MA, Marshall SA, Hayes DM, Kiser ZM et al (2011). Adolescent binge alcohol exposure induces long-lasting partial activation of microglia. Brain Behav Immun 25 Suppl 1: S120–S128.
McKercher SR, Torbett BE, Anderson KL, Henkel GW, Vestal DJ, Baribault H et al (1996). Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J 15: 5647–5658.
Medvinsky A, Dzierzak E (1996). Definitive hematopoiesis is autonomously initiated by the AGM region. Cell 86: 897–906.
Merad M, Manz MG, Karsunky H, Wagers A, Peters W, Charo I et al (2002). Langerhans cells renew in the skin throughout life under steady-state conditions. Nat Immunol 3: 1135–1141.
Meyer-Franke A, Kaplan MR, Pfrieger FW, Barres BA (1995). Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron 15: 805–819.
Miguel-Hidalgo JJ (2009). The role of glial cells in drug abuse. Curr Drug Abuse Rev 2: 72–82.
Miguel-Hidalgo JJ, Wei J, Andrew M, Overholser JC, Jurjus G, Stockmeier CA et al (2002). Glia pathology in the prefrontal cortex in alcohol dependence with and without depressive symptoms. Biol Psychiatry 52: 1121–1133.
Miller AH, Raison CL (2015). The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol 16: 22–34.
Mitchell JM, O'Neil JP, Janabi M, Marks SM, Jagust WJ, Fields HL (2012). Alcohol consumption induces endogenous opioid release in the human orbitofrontal cortex and nucleus accumbens. Sci Transl Med 4: 116ra116.
Mulligan MK, Ponomarev I, Hitzemann RJ, Belknap JK, Tabakoff B, Harris RA et al (2006). Toward understanding the genetics of alcohol drinking through transcriptome meta-analysis. Proc Natl Acad Sci USA 103: 6368–6373.
Narendran R, Lopresti BJ, Mason NS, Deuitch L, Paris J, Himes ML et al (2014). Cocaine abuse in humans is not associated with increased microglial activation: an 18-kDa translocator protein positron emission tomography imaging study with [11C]PBR28. J Neurosci 34: 9945–9950.
Nestler EJ, Hyman SE (2010). Animal models of neuropsychiatric disorders. Nat Neurosci 13: 1161–1169.
Nimmerjahn A, Kirchhoff F, Helmchen F (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308: 1314–1318.
Noble M, Fok-Seang J, Cohen J (1984). Glia are a unique substrate for the in vitro growth of central nervous system neurons. J Neurosci 4: 1892–1903.
North TE, de Bruijn MF, Stacy T, Talebian L, Lind E, Robin C et al (2002). Runx1 expression marks long-term repopulating hematopoietic stem cells in the midgestation mouse embryo. Immunity 16: 661–672.
Northcutt AL, Hutchinson MR, Wang X, Baratta MV, Hiranita T, Cochran TA et al (2015). DAT isn't all that: cocaine reward and reinforcement require Toll-like receptor 4 signaling. Mol Psychiatry 20: 1525–1537.
O'Neill LA, Golenbock D, Bowie AG (2013). The history of Toll-like receptors—redefining innate immunity. Nat Rev Immunol 13: 453–460.
Oades RD, Halliday GM (1987). Ventral tegmental (A10) system: neurobiology. 1. Anatomy and connectivity. Brain Res 434: 117–165.
Olds J (1958). Self-stimulation of the brain; its use to study local effects of hunger, sex, and drugs. Science 127: 315–324.
Olds J, Milner P (1954). Positive reinforcement produced by electrical stimulation of septal area and other regions of rat brain. J Comp Physiol Psychol 47: 419–427.
Orkin SH, Zon LI (2008). Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132: 631–644.
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P et al (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333: 1456–1458.
Pastor IJ, Laso FJ, Romero A, González-Sarmiento R (2005). Interleukin-1 gene cluster polymorphisms and alcoholism in Spanish men. Alcohol Alcohol 40: 181–186.
Peri F, Nüsslein-Volhard C (2008). Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell 133: 916–927.
Perry VH, Hume DA, Gordon S (1985). Immunohistochemical localization of macrophages and microglia in the adult and developing mouse brain. Neuroscience 15: 313–326.
Pfrieger FW, Barres BA (1997). Synaptic efficacy enhanced by glial cells in vitro. Science 277: 1684–1687.
Phillips PEM, Stuber GD, Heien MLAV, Wightman RM, Carelli RM (2003). Subsecond dopamine release promotes cocaine seeking. Nature 422: 614–618.
Piccinini AM, Midwood KS (2010). DAMPening inflammation by modulating TLR signalling. Mediators Inflamm 2010.
Pleil KE, Rinker JA, Lowery-Gionta EG, Mazzone CM, McCall NM, Kendra AM et al (2015). NPY signaling inhibits extended amygdala CRF neurons to suppress binge alcohol drinking. Nat Neurosci 18: 545–552.
Prinz M, Priller J (2014). Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci 15: 300–312.
Qin L, Crews FT (2012). NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. J Neuroinflammation 9: 5.
Ramón y, Cajal S (1906). The structure and connexions of neurons. Nobel Lectures, Physiology or Medicine 1901-1921. Elsevier: Amsterdam. pp 220–253.
Ransohoff RM, Cardona AE (2010). The myeloid cells of the central nervous system parenchyma. Nature 468: 253–262.
Ransohoff RM, Perry VH (2009). Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol 27: 119–145.
Renau-Piqueras J, Zaragoza R, De Paz P, Baguena-Cervellera R, Megias L, Guerri C (1989). Effects of prolonged ethanol exposure on the glial fibrillary acidic protein-containing intermediate filaments of astrocytes in primary culture: a quantitative immunofluorescence and immunogold electron microscopic study. J Histochem Cytochem 37: 229–240.
Risher ML, Fleming RL, Risher WC, Miller KM, Klein RC, Wills T et al (2015a). Adolescent intermittent alcohol exposure: persistence of structural and functional hippocampal abnormalities into adulthood. Alcohol Clin Exp Res 39: 989–997.
Risher ML, Sexton HG, Risher WC, Wilson WA, Fleming RL, Madison RD et al (2015b). Adolescent intermittent alcohol exposure: dysregulation of thrombospondins and synapse formation are associated with decreased neuronal density in the adult hippocampus. Alcohol Clin Exp Res 39: 2403–2413.
Ritz MC, Cone EJ, Kuhar MJ (1990). Cocaine inhibition of ligand binding at dopamine, norepinephrine and serotonin transporters: a structure-activity study. Life Sci 46: 635–645.
Rivest S (2009). Regulation of innate immune responses in the brain. Nat Rev Immunol 9: 429–439.
Robel S, Sontheimer H (2016). Glia as drivers of abnormal neuronal activity. Nat Neurosci 19: 28–33.
Roberto M, Cruz MT, Gilpin NW, Sabino V, Schweitzer P, Bajo M et al (2010). Corticotropin releasing factor-induced amygdala gamma-aminobutyric acid release plays a key role in alcohol dependence. Biol Psychiatry 67: 831–839.
Rocha BA, Fumagalli F, Gainetdinov RR, Jones SR, Ator R, Giros B et al (1998). Cocaine self-administration in dopamine-transporter knockout mice. Nat Neurosci 1: 132–137.
Rolls A, Shechter R, London A, Ziv Y, Ronen A, Levy R et al (2007). Toll-like receptors modulate adult hippocampal neurogenesis. Nat Cell Biol 9: 1081–1088.
Root DH, Mejias-Aponte CA, Zhang S, Wang HL, Hoffman AF, Lupica CR et al (2014). Single rodent mesohabenular axons release glutamate and GABA. Nat Neurosci 17: 1543–1551.
Rosenberg PA, Aizenman E (1989). Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neurosci Lett 103: 162–168.
Rostène W, Kitabgi P, Parsadaniantz SM (2007). Chemokines: a new class of neuromodulator? Nat Rev Neurosci 8: 895–903.
Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW et al (1996). Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16: 675–686.
Saba LM, Flink SC, Vanderlinden LA, Israel Y, Tampier L, Colombo G et al (2015). The sequenced rat brain transcriptome—its use in identifying networks predisposing alcohol consumption. FEBS J 282: 3556–3578.
Saijo K, Glass CK (2011). Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol 11: 775–787.
Salamone JD, Correa M (2012). The mysterious motivational functions of mesolimbic dopamine. Neuron 76: 470–485.
Salter MW, Beggs S (2014). Sublime microglia: expanding roles for the guardians of the CNS. Cell 158: 15–24.
Samokhvalov IM, Samokhvalova NI, Nishikawa S (2007). Cell tracing shows the contribution of the yolk sac to adult haematopoiesis. Nature 446: 1056–1061.
Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R et al (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74: 691–705.
Schafer DP, Lehrman EK, Stevens B (2013). The ‘quad-partite’ synapse: microglia-synapse interactions in the developing and mature CNS. Glia 61: 24–36.
Schafer DP, Stevens B (2013). Phagocytic glial cells: sculpting synaptic circuits in the developing nervous system. Curr Opin Neurobiol 23: 1034–1040.
Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K et al (2012). A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336: 86–90.
Schwarz JM, Hutchinson MR, Bilbo SD (2011). Early-life experience decreases drug-induced reinstatement of morphine CPP in adulthood via microglial-specific epigenetic programming of anti-inflammatory IL-10 expression. J Neurosci 31: 17835–17847.
Schwarz JM, Smith SH, Bilbo SD (2013). FACS analysis of neuronal-glial interactions in the nucleus accumbens following morphine administration. Psychopharmacology (Berl) 230: 525–535.
Scofield MD, Boger HA, Smith RJ, Li H, Haydon PG, Kalivas PW (2015). Gq-DREADD selectively initiates glial glutamate release and inhibits cue-induced cocaine seeking. Biol Psychiatry 78: 441–451.
Scofield MD, Li H, Siemsen BM, Healey KL, Tran PK, Woronoff N et al (2016). Cocaine self-administration and extinction leads to reduced glial fibrillary acidic protein expression and morphometric features of astrocytes in the nucleus accumbens core. Biol Psychiatry 80: 207–215.
Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N et al (2016). Schizophrenia risk from complex variation of complement component 4. Nature 530: 177–183.
Sekine Y, Ouchi Y, Sugihara G, Takei N, Yoshikawa E, Nakamura K et al (2008). Methamphetamine causes microglial activation in the brains of human abusers. J Neurosci 28: 5756–5761.
Shnitko TA, Spear LP, Robinson DL (2016). Adolescent binge-like alcohol alters sensitivity to acute alcohol effects on dopamine release in the nucleus accumbens of adult rats. Psychopharmacology (Berl) 233: 361–371.
Shoaib M, Spanagel R, Stohr T, Shippenberg TS (1995). Strain differences in the rewarding and dopamine-releasing effects of morphine in rats. Psychopharmacology (Berl) 117: 240–247.
Sofuoglu M, Mooney M, Kosten T, Waters A, Hashimoto K (2011). Minocycline attenuates subjective rewarding effects of dextroamphetamine in humans. Psychopharmacology (Berl) 213: 61–68.
Stellwagen D, Malenka RC (2006). Synaptic scaling mediated by glial TNF-alpha. Nature 440: 1054–1059.
Stence N, Waite M, Dailey ME (2001). Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia 33: 256–266.
Sternson SM (2013). Hypothalamic survival circuits: blueprints for purposive behaviors. Neuron 77: 810–824.
Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N et al (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131: 1164–1178.
Stevens CW, Aravind S, Das S, Davis RL (2013). Pharmacological characterization of LPS and opioid interactions at the toll-like receptor 4. Br J Pharmacol 168: 1421–1429.
Swartzwelder HS, Wilson WA, Tayyeb MI (1995). Age-dependent inhibition of long-term potentiation by ethanol in immature versus mature hippocampus. Alcohol Clin Exp Res 19: 1480–1485.
Takayama N, Ueda H (2005). Morphine-induced chemotaxis and brain-derived neurotrophic factor expression in microglia. J Neurosci 25: 430–435.
Takeda K, Akira S (2004). TLR signaling pathways. Semin Immunol 16: 3–9.
Taylor AM, Castonguay A, Ghogha A, Vayssiere P, Pradhan AA, Xue L et al (2016). Neuroimmune regulation of GABAergic neurons within the ventral tegmental area during withdrawal from chronic morphine. Neuropsychopharmacology 41: 949–959.
Taylor SB, Lewis CR, Olive MF (2013). The neurocircuitry of illicit psychostimulant addiction: acute and chronic effects in humans. Subst Abuse Rehabil 4: 29–43.
Theberge FR, Li X, Kambhampati S, Pickens CL, St Laurent R, Bossert JM et al (2013). Effect of chronic delivery of the Toll-like receptor 4 antagonist (+)-naltrexone on incubation of heroin craving. Biol Psychiatry 73: 729–737.
Thomas DM, Walker PD, Benjamins JA, Geddes TJ, Kuhn DM (2004). Methamphetamine neurotoxicity in dopamine nerve endings of the striatum is associated with microglial activation. J Pharmacol Exp Ther 311: 1–7.
Thomas WE (1992). Brain macrophages: evaluation of microglia and their functions. Brain Res Brain Res Rev 17: 61–74.
Tian L, Ma L, Kaarela T, Li Z (2012). Neuroimmune crosstalk in the central nervous system and its significance for neurological diseases. J Neuroinflammation 9: 155.
Trapp BD, Wujek JR, Criste GA, Jalabi W, Yin X, Kidd GJ et al (2007). Evidence for synaptic stripping by cortical microglia. Glia 55: 360–368.
Tremblay MÈ, Lowery RL, Majewska AK (2010). Microglial interactions with synapses are modulated by visual experience. PLoS Biol 8: e1000527.
Tritsch NX, Sabatini BL (2012). Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron 76: 33–50.
Trotta T, Porro C, Calvello R, Panaro MA (2014). Biological role of Toll-like receptor-4 in the brain. J Neuroimmunol 268: 1–12.
Turchan-Cholewo J, Dimayuga FO, Ding Q, Keller JN, Hauser KF, Knapp PE et al (2008). Cell-specific actions of HIV-Tat and morphine on opioid receptor expression in glia. J Neurosci Res 86: 2100–2110.
Ullian EM, Sapperstein SK, Christopherson KS, Barres BA (2001). Control of synapse number by glia. Science 291: 657–661.
Valenzuela CF (1997). Alcohol and neurotransmitter interactions. Alcohol Health Res World 21: 144–148.
Vander Weele CM, Porter-Stransky KA, Mabrouk OS, Lovic V, Singer BF, Kennedy RT et al (2014). Rapid dopamine transmission within the nucleus accumbens: dramatic difference between morphine and oxycodone delivery. Eur J Neurosci 40: 3041–3054.
Volkow ND, Wang GJ, Fowler JS, Logan J, Gatley SJ, Hitzemann R et al (1997). Decreased striatal dopaminergic responsiveness in detoxified cocaine-dependent subjects. Nature 386: 830–833.
Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J (2009). Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci 29: 3974–3980.
Walsh JG, Muruve DA, Power C (2014). Inflammasomes in the CNS. Nat Rev Neurosci 15: 84–97.
Walton MR, Gibbons H, MacGibbon GA, Sirimanne E, Saura J, Gluckman PD et al (2000). PU.1 expression in microglia. J Neuroimmunol 104: 109–115.
Wang JY, Wen LL, Huang YN, Chen YT, Ku MC (2006). Dual effects of antioxidants in neurodegeneration: direct neuroprotection against oxidative stress and indirect protection via suppression of glia-mediated inflammation. Curr Pharm Des 12: 3521–3533.
Wang X, Loram LC, Ramos K, de Jesus AJ, Thomas J, Cheng K et al (2012a). Morphine activates neuroinflammation in a manner parallel to endotoxin. Proc Natl Acad Sci USA 109: 6325–6330.
Wang X, Zhang Y, Peng Y, Hutchinson MR, Rice KC, Yin H et al (2016). Pharmacological characterization of the opioid inactive isomers (+)-naltrexone and (+)-naloxone as antagonists of toll-like receptor 4. Br J Pharmacol 173: 856–869.
Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M et al (2012b). IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol 13: 753–760.
Warner LA, Kessler RC, Hughes M, Anthony JC, Nelson CB (1995). Prevalence and correlates of drug use and dependence in the United States. Results from the National Comorbidity Survey. Arch Gen Psychiatry 52: 219–229.
Watkins LR, Hutchinson MR, Rice KC, Maier SF (2009). The ‘toll’ of opioid-induced glial activation: improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol Sci 30: 581–591.
Weiss F, Lorang MT, Bloom FE, Koob GF (1993). Oral alcohol self-administration stimulates dopamine release in the rat nucleus accumbens: genetic and motivational determinants. J Pharmacol Exp Ther 267: 250–258.
Weiss F, Porrino LJ (2002). Behavioral neurobiology of alcohol addiction: recent advances and challenges. J Neurosci 22: 3332–3337.
Werling DM, Parikshak NN, Geschwind DH (2016). Gene expression in human brain implicates sexually dimorphic pathways in autism spectrum disorders. Nat Commun 7: 10717.
Wilhelm CJ, Guizzetti M (2015). Fetal alcohol spectrum disorders: an overview from the glia perspective. Front Integr Neurosci 9: 65.
Williamson LL, Sholar PW, Mistry RS, Smith SH, Bilbo SD (2011). Microglia and memory: modulation by early-life infection. J Neurosci 31: 15511–15521.
Wu Y, Lousberg EL, Moldenhauer LM, Hayball JD, Coller JK, Rice KC et al (2012). Inhibiting the TLR4-MyD88 signalling cascade by genetic or pharmacological strategies reduces acute alcohol-induced sedation and motor impairment in mice. Br J Pharmacol 165: 1319–1329.
Xanthos DN, Sandkühler J (2014). Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci 15: 43–53.
Xu HT, Pan F, Yang G, Gan WB (2007). Choice of cranial window type for in vivo imaging affects dendritic spine turnover in the cortex. Nat Neurosci 10: 549–551.
Yan H, Li Q, Madison R, Wilson WA, Swartzwelder HS (2010). Differential sensitivity of hippocampal interneurons to ethanol in adolescent and adult rats. J Pharmacol Exp Ther 335: 51–60.
Yang SK, Wang YC, Chao CC, Chuang YJ, Lan CY, Chen BS (2010). Dynamic cross-talk analysis among TNF-R, TLR-4 and IL-1R signalings in TNFalpha-induced inflammatory responses. BMC Med Genomics 3: 19.
Yim HJ, Gonzales RA (2000). Ethanol-induced increases in dopamine extracellular concentration in rat nucleus accumbens are accounted for by increased release and not uptake inhibition. Alcohol 22: 107–115.
Yoon SY, Patel D, Dougherty PM (2012). Minocycline blocks lipopolysaccharide induced hyperalgesia by suppression of microglia but not astrocytes. Neuroscience 221: 214–224.
Zeisel A, Muñoz-Manchado AB, Codeluppi S, Lönnerberg P, La Manno G, Juréus A et al (2015). Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347: 1138–1142.
Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F et al (2014). Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci 17: 400–406.
Zhang XQ, Cui Y, Cui Y, Chen Y, Na XD, Chen FY et al (2012). Activation of p38 signaling in the microglia in the nucleus accumbens contributes to the acquisition and maintenance of morphine-induced conditioned place preference. Brain Behav Immun 26: 318–325.
Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S et al (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34: 11929–11947.
Acknowledgements
This work is supported by NIH grants DA034185 (SBD) and R01DA034185-03S1 (PDR).
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article

Cite this article
Lacagnina, M., Rivera, P. & Bilbo, S. Glial and Neuroimmune Mechanisms as Critical Modulators of Drug Use and Abuse. Neuropsychopharmacol 42, 156–177 (2017). https://doi.org/10.1038/npp.2016.121
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2016.121
This article is cited by
-
Dorsal hippocampal astrocytes mediate the development of heroin withdrawal-enhanced fear learning
Psychopharmacology (2024)
-
Dietary polyphenols drive dose-dependent behavioral and molecular alterations to repeated morphine
Scientific Reports (2023)
-
Methamphetamine-induced region-specific transcriptomic and epigenetic changes in the brain of male rats
Communications Biology (2023)
-
Sex-specific inflammatory and white matter effects of prenatal opioid exposure: a pilot study
Pediatric Research (2023)
-
Toll-like receptor 4 antagonists reduce cocaine-primed reinstatement of drug seeking
Psychopharmacology (2023)
