
Abstract
An increase in the ratio of cellular excitation to inhibition (E/I ratio) has been proposed to underlie the pathogenesis of neuropsychiatric disorders, such as autism spectrum disorders (ASD), obsessive-compulsive disorder (OCD), and Tourette’s syndrome (TS). A proper E/I ratio is achieved via factors expressed in neuron and glia. In astrocytes, the glutamate transporter GLT1 is critical for regulating an E/I ratio. However, the role of GLT1 dysfunction in the pathogenesis of neuropsychiatric disorders remains unknown because mice with a complete deficiency of GLT1 exhibited seizures and premature death. Here, we show that astrocyte-specific GLT1 inducible knockout (GLASTCreERT2/+/GLT1flox/flox, iKO) mice exhibit pathological repetitive behaviors including excessive and injurious levels of self-grooming and tic-like head shakes. Electrophysiological studies reveal that excitatory transmission at corticostriatal synapse is normal in a basal state but is increased after repetitive stimulation. Furthermore, treatment with an N-methyl-D-aspartate (NMDA) receptor antagonist memantine ameliorated the pathological repetitive behaviors in iKO mice. These results suggest that astroglial GLT1 has a critical role in controlling the synaptic efficacy at corticostriatal synapses and its dysfunction causes pathological repetitive behaviors.
Similar content being viewed by others


INTRODUCTION
An increase in the E/I ratio due to increased glutamatergic excitation or to reduced GABAergic inhibition has been proposed to cause neuropsychiatric disorders, such as autism spectrum disorders (ASD), obsessive-compulsive disorder (OCD), and Tourette’s syndrome (TS) (Albin and Mink, 2006; Rubenstein and Merzenich, 2003; Wu et al, 2012). A proper E/I ratio is controlled by factors expressed in neurons and glia. In neurons, scaffolding proteins (eg, postsynaptic density protein 95 (PSD95) or gephyrin) and cell adhesion molecules (eg, neuroligins or neurexins) are critical for controlling the brain’s excitatory state (Choudhury et al, 2012). An E/I ratio is also influenced by surrounding astrocytes, which control the extracellular glutamate level via the glutamate transporters GLAST and GLT1 (in humans, EAAT1 and EAAT2, respectively) (Tanaka et al, 1997; Watase et al, 1998).
Several studies have reported astrocyte dysfunction and impaired glial glutamate uptake in the pathophysiology of many neuropsychiatric disorders (Adamczyk et al, 2011; Molofsky et al, 2012; Szatmari et al, 2007). We previously reported that GLAST knockout mice showed schizophrenia-like phenotypes (Karlsson et al, 2008, 2009) and that GLAST/GLT1 double knockout mice exhibited multiple brain defects that were similar to schizophrenia-associated developmental defects (Aida et al, 2012; Matsugami et al, 2006). Because GLT1 is a major glutamate transporter in the forebrain (Tanaka et al, 1997), we hypothesized that astroglial GLT1 dysfunction has a critical role in the pathogenesis of neuropsychiatric disorders. However, mice with a complete deficiency of GLT1 (GLT1 KO) exhibited seizures and premature death (Tanaka et al, 1997).
To overcome the premature lethality of GLT1 KO mice, we generated inducible knockout mice (iKO mice) in which GLT1 is inactivated in a temporally controlled manner. Here, we show the comprehensive characterization of the inducible GLT1 knockout mice to address whether astroglial glutamate transporter dysfunction contributes to synaptic E/I imbalance, leading to pathogenesis of abnormal behaviors such as pathological repetitive behaviors shared between patients with ASD, OCD, and TS (Ting and Feng, 2011). We further tested the several drugs to suppress the phenotype of iKO mice.
MATERIALS AND METHODS
All research and animal care procedures were approved by the Tokyo Medical and Dental University Animal Care and Use Committee.
Experiment 1: Generation and Validation of Astrocyte-Specific Inducible GLT1 Knockout Mice
Floxed GLT1 (GLT1flox/+) mice were generated by inserting loxP sites into flanking introns of GLT1 exon 4 of embryonic stem cells derived from the 129Sv strain (Cui et al, 2014). Heterozygous GLT1flox/+ mice were then crossed to obtain homozygous GLT1flox/flox mice. To induce astrocyte-specific GLT1 deletion in a temporally controlled manner, we crossed GLT1flox/flox mice with mice expressing an inducible form of Cre (CreERT2) under the control of the endogenous astrocyte-specific GLAST locus (Mori et al, 2006; Saab et al, 2012). Then, the double heterozygous mice were crossed to obtain double homozygous GLASTCreERT2/CreERT2/GLT1flox/flox mice. Finally, GLASTCreERT2/+/GLT1flox/flox mice were generated by crossing GLT1flox/flox mice with GLASTCreERT2/CreERT2/GLT1flox/flox mice.
We induced GLT1 deletion in the GLASTCreERT2/+/GLT1flox/flox (iKO) mice by intraperitoneal administration of tamoxifen (Wako, Osaka, Japan, 100 mg/kg) dissolved in corn oil (Sigma, St. Louis, MO, USA). For control (Ctrl) mice, we intraperitoneally injected corn oil into the littermate GLASTCreERT2/+/GLT1flox/flox mice. Injections were performed once at postnatal day (P) 1, once a day for consecutive 5 days from P19, or once a day for consecutive 5 days from 12 weeks old to investigate the effect of developmental stage-specific GLT1 deletion.
GLT1 protein levels in Ctrl and iKO mice were investigated by GLT1 immunohistochemistry at 4 weeks post injection (wpi) for P1 injection, 8 wpi for P19 and adult injections as well as GLT1 western blot analysis at 2, 3, and 6 wpi for P19 injections as described in Supplementary Information.
To investigate the astrocyte specificity of CreERT2-mediated recombination, GLASTCreERT2/+ mice were crossed with ROSACAG−TdTomato/+ reporter mice (Madisen et al, 2010). Tamoxifen was intraperitoneally injected into GLASTCreERT2/+/ROSACAG−TdTomato/+ mice once a day for consecutive 5 days from P19. Eight weeks later, the reporter expressions were analyzed by immunohistochemistry with GFAP, S100β, and NeuN antibodies as described in Supplementary Information.
Experiment 2: Effects of GLT1 Deletion on Neurodegeneration
For brain wet weight measurement, Ctrl (n=6) and iKO (n=8) mice (8- to 9-week-old female) were killed with pentobarbital (100 mg/kg, i.p.). The weights of brain rostral to the medulla were measured.
For neuronal count, NeuN-immunostained neuronal profiles in layer 2/3 in somatosensory cortex, thalamus, dorsal striatum, and CA1 in hippocampus from 4-month-old Ctrl (n=3) and iKO mice (n=3) were counted. The number of NeuN-positive cells within 200 × 200 μm regions on each confocal image was counted using the cell counter plug-in from ImageJ software (NIH, Bethesda, MD) under the blind condition for the tamoxifen treatment. Every fourth section was counted (3–4 sections per mice) and quantified as the number of NeuN-positive cells per square millimeters.
To investigate gliosis/scarring in iKO mice, we performed immunohistochemistry with anti-GFAP and anti-IbaI antibodies on sections of layer 2/3 in somatosensory cortex, thalamus, dorsal striatum, and CA1 in hippocampus from 4-month-old Ctrl (n=3) and iKO mice (n=3) as described in Supplementary Information.
Experiment 3: Effects of GLT1 Deletion on Behaviors
Ctrl and iKO mice were sequentially subjected to a series of behavioral tests with more than 3 days interval between experiments. Adult (2–6 months old) male mice were used for the behavior analyses. The observers were blinded to the condition of the tamoxifen treatment and genotype.
Grooming and tic-like movements in a novel environment
The 4-month-old Ctrl (n=7) and iKO mice (n=8) were placed in a novel environment (acrylic cylinders; diameter, 12 cm; height, 22 cm; O’Hara, Tokyo, Japan)) and the behaviors were video recorded for 10 min. The total amount of time spent in grooming and the total number of tic-like movements defined as any very brief isolated head and/or body jerk or shake (Nordstrom and Burton, 2002) were manually measured. The 8-month-old male wild-type (n=10) and EAAC1 knockout mice (n=13) (Peghini et al, 1997) on C57BL/6 genetic background were also used.
Grooming in a familiar environment
The 3-month-old Ctrl (n=6) and iKO mice (n=6) were placed in new cages and habituated for 24 h. Then, the behaviors were video recorded for 2 h between 0900 and 1100 h. The total amount of time spent grooming was manually measured.
Hot plate test
The 7-month-old Ctrl (n=7) and iKO mice (n=8) were placed on a 55.0 °C hot plate (O’Hara), and the latency to the first hind-paw response (hind-paw lick, standing or jumping) was manually recorded.
Anxiety-like behaviors
Anxiety-like behaviors of Ctrl and iKO mice were examined using the elevated plus maze (4 months old, n=22 for Ctrl and 27 for iKO mice), light-dark box (4 months old, n=21 for Ctrl and 29 for iKO mice), and open field (3 months old, n=22 for Ctrl and 29 for iKO mice) tests. Full methods are described in Supplementary Information.
Social behaviors
Social behaviors of Ctrl and iKO mice were measured by reciprocal social interaction test (5 months old, n=7 for Ctrl and 9 for iKO mice) and three-chamber social interaction test (6 months old, n=7 for each genotype). Full methods are described in Supplementary Information.
Experiment 4: Effects of GLT1 Deletion on Seizure Susceptibility and Electroencephalogram.
Susceptibility to kainate-induced seizures
Seizures were induced by intraperitoneal administration of kainic acid (30 mg/kg, Sigma) dissolved in saline into 8-week-old Ctrl (n=8) and iKO mice (n=12). Seizure behaviors were recorded by video for 1 h and scored on a 0–7 rating scale as previously described (Bateup et al, 2013). Maximum seizure severity was recorded for each 5-min interval for each mouse for 60 min after kainic acid injection. The observers were blinded to the condition of the tamoxifen treatment.
Simultaneous video and electroencephalogram recording
We implanted a Teflon-coated silver wire above the primary somatosensory cortex (1.5 mm posterior and 1.5 mm lateral to the bregma) and a stainless screw electrode above the cerebellum as a reference electrode for EEG to iKO mice (4 to 5 months old, n=4) under 2% isoflurane anesthesia. Animals were transferred to the recording chamber in a sound-attenuating box after a recovery period of at least 5 days. Signals were amplified using a preamplifier (TLC2774c, Texas Instruments, Dallas, TX) and a main amplifier (EX4-400, Dagan) and sampled at 2000 Hz (final gain, 2000; band-pass filter, 0.1–1000 Hz) simultaneously with video monitoring of the mouse behaviors (AQ-VU, TEAC, Tokyo, Japan).
Experiment 5: Effects of GLT1 Deletion on Corticostriatal Synaptic Transmission
c-Fos mappings
Ctrl (n=8) and iKO (n=9) male mice (4–5 months old) were placed in new cages for 15 min and then were anesthetized and perfused with 4% PFA. In situ hybridization was performed as previously described (Shimogori et al, 2010). Full methods are described in Supplementary Information.
Western blotting of synaptic molecules
For preparation of crude synaptosomal fractions, 16-week-old Ctrl (n=3) and iKO mice (n=3) were used. Full methods are described in Supplementary Information.
Electrophysiology
Sagittal brain slices containing the cortex and the striatum (300 μm thick) were prepared from 3- to 4-month-old Ctrl (n=8) and iKO (n=10) mice. For repetitive stimulation, 3- to 4-month-old Ctrl (n=5) and iKO mice (n=4) were used. Briefly, whole-cell recordings were made from medium spiny neurons in the anterior region of the striatum using an upright microscope (BX50WI, Olympus) equipped with an infrared CCD camera system (Hamamatsu Photonics). Membrane currents were recorded with an EPC9/2 amplifier (HEKA Electronik). To stimulate corticostriatal axons, two glass micropipettes filled with normal saline were placed in the cerebral cortex or in the underlying white matter. The normal bath solution was supplemented with picrotoxin (100 μM, Tocris) for the measurement of the AMPA/NMDA ratio and the paired-pulse ratio. AMPA-mediated EPSCs were measured at a holding potential of −70 mV. When NMDA-mediated EPSCs were recorded, the membrane potential was held at +50 mV, and the amplitude at 60 ms after the stimulus was measured as the NMDA-mediated EPSC. Repetitive stimulations (10 Hz, 100 pulses) were applied to corticostriatal fibers and the amplitudes of AMPA receptor-mediated EPSCs relative to the amplitudes of the first response were measured. Full methods are described in Supplementary Information.
Microdialysis
Ctrl (n=11) and iKO (n=5) mice (4–5 months old) were used for microdialysis experiment. Full methods are described in Supplementary Information.
Experiment 6: Effects of Desloratadine, Fluoxetine, and Memantine on Repetitive Behaviors
Desloratadine
In the first week, the 6-month-old male Ctrl (n=8) and iKO mice (n=7) were orally administered saline containing 5% DMSO once a day for 5 days. Grooming behaviors were recorded 1 h after the final saline administration. In the second week, the same mice were orally administered desloratadine (5 mg/kg, TCI, Tokyo, Japan), a blood–brain barrier impermeable antihistamine, dissolved in saline containing 5% DMSO once a day for 5 days as previously described (Lagerström et al, 2010). Grooming behaviors were recorded 1 h after the final administration of desloratadine.
Fluoxetine
In the first week, the 7- to 8-month-old male Ctrl (n=10) and iKO (n=7) mice were intraperitoneally injected with saline once a day for 6 days. Grooming and tic-like movement behaviors were recorded 1 h after the final saline injection. Three weeks later, the same mice were intraperitoneally injected with fluoxetine (5 mg/kg, TCI), a selective serotonin reuptake inhibitor (SSRI), dissolved in saline once a day for 6 days (Welch et al, 2007). Grooming behaviors were recorded 1 h after the final injection of fluoxetine. The 4- to 5-month-old male iKO mice were divided into two groups (n=9 each) and treated water or fluoxetine (18 mg/kg/day) dissolved in water delivered ad libitum in the drinking water for 3 weeks as described previously (Shmelkov et al, 2010). Grooming behaviors were recorded on 21th day.
Memantine
The 3- to 5-month-old male and female Ctrl and iKO mice were divided into two groups (n=8 for saline treatment in Ctrl and iKO mice, n=9 for memantine treatment in Ctrl mice, and n=7 for memantine treatment in iKO mice). Each group was intraperitoneally injected with saline or memantine (10 mg/kg, Sigma), an NMDA receptor inhibitor, dissolved in saline. Grooming and tic-like movement behaviors were recorded 30 min after the injections. On the next day, open field tests were performed 30 min after the injections of saline or memantine.
Statistical Analysis
All data are presented as the mean±SEM. Statistical methods were described in the figure legends for each data set. Briefly, Student’s t-tests and Mann–Whitney’s u-tests were used to compare differences between any two groups. Two-way repeated measures ANOVA with post hoc t-test were used for the drug treatments, three-chamber social interaction tests, and grooming development analyses. Two-way ANOVA with Holm-Sidak’s post hoc tests was used for the electrophysiological analysis. Mann–Whitney’s u-test was used for seizure scoring. Statistical significance was set at p<0.05.
Experimental details of mouse characterization by behavioral, electrophysiological, biochemical, and immunohistochemical analyses are described in Supplementary Information.
RESULTS
Inducible Deletion of GLT1 in Astroglia
To generate inducible GLT1 knockout mice in which GLT1 is inactivated in a temporally controlled manner, we crossed mice carrying a loxP-flanked GLT1 allele (GLT1flox/flox) (Cui et al, 2014) with mice expressing an inducible form of Cre (CreERT2) under the astrocyte-specific, endogenous GLAST promoter, that are heterozygous for GLAST locus (Mori et al, 2006; Saab et al, 2012) (Figure 1a). We injected tamoxifen (Tmx) into GLASTCreERT2/+/GLT1flox/flox mice (iKO, Supplementary Figure S1) during the neonatal (P1 for 1day, neonatal-iKO), adolescent (P19 for 5 days, adolescent-iKO), or adult stage (12 weeks old for 5 days, adult-iKO) and examined the ablation of GLT1 after 4 weeks for neonatal-iKO or 8 weeks for adolescent- and adult-iKO mice (Figure 1a,Supplementary Figures S1 and S2). Control mice (Ctrl) were littermate GLASTCreERT2/+/GLT1flox/flox mice treated with corn oil, thus the effect of GLT1 deletion was tested on GLAST heterozygous genetic background (Figure 1a, Supplementary Figures S1 and S2). In neonatal-iKO mice, the GLT1 protein was almost completely deleted (Supplementary Figure S2a), and these mice exhibited premature death similar to GLT1 KO mice (Tanaka et al, 1997) (data not shown). In adult-iKO mice, only a mild reduction of GLT1 was observed (Supplementary Figure S2b), and these mice showed no behavioral abnormalities (data not shown).

Inducible ablation of GLT1 in adolescent mice. (a) Generation of adolescent-GLASTCreERT2/+/GLT1flox/flox mice (iKO) or control (Ctrl) mice by tamoxifen (Tmx) or oil injections. (b) Western blot analysis of GLT1 of the cerebral cortex, thalamus and striatum from postnatal day (P) 63 mice (n=3). Monomer bands of GLT1, or β-Actin are shown. GLT1 band intensities were normalized with those of β-actin. All data are presented as the mean±SEM. Student’s t-tests were used to compare GLT1 protein levels of Ctrl and iKO mice for each brain region. *P<0.05, ***P<0.005. (c) GLT1 immunohistochemistry. Boxed regions were enlarged in the right panels. Scale bar: 1 mm, and 100 μm for inset. (d) Facial lesions in 12- to 16-week-old male iKO mice.
In adolescent-iKO mice, western blot analysis of the GLT1 protein revealed a 60–80% reduction in cortex, striatum, and thalamus compared with control littermates (p=0.000384 for cerebral cortex, p=0.00361 for thalamus, and p=0.025 for striatum; Figure 1b), consistent with the immunohistochemistry results for GLT1 (Figure 1c). Under the Tmx treatment condition, almost all of the astrocytes throughout the brain were targeted, as shown by the reporter mice (Supplementary Figure S3), suggesting that GLT1 was specifically reduced in the astrocytes in adolescent-iKO mice. In contrast to GLT1 KO mice, the adolescent-iKO mice did not have lethal seizures and survived to adulthood (Tanaka et al, 1997). The brain wet weight was comparable between Ctrl and the adolescent-iKO mice (p=0.183; Supplementary Figure S4). Moreover, in contrast to GLT1 KO mice (Kiryk et al, 2008; Tanaka et al, 1997), there was no difference in the number of NeuN-positive cells between Ctrl and adolescent-iKO mice (p=0.794 for cerebral cortex, p=0.597 for thalamus, p=0.609 for striatum, and p=0.464 for hippocampus; Supplementary Figure S5). The adolescent-iKO mice did not show any signs of gliosis/scarring (Supplementary Figure S6). These results suggest that the brain is grossly intact in the adolescent-iKO mice. From 8 to 9 weeks old, the adolescent-iKO mice started to develop skin lesions on their neck (Figure 1d).
Adolescent-iKO Mice Showed Pathological Repetitive Behaviors
We hypothesized that the skin lesions could be the result of excessive self-grooming because they were present in animals housed alone. Moreover, no lesions were found in the control mice housed in the same cage with the adolescent-iKO mice (data not shown). The adolescent-iKO (hereafter, iKO) mice were often observed engaged in self-grooming. We assessed the grooming behavior and found a significant increase in the duration of grooming events in iKO mice compared with the control mice both in a novel (p=0.00268; Figure 2a and Supplementary Video S1) and familiar environment (p=0.00752; Figure 2b). Next, we examined the possibility that excessive grooming might be caused by peripheral defects, such as cutaneous inflammation, nociceptive defects or chronic itching. However, histological analysis of the facial skin from pre-lesion iKO mice did not reveal evidence of lymphocytic or granulocytic infiltration (Figure 2c). Moreover, no differences were observed in pain sensitivity between the control and iKO mice as assessed by the hot plate test (p=0.279; Figure 2d). To investigate whether a chronic itch sensation was the cause of excessive grooming, we orally administered the blood–brain barrier impermeant antihistamine desloratadine to the mice (Lagerström et al, 2010). However, this treatment had no effect on excessive grooming in the iKO mice (main effect of genotype, F (1, 26)=36.8, p=2.1E−6; main effect of treatment, F (1, 26)=0.6, p=0.45; interaction of genotype × treatment, F (1, 26)=2.1, p=0.16; Figure 2e).

Excessive repetitive behaviors, and normal anxiety levels and social behavior in iKO mice. (a, b) Durations of self-grooming in Ctrl and iKO mice in a novel environment (n=7 for Ctrl and 8 for iKO mice, a) and in the home cage (n=6, b). (c) Hematoxylin-eosin (HE) staining of neck skin from Ctrl and pre-lesion iKO mice. Scale bar: 500 μm. (d) Hot plate test (55 °C, n=7 for Ctrl and 8 for iKO mice). (e) Durations of self-grooming in Ctrl (n=8) and iKO (n=7) mice treated with saline (blue bar) or desloratadine (DL, red bar). (f–k) Behaviors analyses. (f) Number of tic-like movements in a novel environment (n=7 for Ctrl and 8 for iKO mice). (g) Elevated plus maze test (n=22 for Ctrl and 27 for iKO mice). (h) Light-dark box test (n=21 for Ctrl and 29 for iKO mice). (i) Open field test (n=22 for Ctrl and 29 for iKO mice). (j) Reciprocal social interaction test (n=7 for Ctrl and 9 for iKO mice). (j) Three-chamber social interaction test (n=7). All data are presented as the mean±SEM. Statistical significance was calculated by Student’s t-test in (a, b, d and j), by Mann–Whitney’s u-tests in (f–i), and by two-way repeated measures ANOVA with post hoc t-test in (e) and (k). *P<0.05, **P<0.01, ***P<0.005.
These results showed no obvious peripheral defects that would indicate a cause for excessive grooming, raising the possibility that the iKO mice displayed excessive and injurious self-grooming behavior caused by brain dysfunction. Because excessive repetitive behaviors are observed in patients with TS, OCD, and ASD (Albin and Mink, 2006; Langen et al, 2011; Ting and Feng, 2011), we examined additional behavior abnormalities, such as tic-like movements, increased anxiety, and social impairment related to these diseases, respectively. Although almost no tic-like movements such as brief isolated head shake (Macrì et al, 2013) were observed in Ctrl mice, iKO mice showed a significant increase in the number of tic-like movements (p<0.01; Figure 2f and Supplementary Video S2). We next examined anxiety-like behaviors using the elevated plus maze, light-dark box, and open field tests. These were comparable between iKO and control mice (p=0.351 and z=0.932; Figure 2g, p=0.723 and z=−0.354; Figures 2h, p=0.0664 and z=−1.835; Figure 2i). We next examined social behaviors using the reciprocal social interaction test and the three-chamber test. There were no significant differences in time spent interacting with other animals (p=0.447; Figure 2j) or in time spent in each chamber of the three-chamber test (main effect of genotype, F (1, 24)=0.47, p=0.499; main effect of object, F (1, 24)=23.9, p=5.5E−5; interaction of genotype × object, F (1, 24)=0.223, p=0.641. t-test: time in close interaction in Ctrl mice compared between mouse cage and empty cage, p=0.0188; time in close interaction in iKO mice compared between mouse cage and empty cage, p=0.0396; Figure 2k) between control and iKO. Together, these findings suggest that the iKO mouse shows pathological repetitive behaviors without increased anxiety or social abnormality.
Although a 60–80% reduction of the GLT1 protein was achieved by 3 weeks post injection (wpi) of tamoxifen in the iKO mice (p=0.839, 0.048, and 0.0003 for 2, 3, and 6 wpi cerebral cortex; p=0.161, 0.047, and 0.003 for 2, 3, and 6 wpi thalamus; p=0.652, 0.0008, and 0.025 for 2, 3, and 6 wpi striatum; Supplementary Figure S7a), we did not observe any overt excessive grooming in the iKO mice at 4 wpi. However, by 5 wpi, the iKO mice exhibited excessive grooming (main effect of genotype, F (1, 40)=15.6, p=0.0003; main effect of time course, F (1, 40)=8.4, p=0.0059; interaction of genotype × time course, F (1, 40)=7.9, p=0.0076; t-test: Ctrl and iKO mice at 4 wpi, p=0.13; Ctrl and iKO mice at 5 wpi, p=0.0018; Supplementary Figure S7b). This finding suggests that the loss of astroglial GLT1 led to excessive grooming through chronic circuit changes.
Neither GLAST KO (Karlsson et al, 2008, 2009, 2012) nor EAAC1 KO mice show excessive repetitive behaviors (p=0.929 for grooming; Supplementary Figure S8a, and p=1.0 for tic-like movements; Supplementary Figure S8b). Thus, our data indicate that a loss of major glutamate transporter GLT1 in astrocytes triggers pathological repetitive behaviors in mice.
iKO Mice Exhibited Altered Synaptic Transmission Efficacy at Corticostriatal Synapses
To investigate whether iKO mice show a hyperexcitability phenotype, we first examined the susceptibility to kainate-induced seizures. The iKO mice exhibited increased seizure sensitivity demonstrated by a higher percentage of mice with a maximum seizure score of 7 (mortality) during the test period (p<0.01; Figure 3a). These results suggest that iKO mice show a hyperexcitability phenotype, even in the absence of spontaneous seizures. To exclude the possibility that pathological repetitive behaviors in iKO mice are simply a manifestation of seizure discharge, we performed simultaneous video and electroencephalogram (EEG) recordings from iKO mice under freely moving condition (Figure 3b). No epileptiform discharges were observed in iKO mice both during resting states, and grooming and tic-like movement bouts. These results indicate that seizure is not the cause of pathological repetitive behaviors in iKO mice.

Increased seizure severity in iKO mice. (a) Seizure severity was tested in 8-week-old-Ctrl and iKO mice (n=8 and 12, respectively) by intraperitoneal administration of 30 mg/kg kainic acid. Higher maximum seizure scores represent more severe seizure status; a score of 7 indicates mortality. Statistical significance was calculated by Mann–Whitney’s u-test. **P<0.01. (b) EEG recordings from iKO mice (n=4). A representative EEG trace from somatosensory cortex is shown (upper panel). Lines represent periods of grooming bouts. Arrows indicate timing of tic-like movements. EEG traces around grooming onsets are enlarged in lower panels. Lines with gray boxes represent periods of grooming bouts. iKO mice do not show any epileptiform discharge both during resting states and grooming bouts.
Next, we determined which brain regions might be responsible for the repetitive behaviors observed in the iKO mice by measuring expression of c-fos, a marker of neural activity. We found that the number of c-fos positive cells in the striatum exhibited a tendency to increase in the iKO mice (p=0.136; Figure 4a), although the difference did not reach statistical significance. Other brain regions, such as the medial prefrontal cortex (mPFC) and thalamus, did not show an increase in the number of c-fos positive cells (p=0.535 for mPFC and p=0.796 for thalamus; Supplementary Figure S9). Therefore, we focused our analyses on striatal neurons and corticostriatal synapses. We conducted biochemical analyses of striatal synapses in the iKO and control mice and found no significant differences in the levels of glutamate receptors (NR1, NR2A, NR2B, GluR1, and GluR2), PSD95, and synaptophysin (SYP) in striatal P2 crude synaptosomal fractions and other glutamate transporters (GLAST and EAAC1) in striatum homogenates between the iKO and control mice (p=0.2 for NR1, p=0.557 for NR2A, p=0.499 for NR2B, p=0.552 for GluR1, p=0.594 for GluR2, p=0.420 for PSD95, p=0.966 for SYP, p=0.848 for GLAST, and p=0.72 for EAAC1; Figure 4b and Supplementary Figure S10).

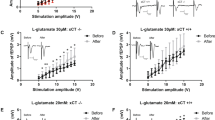
Altered synaptic transmission efficacy at corticostriatal synapses in iKO mice. (a) c-fos mapping in the striatum (n=8 for Ctrl and 9 for iKO mice). c-Fos positive cells within each boxed region (red) were counted. Statistical significance was calculated by Student’s t-test. Scale bars: 500 μm. (b) Western blot analysis of 16-week-old striatum crude synaptosomal fractions (n=3). Glutamate receptors (NR1, NR2A, NR2B, GluR1, and GluR2), postsynaptic density protein-95 (PSD95), synaptophysin (SYP), or β-actin were shown. Statistical significance was calculated by Student’s t-test. (c) Representative traces of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-D-aspartate (NMDA) receptor-mediated excitatory postsynaptic currents (EPSCs) from medium spiny neurons (MSNs). Amplitudes were measured at the peaks (leftward arrows for AMPA EPSCs and downward arrows for NMDA EPSCs) (n=8 for Ctrl and 10 for iKO mice). Interpulse intervals: 50 ms. Statistical significance was calculated by Student’s t-test and Mann–Whitney’s u-tests. (d) (Top) Representative traces of EPSCs from the MSNs during prolonged repetitive stimulation (10 Hz, 100 pulses). Only responses 1–5 (black) and 21–25 (red) are shown. (Bottom) The average normalized response amplitudes of EPSCs (n=5 for Ctrl and 4 for iKO mice). Each point represents the average of 10 consecutive responses. Statistical significance was calculated by two-way ANOVA with Holm-Sidak’s post hoc tests. ***P<0.001.
Next, we examined corticostriatal synaptic transmission in acute slices from 3- to 4-month-old animals to elucidate the functional consequences of the reduced glial glutamate transporters. We did not observe any difference in the 10–90% rise time and the decay time constant of either the AMPA or NMDA receptor-mediated excitatory postsynaptic currents (EPSCs) between the control and iKO mice (p=1.0 for the rise time of AMPA EPSCs, p=0.894 for the decay time constant of AMPA EPSCs, and p=0.991 for the decay time constant of NMDA EPSCs; Figure 4c). Furthermore, the AMPA/NMDA ratio was not altered in the iKO mice when compared with the control mice (p=0.606; Figure 4c). The paired pulse ratio of evoked AMPA receptor-mediated EPSCs in the iKO mice was not significantly different from that of the control mice at 50 ms interpulse interval (no significance, Figure 4c). These results indicate that the basal properties of corticostriatal synaptic transmission were not altered in the iKO mice. Consistent with these findings, extracellular glutamate levels measured by microdialysis were not altered in the iKO mice (p=0.726; Supplementary Figure S11).
We next examined synaptic responses to a prolonged repetitive stimulation that triggers a massive glutamate release. In the control and iKO mice, the normalized EPSC amplitude decreased gradually during prolonged repetitive stimulation. Notably, the depression of the normalized EPSC amplitude was significantly weaker in the iKO mice (F (2, 150)=33.2263, p<0.001; Holm-Sidak’s post hoc tests: p=0.033; Figure 4d). These data suggest that prolonged repetitive stimulation, but not a single episode of stimulation, leads to corticostriatal glutamatergic hyperactivity in iKO mice. Together, these findings indicate that corticostriatal synaptic response was elevated during repeated stimulation in iKO mice compared with control mice.
Memantine Treatment Attenuated the Pathological Repetitive Behaviors in iKO Mice
Because SSRIs are the first-line treatment for repetitive behaviors in OCD and ASD (Fineberg et al, 2012), we tested the effect of fluoxetine, an SSRI, treatment on repetitive behaviors in the iKO mice. Fluoxetine treatment for 6 days (Ahmari et al, 2013; Welch et al, 2007) reduced neither excessive grooming (main effect of genotype, F (1, 30)=186.8, p=2.0E−14; main effect of treatment, F (1, 30)=6.3, p=0.018; interaction of genotype × treatment, F (1, 30)=6.0, p=0.02; t-test: Ctrl mice treated with SSRI compared with Ctrl mice treated with saline, p=0.296; iKO mice treated with SSRI compared with iKO mice treated with saline, p=0.0261; Figure 5a) nor tic-like movements (main effect of genotype, F (1, 30)=119.0, p=5.8E−12; main effect of treatment, F (1, 30)=0.09, p=0.76; interaction of genotype × treatment, F (1, 30)=0.21, p=0.65; Figure 5b) in iKO mice. Chronic administration of fluoxetine for 3 weeks also had no effect on repetitive behaviors in iKO mice (data not shown). Next, we tried to identify effective agents that rescue repetitive behaviors in iKO mice. Given that repetitive behaviors in iKO mice emerge from the corticostriatal glutamatergic hyperactivity caused by decreased GLT1 activity, we reasoned that repetitive behaviors in iKO mice could be rescued by inhibiting glutamatergic transmission. To test this idea, we treated iKO with memantine, an NMDA receptor inhibitor used for the treatment of patients with Alzheimer’s disease. Treatment of the iKO mice with memantine led to a drastic and immediate reduction in the duration of grooming (main effect of genotype, F (1, 28)=61.2, p=1.6E−8; main effect of treatment, F (1, 28)=47.9, p=1.6E−7; interaction of genotype × treatment, F (1, 28)=37.9, p=1.2E−6; t-test: Ctrl mice treated with memantine compared with Ctrl mice treated with saline, p=0.171; iKO mice treated with memantine compared with iKO mice treated with saline, p=0.0000931; Figure 5c) and the number of tic-like movements (main effect of genotype, F (1, 28)=84.8, p=5.7E−10; main effect of treatment, F (1, 28)=14.0, p=0.00085; interaction of genotype × treatment, F (1, 28)=13.5, p=0.001; t-test: Ctrl mice treated with memantine compared with Ctrl mice treated with saline, p=0.195; iKO mice treated with memantine compared with iKO mice treated with saline, p=0.00436; Figure 5d) compared with the saline-treated iKO mice. Memantine treatment did not affect total activities in the iKO mice (main effect of genotype, F (1, 28)=2.45, p=0.13; main effect of treatment, F (1, 28)=2.78, p=0.11; interaction of genotype × treatment, F (1, 28)=5.05, p=0.032; t-test: Ctrl mice treated with memantine compared with Ctrl mice treated with saline, p=0.00922; iKO mice treated with memantine compared with iKO mice treated with saline, p=0.673; Figure 5e). Thus, memantine treatment attenuated the pathological repetitive behaviors in the iKO mice.

Memantine rescued excessive grooming and tic-like movements in the iKO mice. (a, b) Durations of self-grooming (a) and number of tic-like movements (b) in the Ctrl (n=10) and iKO (n=7) mice after 6-day saline (blue bar) or fluoxetine (SSRI, red bar) treatment. (c–e) Durations of self-grooming (c), number of tic-like movements (d), or traveled distance in open field tests (e) in the Ctrl and iKO mice after 30-min saline (blue bar, n=8) or memantine (red bar, n=9 for Ctrl, and 7 for iKO mice) treatment. Statistical significance was calculated by two-way repeated measure ANOVA with post hoc t-test. *P<0.05, **P<0.01, ***P<0.005.
DISCUSSION
In mammalian forebrain, three glutamate transporters including astroglial GLT1 and GLAST, and neuronal EAAC1, are expressed. Although there are many studies about behavioral phenotypes of GLAST- and EAAC-deficient mice (Karlsson et al, 2008, 2009, 2012; Peghini et al, 1997; Watase et al, 1998), early postnatal lethality in GLT1 KO mice hindered their comprehensive behavioral analysis. To overcome the premature lethality of GLT1 KO mice, we generated GLT1 iKO mice that exhibit a 60–70% decrease in GLT1 protein levels. GLT1 iKO mice showed pathological repetitive behaviors, whereas neither GLAST nor EAAC1 KO mice show excessive repetitive behaviors. This may be because GLT1 accounts for more than 90% of glutamate uptake in forebrain (Tanaka et al, 1997).
Excessive grooming in iKO mice is similar to that previously observed in mouse models of OCD, such as Sapap3- and Slitrk5-deficient mice (Shmelkov et al, 2010; Welch et al, 2007). The syntactic complex grooming chain patterns (Berridge et al, 2005) are proposed to resemble sequential super-stereotypy due to the overly rigid sequential patterns of action that are observed in OCD (Wang et al, 2009). Head-shakes occur in rodents following administration of various drugs, including the selective serotonin 5-HT2A/2C agonist 1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane (DOI) (Gaynor and Handley, 2001). Head-shakes are also observed in a model of comorbid Tourette’s syndrome and OCD, ‘DICT-7’ transgenic mouse expressing the intracellular A1 subunit of cholera toxin within a cortico-limbic subset of dopamine D1-receptor expressing neurons. These movements have been proposed as a putative model of human motor tics (Handley and Dursun, 1992; Nordstrom and Burton, 2002; Swerdlow and Sutherland, 2005).
Overgrooming and tic-like movements in these mutant mice are directly or indirectly linked with abnormal corticostriatal synaptic function, which has been implicated in patients with OCD (Nordstrom and Burton, 2002; Shmelkov et al, 2010; Welch et al, 2007). Furthermore, repeated corticostriatal stimulation using optogenetics has been shown to trigger excessive grooming in mice (Ahmari et al, 2013). Although the basal corticostriatal excitatory neurotransmission was not altered, we found the reduced attenuation of EPSCs in the iKO mice under the prolonged repetitive stimulation that triggers a massive glutamate release. This attenuation of EPSCs in the striatum during repetitive stimulation is thought to reflect a presynaptic cycling process in which depleted docked vesicles are replenished by reserve pool vesicles (Hoshina et al, 2013). Since there is no difference in paired-pulse ratio in the striatum between iKO and control mice (Figure 4c), the presynaptic functions at corticostriatal synapses are intact in iKO mice. Therefore, the reduced attenuation of EPSCs in the striatum of iKO mice can be explained by increased extracellular glutamate concentration due to insufficient clearance of glutamate from the synaptic cleft. In previous mouse models of OCD, dysfunction of neuron-specific proteins such as Sapap3 and Slitrk5 led to repetitive behavior through abnormal corticostriatal neurotransmission.(Shmelkov et al, 2010; Welch et al, 2007), In contrast, GLT1 iKO mice exhibit pathological repetitive behaviors due to dysfunction of an astroglial membrane protein, suggesting that glial dysfunction may be involved in the pathophysiology of pathological repetitive behaviors.
The SSRIs are the first-line treatment for repetitive behaviors in OCD (Fineberg et al, 2012). Although we tested both 6 days and 3 weeks fluoxetine treatment, the pathologic repetitive behaviors in GLT1 iKO mice were not ameliorated. Rather, grooming in GLT1 iKO mice was worsened by 6 days fluoxetine treatment. This effect can be explained by the interaction of serotonin and glutamate. Cai et al (2013) demonstrated that serotonin potentiated glutamatergic transmission by increasing the function of postsynaptic AMPARs (Cai et al, 2013). In GLT1 iKO mice, increased serotonin by fluoxetine treatment might potentiate corticostriatal synaptic transmission more efficiently than control mice, resulting in the enhancement of grooming. Since a substantial minority of OCD patients fail to respond to SSRIs, the molecular mechanisms underlying repetitive behaviors in OCD are heterogeneous. The pathological repetitive behaviors in GLT1 iKO mice might recapitulate repetitive behaviors cause by glutamatergic overactivity. Also, alpha-2 agonists or the atypical antipsychotics are the first-line treatment for tics in TS patients, these are often ineffective and frequently associated with side effects (Cavanna and Seri, 2013; Shprecher et al, 2014; Singer et al, 2010). We showed that memantine immediately and efficiently attenuated the pathological repetitive behaviors in the iKO mice. Although it is not used as standard care for OCD, a report of a controlled trial has suggested the possibility that memantine might reduce OCD symptoms (Ghaleiha et al, 2013). Thus, our findings raise the possibility that memantine may be a novel therapeutic candidate for patients with the pathologic repetitive behaviors.
We also showed memantine-induced enhanced locomotion in Ctrl mice as previously described (Gilmour et al, 2009; Hiyoshi et al, 2014), and its loss in iKO mice. Memantine is known to preferentially inhibit NMDA receptors containing NR2C and NR2D subunits (Kotermanski and Johnson, 2009). The NR2D-containing NMDA receptors are specifically expressed in GAD67-positive GABAergic inhibitory neurons throughout the mouse brain (Yamasaki et al, 2014). Thus, memantine treatment might specifically inhibit the excitation of inhibitory GABAergic interneurons, leading to hyperexcitation of glutamatergic neurons (disinhibition) and enhanced locomotor activity in Ctrl mice. In iKO mice, the increased synaptic excitability might reduce inhibitory effect of memantine on NR2D-containing NMDA receptors, resulting in impaired disinhibition of glutamatergic neurons, which in turn could cause the loss of memantine enhancement of locomotor activity.
FUNDING AND DISCLOSURE
This work was supported by the Strategic Research Program for Brain Sciences (SRPBS) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT, to KT and MK). Additional support was provided by a Grant-in-Aid for Science Research (22700328 to TA) from MEXT, grants to TA from The Moritani Scholarship Foundation, a grant to JY from the Medical Research Institute (MRI), of TMDU, and the Joint Usage/Research Program of the MRI of TMDU to KFT. The authors declare no conflict of interest.
References
Adamczyk A, Gause CD, Sattler R, Vidensky S, Rothstein JD, Singer H et al (2011). Genetic and functional studies of a missense variant in a glutamate transporter, SLC1A3, in Tourette syndrome. Psychiatr Genet 21: 90–97.
Ahmari SE, Spellman T, Douglass NL, Kheirbek MA, Simpson HB, Deisseroth K et al (2013). Repeated cortico-striatal stimulation generates persistent OCD-like behavior. Science 340: 1234–1239.
Aida T, Ito Y, Takahashi YK, Tanaka K (2012). Overstimulation of NMDA receptors impairs early brain development in vivo. PLoS ONE 7: e36853.
Albin RL, Mink JW (2006). Recent advances in Tourette syndrome research. Trends Neurosci 29: 175–182.
Bateup HS, Johnson CA, Denefrio CL, Saulnier JL, Kornacker K, Sabatini BL (2013). Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 78: 510–522.
Berridge KC, Aldridge JW, Houchard KR, Zhuang X (2005). Sequential super-stereotypy of an instinctive fixed action pattern in hyper-dopaminergic mutant mice: a model of obsessive compulsive disorder and Tourette’s. BMC Biol 3: 4.
Cai X, Kallarackal AJ, Kvarta MD, Goluskin S, Gaylor K, Bailey AM et al (2013). Local potentiation of excitatory synapses by serotonin and its alteration in rodent models of depression. Nat Neurosci 16: 464–472.
Cavanna AE, Seri S (2013). Tourette’s syndrome. BMJ 347: f4964.
Choudhury PR, Lahiri S, Rajamma U (2012). Glutamate mediated signaling in the pathophysiology of autism spectrum disorders. Pharmacol Biochem Behav 100: 841–849.
Cui W, Mizukami H, Yanagisawa M, Aida T, Nomura M, Isomura Y et al (2014). Glial dysfunction in the mouse Habenula causes depressive-like behaviors and sleep disturbance. J Neurosci 34: 16273–16285.
Fineberg NA, Brown A, Reghunandanan S, Pampaloni I (2012). Evidence-based pharmacotherapy of obsessive-compulsive disorder. Int J Neuropsychopharmacol 15: 1173–1191.
Gaynor CM, Handley SL (2001). Effects of nicotine on head-shakes and tryptophan metabolites. Psychopharmacology (Berl) 153: 327–333.
Ghaleiha A, Entezari N, Modabbernia A, Najand B, Askari N, Tabrizi M et al (2013). Memantine add-on in moderate to severe obsessive-compulsive disorder: randomized double-blind placebo-controlled study. J Psychiatr Res 47: 175–180.
Gilmour G, Pioli EY, Dix SL, Smith JW, Conway MW, Jones WT et al (2009). Diverse and often opposite behavioural effects of NMDA receptor antagonists in rats: implications for “NMDA antagonist modelling” of schizophrenia. Psychopharmacology (Berl) 205: 203–216.
Handley S, Dursun S (1992). Serotonin and Tourette’s syndrme: movement such as head-shakes and wet-dog shakes may model human tics. Adv Biosci 85: 235–253.
Hiyoshi T, Kambe D, Karasawa J, Chaki S (2014). Differential effects of NMDA receptor antagonists at lower and higher doses on basal gamma band oscillation power in rat cortical electroencephalograms. Neuropharmacology 85: 384–396.
Hoshina N, Tanimura A, Yamasaki M, Inoue T, Fukabori R, Kuroda T et al (2013). Protocadherin 17 regulates presynaptic assembly in topographic corticobasal Ganglia circuits. Neuron 78: 839–854.
Karlsson R-M, Adermark L, Molander A, Perreau-Lenz S, Singley E, Solomon M et al (2012). Reduced alcohol intake and reward associated with impaired endocannabinoid signaling in mice with a deletion of the glutamate transporter GLAST. Neuropharmacology 63: 181–189.
Karlsson R-M, Tanaka K, Heilig M, Holmes A (2008). Loss of glial glutamate and aspartate transporter (excitatory amino acid transporter 1) causes locomotor hyperactivity and exaggerated responses to psychotomimetics: rescue by haloperidol and metabotropic glutamate 2/3 agonist. Biol Psychiatry 64: 810–814.
Karlsson R-M, Tanaka K, Saksida LM, Bussey TJ, Heilig M, Holmes A (2009). Assessment of glutamate transporter GLAST (EAAT1)-deficient mice for phenotypes relevant to the negative and executive/cognitive symptoms of schizophrenia. Neuropsychopharmacology 34: 1578–1589.
Kiryk A, Aida T, Tanaka K, Banerjee P, Wilczynski GM, Meyza K et al (2008). Behavioral characterization of GLT1 (+/-) mice as a model of mild glutamatergic hyperfunction. Neurotox Res 13: 19–30.
Kotermanski SE, Johnson JW (2009). Mg2+ imparts NMDA receptor subtype selectivity to the Alzheimer’s drug memantine. J Neurosci 29: 2774–2779.
Lagerström MC, Rogoz K, Abrahamsen B, Persson E, Reinius B, Nordenankar K et al (2010). VGLUT2-dependent sensory neurons in the TRPV1 population regulate pain and itch. Neuron 68: 529–542.
Langen M, Durston S, Kas MJH, Engeland H van, Staal WG (2011). The neurobiology of repetitive behavior: …and men. Neurosci Biobehav Rev 35: 356–365.
Macrì S, Onori MP, Roessner V, Laviola G (2013). Animal models recapitulating the multifactorial origin of Tourette syndrome. Int Rev Neurobiol 112: 211–237.
Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala H a, Gu H et al (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13: 133–140.
Matsugami TR, Tanemura K, Mieda M, Nakatomi R, Yamada K, Kondo T et al (2006). Indispensability of the glutamate transporters GLAST and GLT1 to brain development. Proc Natl Acad Sci USA 103: 12161–12166.
Molofsky A V, Krencik R, Krenick R, Ullian EM, Ullian E, Tsai H et al (2012). Astrocytes and disease: a neurodevelopmental perspective. Genes Dev 26: 891–907.
Mori T, Tanaka K, Buffo A, Wurst W, Kühn R, Götz M (2006). Inducible gene deletion in astroglia and radial glia-a valuable tool for functional and lineage analysis. Glia 54: 21–34.
Nordstrom EJ, Burton FH (2002). A transgenic model of comorbid Tourette’s syndrome and obsessive-compulsive disorder circuitry. Mol Psychiatry 7: 617–625.
Peghini P, Janzen J, Stoffel W (1997). Glutamate transporter EAAC-1-deficient mice develop dicarboxylic aminoaciduria and behavioral abnormalities but no neurodegeneration. EMBO J 16: 3822–3832.
Rubenstein JLR, Merzenich MM (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav 2: 255–267.
Saab AS, Neumeyer A, Jahn HM, Cupido A, Šimek AAM, Boele H-J et al (2012). Bergmann glial AMPA receptors are required for fine motor coordination. Science 337: 749–753.
Shimogori T, Lee DA, Miranda-Angulo A, Yang Y, Wang H, Jiang L et al (2010). A genomic atlas of mouse hypothalamic development. Nat Neurosci 13: 767–775.
Shmelkov S V, Hormigo A, Jing D, Proenca CC, Bath KG, Milde T et al (2010). Slitrk5 deficiency impairs corticostriatal circuitry and leads to obsessive-compulsive-like behaviors in mice. Nat Med 16: 598–602.
Shprecher DR, Schrock L, Himle M (2014). Neurobehavioral aspects, pathophysiology, and management of Tourette syndrome. Curr Opin Neurol 27: 484–492.
Singer HS, Morris C, Grados M (2010). Glutamatergic modulatory therapy for Tourette syndrome. Med Hypotheses 74: 862–867.
Swerdlow NR, Sutherland AN (2005). Using animal models to develop therapeutics for Tourette Syndrome. Pharmacol Ther 108: 281–293.
Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu X-Q et al (2007). Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet 39: 319–328.
Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K et al (1997). Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276: 1699–1702.
Ting JT, Feng G (2011). Neurobiology of obsessive-compulsive disorder: insights into neural circuitry dysfunction through mouse genetics. Curr Opin Neurobiol 21: 842–848.
Wang L, Simpson HB, Dulawa SC (2009). Assessing the validity of current mouse genetic models of obsessive-compulsive disorder. Behav Pharmacol 20: 119–133.
Watase K, Hashimoto K, Kano M, Yamada K, Watanabe M, Inoue Y et al (1998). Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur J Neurosci 10: 976–988.
Welch JM, Lu J, Rodriguiz RM, Trotta NC, Peca J, Ding J-D et al (2007). Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature 448: 894–900.
Wu K, Hanna GL, Rosenberg DR, Arnold PD (2012). The role of glutamate signaling in the pathogenesis and treatment of obsessive-compulsive disorder. Pharmacol Biochem Behav 100: 726–735.
Yamasaki M, Okada R, Takasaki C, Toki S, Fukaya M, Natsume R et al (2014). Opposing role of NMDA receptor GluN2B and GluN2D in somatosensory development and maturation. J Neurosci 34: 11534–11548.
Acknowledgements
We thank Dr Masahiko Watanabe (Hokkaido University) for providing antibodies, Ms. Harumi Ishikubo, Ms. Masako Hidaka, Ms. Kaori Sugiyama, and Ms. Risa Imahashi (MRI, TMDU), and Dr Naofumi Uesaka (University of Tokyo) for their technical support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Aida, T., Yoshida, J., Nomura, M. et al. Astroglial Glutamate Transporter Deficiency Increases Synaptic Excitability and Leads to Pathological Repetitive Behaviors in Mice. Neuropsychopharmacol 40, 1569–1579 (2015). https://doi.org/10.1038/npp.2015.26
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2015.26
This article is cited by
-
Astrocytes in human central nervous system diseases: a frontier for new therapies
Signal Transduction and Targeted Therapy (2023)
-
Astrocytic Glutamate Transporters and Migraine
Neurochemical Research (2023)
-
The role of excitatory amino acid transporter 2 (EAAT2) in epilepsy and other neurological disorders
Metabolic Brain Disease (2023)
-
Memantine/Aripiprazole Combination Alleviates Cognitive Dysfunction in Valproic Acid Rat Model of Autism: Hippocampal CREB/BDNF Signaling and Glutamate Homeostasis
Neurotherapeutics (2023)
-
Calcineurin Signalling in Astrocytes: From Pathology to Physiology and Control of Neuronal Functions
Neurochemical Research (2023)
