
Abstract
Systemic doses of the psychotomimetic ketamine alter the spectral characteristics of hippocampal and prefrontal cortical network activity. Using dynamic causal modeling (DCM) of cross-spectral densities, we quantify the putative synaptic mechanisms underlying ketamine effects in terms of changes in directed, effective connectivity between dorsal hippocampus and medial prefrontal (dCA1-mPFC) cortex of freely moving rats. We parameterize dose-dependent changes in spectral signatures of dCA1-mPFC local field potential recordings, using neural mass models of glutamatergic and GABAergic circuits. Optimizing DCMs of theta and gamma frequency range responses, model comparisons suggest that both enhanced gamma and depressed theta power result from a reduction in top-down connectivity from mPFC to the hippocampus, mediated by postsynaptic NMDA receptors (NMDARs). This is accompanied by an alteration in the bottom-up pathway from dCA1 to mPFC, which exhibits a distinct asymmetry: here, feed-forward drive at AMPA receptors increases in the presence of decreased NMDAR-mediated inputs. Setting these findings in the context of predictive coding suggests that NMDAR antagonism by ketamine in recurrent hierarchical networks may result in the failure of top-down connections from higher cortical regions to signal predictions to lower regions in the hierarchy, which consequently fail to respond consistently to errors. Given that NMDAR dysfunction has a central role in pathophysiological theories of schizophrenia and that theta and gamma rhythm abnormalities are evident in schizophrenic patients, the approach followed here may furnish a framework for the study of aberrant hierarchical message passing (of prediction errors) in schizophrenia—and the false perceptual inferences that ensue.
Similar content being viewed by others


INTRODUCTION
Coordinated oscillations in membrane potentials provide a basis through which information can be integrated across neuronal populations at multiple timescales (Varela et al, 2001; Fries, 2005). For example, numerous investigations of prefrontal and hippocampal circuits in both rodents and humans highlight the importance of theta (4–10 Hz) and gamma (32–150 Hz) rhythms and their interaction for the successful integration of both spatial and non-spatial information during goal-directed behaviors (Jones and Wilson, 2005; Sirota et al, 2008; Anderson et al, 2010; Benchenane et al, 2010; Hyman et al, 2010; Womelsdorf et al, 2010; Brockmann et al, 2011). These oscillatory rhythms emerge from interactions between excitatory glutamatergic and inhibitory GABAergic neurotransmission, which are in turn subject to behavior-dependent neuromodulation. Hence, as features of circuit activity measurable in both animal models and in clinical neurophysiology, they offer a translational bridge between molecular-synaptic substrates and associated behavior. Dynamic causal modeling (DCM) is also a powerful means to interpret the fMRI data increasingly available from animal and human studies, for example, Gass et al (2014).
It is rarely possible to directly measure synaptic effects and neural network effects simultaneously in conscious subjects. Here, we therefore apply DCM (Moran et al, 2011) to infer the synaptic basis of ketamine-induced theta–gamma derangements in the interconnected neural circuits of the rat hippocampus and prefrontal cortex. Given that ketamine’s effects at NMDA receptors (NMDARs) are known to impact downstream receptor signaling—for example, by enhancing AMPA-mediated responses (Moghaddam et al, 1997)—we used DCM in order to identify multivariate changes in the circuit. Specifically, we applied different DCMs for cross-spectral densities (Friston et al, 2012) to characterize hippocampal and prefrontal neural network oscillations in terms of biologically realistic models comprising interacting neural ensembles. Applying and comparing different models allows one to estimate the most plausible changes to synaptic parameters following ketamine administration. For example, a model could allow ketamine to alter prefrontal NMDARs on inhibitory cell populations, or excitatory cell populations, or both; using spectral analysis of local field potential data to provide probabilistic evidence for these competing scenarios, the most likely changes under ketamine can be deduced for an active prefrontal–hippocampal circuit. In other words, estimates of synaptic responses within each neuronal ensemble (Moran et al, 2011), the effective connectivity between ensembles in interconnected brain regions (Friston et al, 2012), and how these parameters are altered under pharmacological challenge (Moran et al, 2011) can be inferred, given a realistic biophysical model, and empirical data. Importantly, as DCM has been widely applied in human neuroimaging studies (Friston et al, 2013), the approach presented here is amenable to human schizophrenic patient studies, where similar oscillatory abnormalities have been observed (Basar-Eroglu et al, 2007; Uhlhaas and Singer, 2010).
The disconnection hypothesis of schizophrenia (Friston, 1998) pointed to abnormalities in the modulation of NMDAR-dependent plasticity (connectivity) by dopamine and/or acetylcholine. Individual differences in the expression of this pathophysiology may explain the spectrum of symptoms, clinical outcome, and treatment response across schizophrenic patients (Stephan et al, 2009). From a predictive coding perspective (Dolan et al, 1999; Corlett et al, 2007; Fletcher and Frith, 2009; Adams et al, 2013), a malfunction of neuromodulation (eg, dopaminergic transmission) is expected to cause a shift in the relative precision of regional inputs, resulting in a predominance of bottom-up, sensory-driven signals, which report sensory prediction errors (Adams et al, 2013). This shift has been proposed as an early (prodromal) deficit that underlies abnormal learning over longer-time periods, in order to accommodate (explain away) unexpected bottom-up inputs (Corlett et al, 2011). Ketamine has been shown to disrupt neuronal connections responsible for reporting prediction errors (Schmidt et al, 2013) and has been proposed as a model of early-stage disease processes (Corlett et al, 2007).
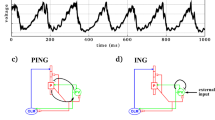
Recent developments in DCM provide biophysical models with the necessary architecture to test these predictive coding theories (Bastos et al, 2012). The models now incorporate separate populations of pyramidal cells to represent the main source of forward and backward connections, respectively (Douglas and Martin, 1991). They also contain glutamatergic receptors of different types (NMDARs and AMPARs; Moran et al, 2011). Thus, sensory-bound and prediction-related signals emanating from lower and higher-level cortical regions can be disentangled. In addition, whether ketamine has distinct effects on prefrontal (top-down) and hippocampal (bottom-up) connectivity can be assessed given the distinct afferent and efferent pyramidal cell model (Figure 1a).

Dynamic causal Modeling. (a) Top: the neural mass model used to represent each source. Three neuronal sub-populations represent each source, with two pyramidal cell populations. Glutamatergic within source (intrinsic: gray arrows) and between source (extrinsic: gray arrows) connections are mediated by AMPARs and NMDARs. Intrinsic GABAergic currents (red arrows) are mediated through GABAA receptors. In our model, comparison of 10 possible effects of ketamine were tested (blue text): all models allowed for extrinsic NMDA and AMPA mediation. Models 1–5 allowed extrinsic modulation at pyramidal cells, whereas models 6–10 allowed for extrinsic modulations at pyramidal cells and interneurons. Within these groups, models were tested which had ketamine effecting either inhibitory connections intrinsically (models 1, 2, 6, and 7) or local feedback inhibition onto particular cells types (models 3, 4, 5, 8, 9, and 10). All models included intrinsic ketamine effects at NMDARs. Bottom: tetrode electrodes in dorsal CA1 and medial PFC were analyzed using DCM. Cross-spectral densities were computed and served as data features to optimize a network model comprising a multi-layered hippocampal formation and mPFC. (b) Bayesian model comparison was used to assess the intrinsic modulation that best described ketamine-induced changes in spectral (theta and gamma) responses. Model 5, the winning model included parametric dose effects at intrinsic NMDAR channels and of local feedback inhibition at the output pyramidal cell layer (in dCA1 in HPC and deep layers of mPFC). Extrinsic connections in model 5 targeted the pyramidal sub-populations only. (c) Locomotion measured for five animals in the open-field arena over four dose levels (vehicle, 2, 4, 8 mg/kg). Significant increases in locomotive activity were evident at 8 mg/kg compared with a dose level of 2 mg/kg and compared with vehicle; **P<0.05 corrected Wilcoxon rank sum test.
Here, we examine the synaptic mechanisms of alterations in CA1-mPFC population activity under ketamine in behaving rats. Using high temporally and spatially resolved local field potential data, we investigate whether our models can identify ketamine-induced changes in spectral fingerprints and propose a plausible circuit reorganization underlying these data features.
MATERIALS AND METHODS
Electrophysiological Recordings
Experiments were performed on 10 adult male Long–Evans rats (320–450 g; Harlan, UK), and were conducted in accordance with the Animals (Scientific Procedures) Act, 1986 (UK) and the University of Bristol Ethics Committee. Rats were anesthetized with isoflurane and implanted with arrays of adjustable tetrodes targeted to the mPFC (+3.2 mm, +0.6 mm relative to bregma) and ipsilateral CA1 of the dorsal hippocampus (−3.6 mm, +2.0 mm). Following recovery, tetrodes were advanced daily over the course of approximately 7 days until they were at the desired depth based on neurophysiological hallmarks (pyramidal cell spiking and ripple activity for CA1). Recording locations were verified in a subset of animals by post-mortem electrolytic lesioning of tissue around the tip of each tetrode.
Before recording, rats were habituated to the recording environment, a walled circular arena of 1.5 m diameter. During recording, tetrodes were connected to a Digital Lynx recording system (Neuralynx, MT) via a HS-36 unity gain headstage and counterbalanced fine-wire tether. Differential recordings of LFP (sampled at 2.034 kHz per channel and filtered between 1 and 475 Hz) and unit activity (sampled at 32 kHz and filtered between 600 and 6000 Hz) were made using Cheetah software (Neuralynx, MT). mPFC recordings were referenced against electrodes implanted in superficial areas of the mPFC that exhibited no large amplitude spiking activity, whereas dCA1 signals were referenced against electrodes positioned in the overlying white matter. Ground wires were connected to screws attached to the skull over the cerebellum.
Ketamine Administration
Following connection to the recording system, baseline recordings were made for at least 30 min before intraperitoneal administration of a saline vehicle or (±)-ketamine (Vetalar V, Pfizer Animal Health). Recordings were then continued for 60–90-min post-injection, but we restricted our analyses to 5-min epochs recorded 10–15 min after each injection, when ketamine had its peak effects on LFP power. Rats were allowed to move freely in the familiar circular open field throughout. Five animals received only saline vehicle plus an 8 mg/kg ketamine dose; a further five animals received saline vehicle and ketamine at doses of 2, 4, 8, and 30 mg/kg. For all animals, gaps of at least 48 h were imposed between injections and the order in which different doses were received was randomized. For the current analysis, we collapsed within dose levels (across animals) after spectral estimation and model a single grand-averaged data set.
Data Pre-Processing: Evaluating Cross-Spectral Densities
For each recording, LFP analyses were performed on data recorded from single channels in mPFC and CA1. For the mPFC, this channel was selected on the basis of its projected depth calculated from the number of turns the tetrode was advanced, whereas the CA1 channel was selected based on maximal ripple amplitude.
From each tetrode channel, we extracted contemporaneous prefrontal and hippocampal time series. These were first downsampled to 200 Hz and bandpass filtered from 1 to 85 Hz using the preprocessing tools in SPM12 http://www.fil.ion.ucl.ac.uk/spm/. For each animal, data were then organized into 10-s epochs. From these epochs, we constructed grand-averaged spectral responses. In the gamma band, we obtained complex cross-spectra: from 40 to 75 Hz at 1-Hz resolution for vehicle, 2, 4, 8, and 30 mg/kg. In the theta band, we obtained complex cross-spectra: from 2 to 10 Hz at 1-Hz resolution. We obtained these for vehicle, 2 and 4 mg/kg doses to minimize the confound of locomotive effects contributing to theta power changes (see Results section). These formed the data features for the subsequent inversion of DCMs.
DCM of Cross-Spectra in Prefrontal-Hippocampal Circuit
DCM uses a biophysical model of neuronal responses based on neural mass models (David et al, 2006; Marreiros et al, 2009) to predict electrophysiological data. Synaptic responses are modeled within hippocampal and prefrontal regions (at pyramidal cells and inhibitory interneurons) and between prefrontal and hippocampal regions (at afferent pyramidal cells). Model inversion means optimizing the parameters of a particular model (Figure 1a) to best predict a given data set. We apply model inversion to the cross-spectral data features described above to optimize the parameters of different receptor-mediated synaptic responses. Model inversion returns the optimized neural mass model parameters and also an approximation to the probability of the model itself. This approximation (given uniform prior probabilities over models) is used to directly evaluate the overall circuit architecture that best explains the data at different doses of ketamine.
We tested 10 possible model architectures for generating theta and gamma spectra using an adapted ‘CMM_NMDA’ neural mass model as implemented in the DCM suite in SPM12 (http://www.fil.ion.ucl.ac.uk/spm/). All DCMs used conductance-based neural mass models representing hippocampal and prefrontal sources (David et al, 2004; Moran et al, 2011) with three cell sub-populations. For both sources, two pyramidal cell populations and one population of inhibitory interneurons were used (with pyramidal cells representing superficial and deep layer ensembles in PFC and perforant path, CA3 and CA1 ensembles in HPC) (Figure 1a).
In summary, we tested two broad types of extrinsic architectures in which extrinsic connections (ie, afferent input originating outside mPFC or dCA1) targeted both pyramidal cells and inhibitory interneurons, or just pyramidal cells. Within these two families, we allowed ketamine to modulate NMDARs targeted by extrinsic and intrinsic connections, in addition to modulating one of five local feedback inhibitory connections.
Differences in model architectures were expressed as different extrinsic (between region) connections and different possible ketamine modulations intrinsic to each region. Specifically, given that neurons in CA1 send direct projections to mPFC (Swanson 1981), whereas deep cells in mPFC excite CA1 indirectly via a polysynaptic (perforant) pathway, five models (models 1–5) were considered, where only pyramidal cells were excited by extrinsic glutamatergic connections. In contrast, models 6–10 were constructed such that the glutamatergic long-range connections excited both pyramidal cells and inhibitory interneurons in the respective target region (Tierney et al, 2004; Figure 1a). In all models, extrinsic connections affected both AMPARs and NMDARs, which we modeled separately.
We also tested competing mechanisms for how ketamine would affect synaptic processes intrinsic to the two regions. Although all our models considered a parametric effect of ketamine dose on intrinsic NMDARs, we tested whether ketamine also affected local inhibitory dynamics (red arrows, Figure 1a). Specifically, models 1 and 6 allowed for ketamine-induced changes in feed-forward inhibition from GABAergic interneurons to output pyramidal cells. Models 2 and 7 allowed for ketamine-induced changes in feed-back inhibition from GABAergic interneurons to input pyramidal cells. The remaining models represented changes in local feedback inhibition. These feedback inhibitory connections onto pyramidal cell populations represent the inverse gain or signal-to-noise ratio (SNR) of each sub-population, where high values represent low SNR and diminished correlated activity among constituent neurons. Models 3 and 8 tested for ketamine effects on local feedback inhibition at input pyramidal cells, models 4 and 9 tested for ketamine effects on local feedback inhibition at inhibitory interneurons, and finally, models 5 and 10 tested for ketamine effects on local feedback inhibition at the output pyramidal cell population.
Our lead field (the mapping from membrane potentials to channel recordings) was constructed with a priori weights such that output cells—representing dCA1 and deep pyramidal cells in mPFC—contributed 100% of the recorded signal.
Bayesian Model Inversion and Selection
Model inversion involved fitting each competing model to the cross spectral densities in the theta and gamma frequency domains. A standard variational (Laplace) Bayesian scheme was used to approximate the conditional density over parameters by maximizing a negative free energy bound on log-model evidence (Friston et al, 2007). This inversion was used to assess which models (1–10) best explained the theta and gamma changes. The negative free energy was used for model selection using a fixed effects analysis to pool evidence from the theta and gamma responses (Stephan et al, 2009). Given the winning model, the posterior densities of the ketamine-dependent modulations were assessed for their consistency in describing theta and gamma changes. Below, we report the parametric modulations, that is, modulations weighted by the dose of ketamine at each recording, in terms of the posterior density over the direction of the drug effect. In summary, the effects of ketamine were modeled with eight parameters; modulations of extrinsically driven AMPA and NMDA responses (four modulatory parameters), intrinsically driven NMDA responses (two parameters), and intrinsically driven GABAergic/gain responses (two parameters).
RESULTS
Behavior in Open Field
Locomotor activity levels in the open-field arena were measured from video during the 5-min post-injection recording periods. Increases in movement speed over four dose levels (vehicle, 2, 4, 8 mg/kg; n=5) were observed, but significant increases in locomotive activity were only evident following 8 mg/kg (median speed 11.4±2.1 m/s) compared with a dose level of 2 mg/kg (5.0±1.2 m/s) and vehicle (5.0±0.7 m/s) P<0.05 corrected Wilcoxon rank sum test. These behavioral effects are presented in Figure 1c and illustrate progressive increases in locomotion with increasing ketamine dose.
Model Structure for Theta and Gamma Cross-Spectra
We investigated spectral responses in mPFC and dorsal hippocampus in the theta and gamma frequency ranges, as oscillations at these frequencies and in these structures are consistently implicated in schizophrenia—and have been previously shown to be modified by ketamine (Lazarewicz et al, 2010; Uhlhaas, 2013). Ketamine administration simultaneously (1) enhanced gamma activity, with effects observed throughout the network but with greater power changes in mPFC (Figure 2a) and (2) reduced theta activity throughout the network, particularly in the hippocampal formation, culminating in profoundly reduced dCA1-mPFC cross-spectral density in the theta frequency range (Figure 2b).

Cross-spectral densities and model fits. (a) Grand-averaged data (n=10) in the gamma band (dashed line) were computed from 40 to 75 Hz. Ketamine dose (black lines: vehicle, navy lines: 2 mg/kg, cyan lines: 4 mg/kg, green lines: 8 mg/kg, red lines 30 mg/kg) increases gamma power in both auto and cross-spectral measures. DCM fits (full line) recapitulated this key feature of ketamine modulation. (b) Theta band cross-spectral densities from 2 to 10 Hz were computed for vehicle (n=10) and ketamine doses at 2 (n=5) and 4 (n=5) mg/kg (dashed lines). In contrast to gamma, these densities showed a profound decrease in power in both regions, particularly in the hippocampus. DCM fits captured this theta effect (solid line).
To characterize the neuronal mechanisms underlying these changes in cross-spectra, we examined model properties across 40–75 Hz and from 2 to 10 Hz. Models comprising different extrinsic connectivity (Gabbott et al, 2002) and different intrinsic effects of ketamine were inverted. Bayesian model comparison revealed that the best model for explaining the data across theta and gamma ranges was model 5, which proved superior to the next best model (model 1) by a log-Bayes factor>2000 (Kass and Raftery, 1995) corresponding to a posterior model probability of almost 100% (Figure 1b). Model 5 comprised extrinsic connections (eg, glutamatergic afferents to mPFC) that affected only pyramidal cells (models 6–10 consisted of region-to-region connections that affected both pyramidal cells and inhibitory interneurons). Within a region, model 5, allowed for ketamine modulation of local feedback inhibition on output pyramidal cells (in dCA1 and deep layers of mPFC) as well as ketamine modulation of intrinsic NMDA responses (present in all models). Thus, ketamine altered three separable aspects of prefrontal–hippocampal circuitry: reciprocal drive between dCA1 and mPFC pyramidal cells, signaling by output from dCA1 and mPFC pyramidal cells, and NMDAR-mediated drive within dCA1 and mPFC.
The optimal model here—model 5, represented the best balance of accuracy and complexity (Penny et al, 2004), accurately recapitulating ketamine’s key effects on gamma and theta rhythmicity. Specifically, in the gamma band, parameter changes produced power and frequency alterations in auto- and cross-spectral measures (Figure 2a). Enhanced power was reproduced by the modulation of extrinsic and intrinsic responses, while also mimicking the reduced frequency at high (30 mg/kg) doses observed in prefrontal autospectra and cross-spectra. In the theta range, model fits showed similarly strong correspondence to the empirical data (Figure 2b), where spectral power reduced according to the parametric effects on synaptic model parameters. Here the optimization was constrained to doses <8 mg/kg where locomotive effects were not significantly different from vehicle—as theta frequency and power both increase with running speed under physiological conditions (McFarland et al, 1975). We included dose level 4 mg/kg despite some animals exhibiting heightened locomotive activity (Figure 1c). However, our models were computed on pooled data and so the significant 8 mg/kg level was used as a cutoff.
Parametric Effects of Ketamine on Extrinsic Cortico-Cortical Connections
Given that model 5 had been selected on the basis of both theta and gamma frequency-range responses, we next tested for common modulatory effects of ketamine. In other words, we sought parameters that showed a positive or negative modulation for both the theta suppression and gamma enhancement effects. We studied the eight modulatory parameters comprising: (i) extrinsic connections from deep prefrontal pyramidal cells driving HPC AMPARs, (ii) extrinsic connections from CA1 pyramidal cells driving mPFC AMPARs, (iii) extrinsic connections from deep prefrontal pyramidal cells driving HPC NMDARs, (iv) extrinsic connections from CA1 pyramidal cells driving mPFC NMDARs, (v and vi) intrinsic excitatory responses at NMDARs in both regions, and (vii and viii) local feedback inhibition of output cells in CA1 and mPFC. To assess the significance of each modulatory effect, we used the posterior density following Bayesian parameter averaging over both frequency bands. Below, we report effects with a 95% or more posterior confidence. For completeness, we also report the posterior probability for each frequency band to illustrate their consistency.
Given ketamine’s potential NMDA-blocking and AMPA-promoting properties at cortical synapses, we parameterized these effects separately. Consistent (theta and gamma optimized) effects were observed for NMDAR-mediated top-down control. Specifically, the input to the hippocampal source was modulated by ketamine dose at NMDARs only, with a posterior probability of 100% (100% in the gamma range model and 69% in the theta range model). This modulation reduced NMDAR-mediated (Figure 3a) inputs from 100% (vehicle) to 96% (2 mg/kg) and 92% (4 mg/kg) in the theta model and to 78% (2 mg/kg), 62% (4 mg/kg), 38% (8 mg/kg), and 3% (30 mg/kg) in the gamma model (Figure 3a). The Bayes-averaged estimate (Stephan et al, 2009) reflected this top-down NMDAR-mediated reduction, and also revealed a significant (>95% probable) reduction in top-down AMPAR-mediated control, although these were not consistent across data features and emerged from the gamma model only.

Parametric effects of ketamine on cortico-hippocampal connections. (a) Consistent (positive or negative) modulatory effects of theta and gamma optimized parameters were investigated. Overall top-down connections were reduced and bottom-up connections increased (at fast ionotropic receptors) under ketamine. Parametric effects of ketamine that were consistent across theta and gamma ranges, included (top two panels, black bars) NMDAR-mediated responses, which decreased parametrically with dose from HPC to mPFC and from mPFC to HPC. In contrast, AMPAR-mediated forward connection from CA1 to mPFC increased with increasing ketamine dose (bottom panel, green bars). (b) Consistent modulations of intrinsic connections by ketamine. Only hippocampal parameters were consistent across the two data ranges. These exhibited a dose-dependent decrease in intrinsically excited NMDARs (top panel, black bars). This was accompanied by an increase in local feedback inhibition onto CA1 pyramidal cells (red bars). This latter effect is a lumped representation of several putative synaptic mechanisms, including GABAergic effects, dopaminergic neuromodulation, and hyperpolarizing currents.
Conversely, bottom-up connectivity was altered asymmetrically at AMPA and NMDARs (Figure 3a). Here, for the theta and gamma models, the drive to mPFC from CA1 was enhanced at AMPARs but depressed at NMDARs under ketamine. Marked AMPA-based forward enhancements were found for both data ranges; with theta-expressing increases to 136% (2 mg/kg) and 184% (4 mg/kg) relative to 100% vehicle baseline with a posterior probability of 99% and with gamma-expressing increases to 104% (2 mg/kg), 108% (4 mg/kg), 117% (8 mg/kg), and 183% (30 mg/kg) relative to vehicle baseline with a posterior probability of 91% (Figure 3a). Feed-forward connectivity mediated via NMDARs was, in contrast, reduced: reductions were evident in the theta model at 47% (2 mg/kg) and 22% (4 mg/kg) relative to 100% vehicle baseline with a posterior probability of 100%; in the gamma model levels of connectivity dropped to 94% (2 mg/kg), 89% (4 mg/kg), 78% (8 mg/kg), and 40% (30 mg/kg) relative to vehicle baseline with a posterior probability of 98% (Figure 3a).
Parametric Effects of Ketamine on Intrinsic Regional Connections
In order to quantify changes in intrinsic (within region) excitability across drug doses, we performed a similar analysis for the parametric modulation of local NMDAR-mediated transmission and the local feedback inhibition on output pyramidal cells. Consistent modulation was only observed for the hippocampal source (Figure 3b). Here, as expected, the parameter describing parametric effects on intrinsic NMDAR drive was reduced across doses to 61% (2 mg/kg) and 37% (4 mg/kg) relative to 100% vehicle baseline in the theta model and, in the gamma model to 73% (2 mg/kg), 54% (4 mg/kg), 29% (8 mg/kg), and 1% (30 mg/kg). Both effects and their Bayesian parameter average had a posterior probability of 100% (Figure 3b). Downstream from these effects, we observed a consistent upregulation of the local feedback inhibition representing reduced SNR at the output of the hippocampus. In the theta model, the enhancement effect was large, exhibited at a level of 423% (2 mg/kg) and 1793% (4 mg/kg) relative to 100% vehicle baseline with a posterior probability of 100%; and in the gamma model a smaller increase to 108% (2 mg/kg), 116% (4 mg/kg), 136% (8 mg/kg), and 320% (30 mg/kg) with a posterior probability of 0.99 was observed (Figure 3b).
DISCUSSION
Our results implicate a hierarchical asymmetry in cortico-hippocampal circuits induced by ketamine: enhanced gamma and suppressed theta rhythmicity is accompanied by reduced net drive from mPFC to dCA1 and increased dCA1-to-mPFC drive via AMPA receptors. This DCM-derived analysis supports and extends previous models of hippocampal (Lisman and Buzsáki, 2008; Lisman, 2012) and prefrontal (Yang et al, 1999) dysregulation as a factor in the pathophysiology of schizophrenia by linking ketamine’s effects on specific classes of receptors and synapses to network-level oscillatory activity of CA1-mPFC local field potentials.
Aberrant neural oscillations have been suggested to represent a core feature of the pathophysiology of schizophrenia (Uhlhaas and Singer, 2006, 2010; Sun et al, 2011), although the direction and extent of changes in power and covariance across theta and gamma frequencies are highly dependent upon patient, brain region, and behavioral context. Reductions in gamma synchrony among disparate brain areas have been observed in patient populations (Lee et al, 2003; Mulert et al, 2011) and have been proposed as a framework for understanding inappropriate sensory binding (Lee et al, 2003; Uhlhaas and Singer, 2010). Consistent with rodent data, ketamine has been reported to augment gamma oscillations, while reducing lower frequency (1–5 Hz) oscillations in humans (Hong et al, 2009). In light of recent suggestions that stochastic bursts of gamma may act to synchronize neuronal activity (Xing et al, 2012), sustained and elevated gamma power induced by ketamine may constitute ‘noise’, disrupting information processing by pyramidal cell assemblies. This effect may also be reflected by disrupted phase relationships between theta and gamma rhythms following ketamine (Caixeta et al, 2013).
The net effects of systemic ketamine on CA1-mPFC circuits result from a complex array of direct and indirect actions on glutamatergic neurotransmission, alongside associated disruption of GABAergic, dopaminergic, serotonergic, and/or cholinergic mechanisms (Kapur and Seeman, 2001; Moghaddam, 2003; Corlett et al, 2007; Ma et al, 2012; Moghaddam and Krystal, 2012). In particular, the ‘disinhibition’ model of ketamine’s actions points to a reduced NMDAR-mediated drive of GABAergic interneurons, which—together with enhanced glutamate release—triggers pyramidal neuron hyperactivity (Homayoun and Moghaddam, 2007). This model is supported by recent work showing elevated gamma rhythmicity, reduced theta rhythmicity, and attenuated sensitivity to NMDA-R antagonists in mice lacking functional NMDA-R selectively on parvalbumin-expressing interneurons (Korotkova et al, 2010; Carlen et al, 2012). However, we did not find a preferential effect of ketamine on interneurons, instead observing the largest intrinsic effects to occur at pyramidal cells, where ketamine reduced input reliability by boosting local feedback inhibition (Figure 3b).
This apparent discrepancy may arise because chronic NMDA-R knockdown is likely to culminate in different effects on network activity than acute NMDA-R blockade. Also, our models do not attempt to capture the full interneuronal diversity of CA1-mPFC networks. For example, the hippocampus incorporates circuit where local inhibitory processing can exhibit diverse effects and recent work has shown that ketamine differentially impacts theta oscillations along the septotemporal axis of rat hippocampus (Hinman et al, 2013). However, like knockout of NMDA-R on parvalbumin-expressing interneurons, pyramidal neuron-selective ablation of functional NMDA-R also leads to enhanced CA1 gamma rhythms in addition to impaired CA1 pyramidal cell spatial information coding (Cabral et al, 2014; McHugh et al, 1996). These latter results are in keeping with our DCM-based predictions, and suggest that the relative contributions of NMDA-R at different synaptic locations to ketamine-induced activity remain to be fully resolved.
In the hippocampus, we find that a decrease in intrinsic NMDAR-mediated excitation is accompanied by a ketamine-induced enhancement of local inhibitory feedback onto pyramidal cells in CA1. Although the first intrinsic effect is expected given ketamine’s NMDAR-blocking properties, the secondary modulation of output cells—that give rise to ‘bottom-up’ signals to mPFC—is less well established. Notably, it is the strongest ketamine-induced effect we observed. The model parameters determining local feedback strength are an amalgamated representation designed to capture multiple potential mechanisms that underpin synaptic gain. The ketamine-induced increase in these parameters could reflect enhanced local interneuronal input, hyperpolarizing currents (eg, through blockade of HCN1 channels; Chen et al, 2009) or dopaminergic modulation at D2 receptors (Becker et al, 2003), and is consistent with a recent report showing that ketamine can enhance gamma oscillations by slowing the decay kinetics of GABAA receptor-mediated IPSCs (McNally et al, 2011). In our model, enhanced local feedback inhibition would predict impaired coordinated activity amongst coactive pyramidal cells following ketamine—this should be tested directly, but is consistent with reduced correlated activity following knockout of NMDAR from CA1 pyramidal cells (McHugh et al, 1996). Direct measures of dCA1 interneuronal firing rates following ketamine would also be useful in testing our predictions.
Extrinsically, our connectivity results were predicted by theoretical considerations (Adams et al, 2013) and corroborate a recent fMRI-based analysis showing increased cortico-cortical and CA1-PFC interactions following ketamine in rat (Gass et al, 2014). Corlett et al (2011) place the multi-faceted effects of ketamine on network interactions within a predictive coding framework, prescribing a Bayesian model by which synaptic dysfunction can be linked to symptoms of psychotic illness. They argue that ketamine ‘deranges the ability’ of neurons to specify prior expectations about inputs (NMDAR-mediated top-down dysfunction) and leads to over-responding to violations or errors in predictions with new learning (AMPAR-mediated bottom-up dysfunction), instantiating a cycle of disruptive belief formation and false inference. This may be reflected by NMDAR antagonist-induced impulsive behavior (Gastambide et al, 2013). Our results support this mechanistic description, and furthermore show that reduced NMDA effects may also have a role in aberrant hippocampal signaling to prefrontal regions. Our analysis of these acute effects of ketamine may be a more appropriate analogy of the prodromal phase of schizophrenia (Honey et al, 2008) rather than later, at disease manifestation. This is consistent with recent imaging studies in rodents, which also suggest aspects of frontal hyperconnectivity following acute ketamine (Gass et al, 2014; Dawson et al, 2014).
In the neurobiological accounts of hierarchical cortical message passing using predictive coding, backward connections from deep pyramidal cells deliver the prediction of expectations in lower regions, while superficial pyramidal cells (or here, pyramidal cells in the CA1 output region) report prediction errors from these lower regions (Friston, 2005, 2010). In our models, we retain the key elements of this message passing, in particular the distinction between afferent and efferent pyramidal populations. Top-down signals from regions higher in the cortical hierarchy have recently been shown to subserve conscious processing in an analysis of patients suffering disorders of consciousness (Boly et al, 2011). A similar feature emerged under propofol anesthesia in healthy controls, where a necessary mediator of loss-of-consciousness was a decrease in backward cortico-cortical connectivity from frontal to parietal cortices (Boly et al, 2012). The effects observed here share similar connectivity profiles, with increasing doses of ketamine inducing a parametric decrease in top-down connectivity, which emerges with enhanced, fast bottom-up signals. (Kapur, 2003; Corlett et al, 2007; Fletcher and Frith, 2009; Roiser et al, 2009).
In summary, we find that ketamine-induced disruption of local glutamatergic and GABAergic signaling in CA1 and mPFC culminate in attenuation of top-down control accompanied by an amplification of fast ionotropic bottom-up signaling. These synaptic effects may underlie a functional cortico-hippocampal dysconnection that is dominated by overly precise bottom-up prediction error signals. This fits comfortably with a failure to attenuate sensory precision and consequent false inference (and resistance to illusions) that attends the trait abnormalities of schizophrenia (Adams et al, 2013).
FUNDING AND DISCLOSURE
The authors declare that this work was funded in part by Merck & Co., Inc. through a BBSRC-CASE PhD studentship. The authors delcare no conflict of interest.
References
Adams RA, Stephan KE, Brown HR, Frith CD, Friston KJ (2013). The computational anatomy of psychosis. Front Psychiatry 4: 47.
Anderson KL, Rajagovindan R, Ghacibeh GA, Meador KJ, Ding M (2010). Theta oscillations mediate interaction between prefrontal cortex and medial temporal lobe in human memory. Cerebral Cortex 20: 1604–1612.
Basar-Eroglu C, Brand A, Hildebrandt H, Karolina Kedzior K, Mathes B, Schmiedt C (2007). Working memory related gamma oscillations in schizophrenia patients. Int J Psychophysiol 64: 39–45.
Bastos AM, Usrey WM, Adams RA, Mangun GR, Fries P, Friston KJ (2012). Canonical microcircuits for predictive coding. Neuron 76: 695–711.
Becker A, Peters B, Schroeder H, Mann T, Huether G, Grecksch G (2003). Ketamine-induced changes in rat behaviour: a possible animal model of schizophrenia. Prog Neuro-Psychopharmacol Biol Psychiatry 27: 687–700.
Benchenane K, Peyrache A, Khamassi M, Tierney PL, Gioanni Y, Battaglia FP et al (2010). Coherent theta oscillations and reorganization of spike timing in the hippocampal- prefrontal network upon learning. Neuron 66: 921–936.
Boly M, Garrido MI, Gosseries O, Bruno MA, Boveroux P, Schnakers C et al (2011). Preserved feed forward but impaired top-down processes in the vegetative state. Science 332: 858.
Boly M, Moran R, Murphy M, Boveroux P, Bruno MA, Noirhomme Q et al (2012). Connectivity changes underlying spectral EEG changes during propofol-induced loss of consciousness. J Neurosci 32: 7082–7090.
Brockmann MD, Pöschel B, Cichon N, Hanganu-Opatz IL (2011). Coupled oscillations mediate directed interactions between prefrontal cortex and hippocampus of the neonatal rat. Neuron 71: 332–347.
Cabral HO, Vinck M, Fouquet C, Pennartz CM, Rondi-Reig L, Battaglia FP (2014). Oscillatory dynamics and place field maps reflect hippocampal ensemble processing of sequence and place memory under NMDA receptor control. Neuron 81: 402–415.
Caixeta FV, Cornelio AM, Scheffer-Teixeira R, Ribeiro S, Tort AB (2013). Ketamine alters oscillatory coupling in the hippocampus. Sci Rep 3: 2348.
Carlen M, Meletis K, Siegle JH, Cardin JA, Futai K, Vierling-Claassen D et al (2012). A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol Psychiatry 17: 537–548.
Chen X, Shu S, Bayliss DA (2009). HCN1 channel subunits are a molecular substrate for hypnotic actions of ketamine. J Neurosci 29: 600–609.
Corlett P, Honey G, Fletcher PC (2007). From prediction error to psychosis: ketamine as a pharmacological model of delusions. J Psychopharmacol 21: 238.
Corlett PR, Honey GD, Krystal JH, Fletcher PC (2011). Glutamatergic model psychoses: prediction error, learning, and inference. Neuropsychopharmacology 36: 294–315.
Corlett PR, Murray GK, Honey GD, Aitken MRF, Shanks DR, Robbins TW et al (2007). Disrupted prediction-error signal in psychosis: evidence for an associative account of delusions. Brain 130: 2387–2400.
David O, Cosmelli D, Friston KJ (2004). Evaluation of different measures of functional connectivity using a neural mass model. NeuroImage 21: 659–673.
David O, Kiebel S, Harrison L, Mattout J, Kilner J, Friston K (2006). Dynamic causal modeling of evoked responses in EEG and MEG. NeuroImage 30: 1255–1272.
Dawson N, McDonald M, Higham DJ, Morris BJ, Pratt JA (2014). Subanesthetic ketamine treatment promotes abnormal interactions between neural subsystems and alters the properties of functional brain networks. Neuropsychopharmacology 39: 1786–1798.
Dolan RJ, Fletcher P, McKenna P, Friston K, Frith C (1999). Abnormal neural integration related to cognition in schizophrenia. Acta Psychiatrica Scandinavica 99: 58–67.
Douglas RJ, Martin K (1991). A functional microcircuit for cat visual cortex. Physiol 440: 735–769.
Fletcher PC, Frith CD (2009). Perceiving is believing: a Bayesian approach to explaining the positive symptoms of schizophrenia. Nat Rev Neurosci 10: 48–58.
Fries P (2005). A mechanism for cognitive dynamics: neuronal communication through neuronal coherence. Trends Cogn Sci 9: 474–480.
Friston K (2005). A theory of cortical responses. Philos Trans R Soc B Biol Sci 360: 815–836.
Friston K (2010). The free-energy principle: a unified brain theory? Nat Rev Neurosci 11: 127–138.
Friston K, Bastos A, Litvak V, Stephan K, Fries P, Moran R (2012). DCM for complex-valued data: cross-spectra, coherence and phase-delays. NeuroImage. 59: 439–455.
Friston K, Mattout J, Trujillo-Barreto N, Ashburner J, Penny W (2007). Variational free energy and the Laplace approximation. Neuroimage 34: 220–234.
Friston K, Moran R, Seth AK (2013). Analysing connectivity with Granger causality and dynamic causal modelling. Curr Opin Neurobiol 23: 172–178.
Friston KJ (1998). The disconnection hypothesis. Schizophr Res 30: 115–125.
Gabbott P, Headlam A, Busby S (2002). Morphological evidence that CA1 hippocampal afferents monosynaptically innervate PV-containing neurons and NADPH-diaphorase reactive cells in the medial prefrontal cortex (Areas 25/32) of the rat. Brain Res 946: 314–322.
Gass N, Schwarz AJ, Sartorius A, Schenker E, Risterucci C, Spedding M et al (2014). Sub-anesthetic ketamine modulates intrinsic BOLD connectivity within the hippocampal-prefrontal circuit in the rat. Neuropsychopharmacology 39: 895–906.
Gastambide F, Mitchell SN, Robbine TW, Tricklebank MD, Gilmour G (2013). Temporally distinct effects following acute administration of ketamine and phencyclidine in the rat. Eur Neuropsychopharmacol 23: 1414–1422.
Hinman JR, Penley SC, Escabí MA, Chrobak JJ (2013). Ketamine disrupts theta synchrony across the septotemporal axis of the CA1 region of hippocampus. J Neurophysiol 109: 570–579.
Homayoun H, Moghaddam B (2007). NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci 27: 11496–11500.
Honey GD, Corlett PR, Absalom AR, Lee M, Pomarol-Clotet E, Murray GK et al (2008). Individual differences in psychotic effects of ketamine are predicted by brain function measured under placebo. J Neurosci 28: 6295–6303.
Hong LE, Summerfelt A, Buchanan RW, O'Donnell P, Thaker GK, Weiler MA et al (2009). Gamma and delta neural oscillations and association with clinical symptoms under subanesthetic ketamine. Neuropsychopharmacology 35: 632–640.
Hyman JM, Zilli EA, Paley AM, Hasselmo ME (2010). Frontiers: working memory performance correlates with prefrontal-hippocampal theta interactions but not with prefrontal neuron firing rates. Front Integr Neurosci 4: 2.
Jones MW, Wilson MA (2005). Theta rhythms coordinate hippocampal–prefrontal interactions in a spatial memory task. PLoS Biol 3: e402.
Kapur S (2003). Psychosis as a state of aberrant salience: a framework linking biology, phenomenology, and pharmacology in schizophrenia. Am J Psychiatry 160: 13–23.
Kapur S, Seeman P (2001). NMDA receptor antagonists ketamine and PCP have direct effects on the dopamine D (2) and serotonin 5-HT (2) receptors-implications for models of schizophrenia. Mol Psychiatry 7: 837–844.
Kass RE, Raftery AE (1995). Bayes factors. J Am Stat Assoc 90: 773–795.
Korotkova T, Fuchs EC, Ponomarenko A, von Engelhardt J, Monyer H (2010). NMDA receptor ablation on parvalbumin-positive interneurons impairs hippocampal synchrony, spatial representations, and working memory. Neuron 68: 557–569.
Lazarewicz MT, Ehrlichman RS, Maxwell CR, Gandal MJ, Finkel LH, Siegel SJ (2010). Ketamine modulates theta and gamma oscillations. J Cogn Neurosci 22: 1452–1464.
Lee KH, Williams L, Haig A, Gordon E (2003). “Gamma (40 Hz) phase synchronicity” and symptom dimensions in schizophrenia. Cognitive Neuropsychiatry 8: 57–71.
Lee KH, Williams LM, Breakspear M, Gordon E (2003). Synchronous gamma activity: a review and contribution to an integrative neuroscience model of schizophrenia. Brain Res Rev 41: 57–78.
Lisman J (2012). Excitation, inhibition, local oscillations, or large-scale loops: what causes the symptoms of schizophrenia? Curr Opin Neurobiol 22: 537–544.
Lisman J, Buzsáki G (2008). A neural coding scheme formed by the combined function of gamma and theta oscillations. Schizophr Bull 34: 974–980.
Ma J, Tai SK, Leung LS (2012). Septohippocampal GABAergic neurons mediate the altered behaviors induced by n–methyl–D–aspartate receptor antagonists. Hippocampus 22: 2208–2218.
Marreiros A, Kiebel S, Daunizeau J, Harrison L, Friston K (2009). Population dynamics under the Laplace assumption. NeuroImage 44: 701–714.
McFarland WL, Teitelbaum H, Hedges EK (1975). Relationship between hippocampal theta activity and running speed in the rat. J Comp Physiol Psychol 88: 324.
McHugh TJ, Blum KI, Tsien JZ, Tonegawa S, Wilson MA (1996). Impaired hippocampal representation of spce in CA1-specigic NMDAR1 knockout mice. Cell 87: 1339–1349.
McNally JM, McCarley RW, McKenna JT, Yanagawa Y, Brown RE (2011). Complex receptor mediation of acute ketamine application on in vitro gamma oscillations in mouse prefrontal cortex: modeling gamma band oscillation abnormalities in schizophrenia. Neuroscience 199: 51–63.
Moghaddam B (2003). Bringing order to the glutamate chaos in schizophrenia. Neuron 40: 881–884.
Moghaddam B, Adams B, Verma A, Daly D (1997). Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci 17: 2921–2927.
Moghaddam B, Krystal JH (2012). Capturing the angel in “angel dust”: twenty years of translational neuroscience studies of NMDA receptor antagonists in animals and humans. Schizophr Bull 38: 942–949.
Moran RJ, Mallet N, Litvak V, Dolan RJ, Magill PJ, Friston KJ et al (2011). Alterations in brain connectivity underlying beta oscillations in Parkinsonism. PLoS Comput Biol 7: e1002124.
Moran RJ, Stephan KE, Dolan RJ, Friston KJ (2011). Consistent spectral predictors for dynamic causal models of steady-state responses. Neuroimage 55: 1694–1708.
Moran RJ, Symmonds M, Stephan KE, Friston KJ, Dolan RJ (2011). An in vivo assay of synaptic function mediating human cognition. Curr Biol 21: 1320–1325.
Mulert C, Kirsch V, Pascual-Marqui R, McCarley RW, Spencer KM (2011). Long-range synchrony of gamma oscillations and auditory hallucination symptoms in schizophrenia. Int J Psychophysiol 79: 55–63.
Penny W, Stephan K, Mechelli A, Friston K (2004). Comparing dynamic causal models. NeuroImage 22: 1157–1172.
Roiser JP, Stephan KE, den Ouden HE, Barnes TR, Friston KJ, Joyce EM (2009). Do patients with schizophrenia exhibit aberrant salience? Psychological Med 39: 199–209.
Schmidt A, Diaconescu AO, Kometer M, Friston KJ, Stephan KE, Vollenweider FX (2013). Modeling ketamine effects on synaptic plasticity during the mismatch negativity. Cerebral Cortex 23: 2394–2406.
Sirota A, Montgomery S, Fujisawa S, Isomura Y, Zugaro M, Buzsaki G (2008). Entrainment of neocortical neurons and gamma oscillations by the hippocampal theta rhythm. Neuron 60: 683–697.
Stephan KE, Friston KJ, Frith CD (2009). Dysconnection in schizophrenia: from abnormal synaptic plasticity to failures of self-monitoring. Schizophr Bull 35: 509–527.
Stephan KE, Penny WD, Daunizeau J, Moran RJ, Friston KJ (2009). Bayesian model selection for group studies. NeuroImage 46: 1004–1017.
Sun Y, Farzan F, Barr MS, Kirihara K, Fitzgerald PB, Light GA et al (2011). Gamma oscillations in schizophrenia: mechanisms and clinical significance. Brain Res 1413: 98–114.
Swanson L (1981). A direct projection from Ammon's horn to prefrontal cortex in the rat. Brain Res 217: 150–154.
Tierney PL, Dégenètais E, Thierry AM, Glowinski J, Gioanni Y (2004). Influence of the hippocampus on interneurons of the rat prefrontal cortex. Eur J Neurosci 20: 514–524.
Uhlhaas P, Singer W (2006). Neural synchrony in brain disorders: relevance for cognitive dysfunctions and pathophysiology. Neuron 52: 155–168.
Uhlhaas PJ (2013). Dysconnectivity, large-scale networks and neuronal dynamics in schizophrenia. Curr Opin Neurobiol 23: 283–290.
Uhlhaas PJ, Singer W (2010). Abnormal neural oscillations and synchrony in schizophrenia. Nat Rev Neurosci 11: 100–113.
Varela F, Lachaux JP, Rodriguez E, Martinerie J (2001). The brainweb: phase synchronization and large-scale integration. Nat Rev Neurosci 2: 229–239.
Womelsdorf T, Vinck M, Leung LS, Everling S (2010). Selective theta-synchronization of choice-relevant information subserves goal-directed behavior. Front Hum Neurosci 4: 210.
Xing D, Shen Y, Burns S, Yeh CI, Shapley R, Li W (2012). Stochastic generation of gamma-band activity in primary visual cortex of awake and anesthetized monkeys. J Neurosci 32: 13873–13880a.
Yang C. R., Seamans J. K., Gorelova N. (1999). Developing a neuronal model for the pathophysiology of schizophrenia based on the nature of electrophysiological actions of dopamine in the prefrontal cortex. Neuropsychopharmacology 21: 161–194.
Acknowledgements
RJM is supported by Virginia Tech Carilion Research Institute. KJF and RA are supported by the Wellcome Trust. KES acknowledges support by the René and Susanne Braginsky Foundation and KFSP ‘Multiple Sclerosis’. MWJ and AJB were supported by a BBSRC-CASE studentship and the UK Medical Research Council.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Moran, R., Jones, M., Blockeel, A. et al. Losing Control Under Ketamine: Suppressed Cortico-Hippocampal Drive Following Acute Ketamine in Rats. Neuropsychopharmacol 40, 268–277 (2015). https://doi.org/10.1038/npp.2014.184
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2014.184
This article is cited by
-
Multimodal electrophysiological analyses reveal that reduced synaptic excitatory neurotransmission underlies seizures in a model of NMDAR antibody-mediated encephalitis
Communications Biology (2021)
-
The M1/M4 preferring muscarinic agonist xanomeline modulates functional connectivity and NMDAR antagonist-induced changes in the mouse brain
Neuropsychopharmacology (2021)
-
In vivo electrophysiological recordings of the effects of antidepressant drugs
Experimental Brain Research (2019)
-
Estimating Directed Connectivity from Cortical Recordings and Reconstructed Sources
Brain Topography (2019)
-
Subunit-specific NMDAR antagonism dissociates schizophrenia subtype-relevant oscillopathies associated with frontal hypofunction and hippocampal hyperfunction
Scientific Reports (2018)
