
Abstract
Attentional bias to threat is a key endophenotype that contributes to the chronicity of trauma-related psychopathology. However, little is known about the neurobiology of this endophenotype and no known in vivo molecular imaging study has been conducted to evaluate candidate receptor systems that may be implicated in this endophenotype or the phenotypic expression of trauma-related psychopathology that comprises threat (ie, re-experiencing, avoidance, and hyperarousal) and loss (ie, emotional numbing, depression/dysphoria, generalized anxiety) symptomatology. Using the radioligand [11C]OMAR and positron emission tomography (PET), we evaluated the relationship between in vivo cannabinoid receptor type 1 (CB1) receptor availability in the amygdala, and performance on a dot-probe measure of attentional bias to threat, and clinician interview-based measures of trauma-related psychopathology. The sample comprised adults presenting with a broad spectrum of trauma-related psychopathology, ranging from nontrauma-exposed, psychiatrically healthy adults to trauma-exposed adults with severe trauma-related psychopathology. Results revealed that increased CB1 receptor availability in the amygdala was associated with increased attentional bias to threat, as well as increased severity of threat, but not loss, symptomatology; greater peripheral anandamide levels were associated with decreased attentional bias to threat. A mediation analysis further suggested that attentional bias to threat mediated the relationship between CB1 receptor availability in the amygdala and severity of threat symptomatology. These data substantiate a key role for compromised endocannabinoid function in mediating both the endophenotypic and phenotypic expression of threat symptomatology in humans. They further suggest that novel pharmacotherapies that target the CB1 system may provide a more focused, mechanism-based approach to mitigating this core aspect of trauma-related psychopathology.
Similar content being viewed by others

INTRODUCTION
Studies of how neurobiological systems are linked to the transdiagnostic endophenotypic and phenotypic expression of psychopathology (Cuthbert, 2014) are particularly relevant to trauma-related psychopathology, as three of the most common trauma-related disorders—posttraumatic stress disorder (PTSD), major depressive disorder (MDD), and generalized anxiety disorder (GAD)—are highly comorbid and share common transdiagnostic dimensions of threat and loss (ie, dysphoria) symptomatology (Forbes et al, 2011; Forbes et al, 2010; Grant et al, 2008; Zoellner et al, 2014). Trauma-related threat symptomatology includes intrusive thoughts and memories, and hyperarousal symptoms such as sleep disturbance and hypervigilance, whereas trauma-related loss (ie, dysphoria) symptomatology includes emotional numbing and depressive/dysphoric and generalized anxiety symptoms. Elucidation of neurobiological systems implicated in trauma-related endophenotypes can inform etiologic models of trauma-related psychopathology, as well as the development of more targeted, mechanism-based prevention and treatment strategies.
Attentional bias to threat is one of the core endophenotypic characteristics of trauma-related psychopathology (Fani et al, 2012b). Attentional biases to threatening information, such as faces and words, which are often assessed using a dot-probe paradigm, have been found to contribute to and maintain the persistence of trauma-related threat symptomatology, even months to years after trauma exposure (Fani et al, 2012b; Lindstrom et al, 2011; Sveen et al, 2009). Greater attentional bias to threat is also associated with exaggerated fear expression and impaired extinction in individuals with PTSD (Fani et al, 2012b). Hyperarousal symptoms, such as exaggerated startle response during fear learning, in particular, have been found to contribute to attentional bias to threat in symptomatic trauma survivors (Fani et al, 2012b). Recent functional neuroimaging work has implicated increased amygdala activation in relation to attentional bias to threat among individuals with PTSD (Fani et al, 2012a), suggesting that the amygdala modulates the orientation of attention toward and processing of threatening information in this population.
Although cannabinoid type 1 (CB1) receptors are widely distributed in the human brain (Glass et al, 1997a; Herkenham, 1991), they are found in particularly high concentrations in the amygdala, and have been associated with the processing and storage of threat-related memories, as well as the coordination of threat-related behaviors (LeDoux, 2000). Recently, we reported in vivo evidence of abnormal CB1 receptor-mediated endocannabinoid signaling in individuals with PTSD (Neumeister et al, 2013) and suggested that increased CB1 receptor availability may be a molecular adaptation to reduced endocannabinoid availability. In addition to this work, a large body of preclinical studies has found strong support for a major role of the endocannabinoid anandamide and CB1 receptor signaling in the amygdala in modulating stress-induced threat behaviors (for review, see Gunduz-Cinar et al, 2013a). Understanding how key neuroreceptor systems such as CB1 relate to intermediate endophenotypic (ie, attentional bias to threat) and phenotypic expression of trauma-related psychopathology (ie, threat symptomatology) may thus provide insight into molecular targets that could inform the development of mechanism-based treatment approaches. To date, however, human data evaluating this possibility are lacking.
In the current study, we aimed to address this gap in the literature by using the CB1 receptor antagonist radiotracer [11C]OMAR, which measures volume of distribution (VT) linearly related to CB1 receptor availability, to evaluate the relationship between CB1 receptor availability in the amygdala, and objectively assessed attentional bias to threat, and the transdiagnostic and dimensional expression of trauma-related threat and loss symptomatology. To obtain a sample that encompassed the full-dimensional range of study measures (Cuthbert, 2014), we employed an inclusive sampling approach by recruiting a sample of individuals who represented a broad transdiagnostic and dimensional spectrum of trauma-related psychopathology, ranging from healthy, nontrauma-exposed individuals to trauma-exposed individuals with severe trauma-related psychopathology. On the basis of prior work linking CB1 receptor availability in the amygdala to threat processing (Gunduz-Cinar et al, 2013a; LeDoux, 2000; Rodrigues et al, 2004; Rogan et al, 1997) and threat symptomatology (ie, hyperarousal) to attentional bias to threat (Fani et al, 2012b), we hypothesized that greater CB1 receptor availability in the amygdala would be associated with greater attentional bias to threat, as well as increased severity of threat symptomatology, particularly hyperarousal. We then evaluated a mediational model to examine whether attentional bias to threat mediated the relationship between CB1 receptor availability in the amygdala and trauma-related psychopathology.
MATERIALS AND METHODS
Participants
A total of 20 participants were recruited from the Molecular Imaging Program for Mood and Anxiety Disorders at NYU Langone Medical Center. Trauma-exposed participants were referred from NYU-affiliated outpatient psychiatry clinics (n=16) and psychiatrically healthy, nontrauma-exposed participants (n=4) were recruited from the community using public advertisements. Scores on clinician-administered measures of threat and loss symptomatology (see Assessments below) in the sample represented a broad transdiagnostic and dimensional spectrum of trauma-related psychopathology (see Table 1). This sample is thus representative of the broader population of individuals in the community (ie, unaffected individuals), as well as those who present for treatment at an outpatient mood and anxiety disorders clinic (ie, mild-to-severe symptomatology). The New York University Institutional Review Board, Yale University School of Medicine Human Investigation Committee, Yale University Magnetic Resonance Research Center, and Yale–New Haven Hospital Radiation Safety Committee approved this study. All participants provided written informed consent.
Assessments
Lifetime traumatic events were assessed using the Traumatic Life Events Questionnaire (TLEQ) and psychiatric diagnoses were established using DSM-IV-TR criteria and the Structured Clinical Interview for DSM-IV (SCID) that was administered by an experienced master- or doctoral-level psychiatric clinician. Only traumatic events that met criteria A1 and A2 for a DSM-IV-TR-based diagnosis of PTSD were counted toward participants’ trauma histories in this study. Nontrauma-exposed healthy adults did not report any trauma exposures on the TLEQ and did not have any lifetime psychiatric diagnosis, including substance abuse or dependence or nicotine dependence. Severity of trauma-related threat and loss symptomatology was assessed using the Clinician-Administered PTSD Scale for DSM-IV (CAPS); the Hamilton Rating Scale for Depression (HAM-D) to assess depressive symptoms; and the Hamilton Rating Scale for Anxiety (HAM-A) to assess nonspecific anxiety symptoms. Scores on these structured clinician-administered measures of trauma-related psychopathology represented a transdiagnostic and dimensional spectrum of trauma-related psychopathology, ranging from nontrauma-exposed asymptomatic adults to trauma-exposed adults with severe trauma-related psychopathology (see Table 1).
All participants were evaluated by physical examination, electrocardiogram, standard blood chemistry, hematology laboratory testing, toxicology testing, and urinalysis. All but two participants were psychotropic medication naive, and two took antidepressants for less than a week before the study but were medication free for at least 6 months before the study. Participants with significant medical or neurologic conditions, with substance abuse within 12 months of the scan, lifetime history of intravenous substance dependence, or with history of head injury that involved loss of consciousness were excluded from the study. Lifetime cannabis abuse/dependence was an exclusion criterion, and occasional cannabis users were eligible to participate but not if they had used cannabis within 12 months of the scan. The absence of substance use was determined by self-report and confirmed by the results of urine toxicology and breathalyzer tests at screening and on the days when magnetic resonance imaging (MRI) and positron emission tomography (PET) scans were conducted. The medical and psychiatric evaluation was followed by MRI and a resting-state PET scan on a High Resolution Research Tomograph (HRRT) PET scanner (Siemens/CTI, Knoxville, TN) with the CB1-selective radioligand [11C]OMAR (Horti et al, 2006). To obtain plasma anandamide levels, blood samples were collected at the time of tracer injection and processed immediately after collection in the laboratory that is adjacent to the scan room and frozen at −80 °C until analyzed, as previously described (Neumeister et al, 2013).
PET and MRI Acquisition and Modeling
[11C]OMAR was prepared in high specific activity (111±63 MBq/nmol at end of synthesis). The radiotracer (injected dose: 492±155 MBq, injected mass: 0.03±0.03 μg/kg) was infused over 1 min through the antecubital vein. The radioactivity concentration in blood from the radial artery was measured continuously using an automated system (PBS101, Veenstra Instruments, Joure, The Netherlands) for the first 7 min after radiotracer administration and manually drawn and counted thereafter. Discrete samples were acquired at selected times and measured on a gamma counter (Wizard 1480, Perkin-Elmer, Waltham, MA) to determine radioactivity concentration in whole blood and plasma. Five discrete blood samples (5, 15, 30, 60 and 90 min) were analyzed for the fraction of unchanged [11C]OMAR and its radiometabolites using a column-switching high-pressure liquid chromatography method (Hilton et al, 2000). The fraction of tracer unbound to plasma proteins was determined in triplicate by ultrafiltration. Listmode emission data were collected for 120 min after radiotracer administration using the HRRT (Siemens Medical Systems, Knoxville, TN), a dedicated brain PET scanner with spatial resolution better than 3 mm. Head motion was measured using the Polaris Vicra optical tracking system (Northern Digital, Waterloo, ON, Canada) and incorporated into PET image reconstruction with all corrections (Carson et al, 2003). The dynamic image sequences had 33 frames with the following number and duration: 6 × 30 s, 3 × 1 min, 2 × 2 min, and 22 × 5 min. The PET images were registered to subject-specific T1-weighted magnetic resonance images (256 × 256 × 176 grid of 1 mm isotropic voxels) acquired on a 3 Tesla Trio imaging system (Siemens Medical Systems, Erlangen, Germany). Anatomical MR images were in turn nonlinearly registered to an MR template where regions of interest (ROIs) were defined (Tzourio-Mazoyer et al, 2002). Regional time activity curves (TACs) were extracted from the dynamic PET data and analyzed using the multilinear analysis method (Ichise et al, 2002) with metabolite-corrected arterial input functions and cutoff time t*=30 min. The kinetic analysis yielded regional estimates of total volume of distribution (VT), the equilibrium ratio of radioligand concentration in tissue relative to arterial plasma (Innis et al, 2007) that is directly proportional to CB1 receptor availability. Additional details regarding PET and MRI acquisition and modeling are provided elsewhere (Neumeister et al, 2013). Although [11C]OMAR VT values in the amygdala were of primary interest in this study based on prior research (Gunduz-Cinar et al, 2013a; LeDoux, 2000; Rodrigues et al, 2004; Rogan et al, 1997); (Fani et al, 2012b), we also explored how [11C]OMAR VT values in other brain regions, including the caudate, putamen, pallidum, cerebellum, semiovale, hippocampus, hypothalamus, insula, anterior and posterior cingulate cortex, and occipital, parietal, temporal, and frontal cortices, were related to attentional bias to threat.
Dot-Probe Task
The dot-probe task (Bar-Haim et al, 2007; MacLeod et al, 1986) measures attentional biases toward or away from threatening stimuli. The dot-probe task was composed of 160 trials. Each trial started with a fixation cross (‘+’) presented in the center of the screen for 500 ms. When the fixation cross disappeared, two words in 12-pt Arial font immediately appeared in the center of the screen for 500 ms, one above and one below the location of the fixation cross, separated by 1.5 cm. Following the presentation of the words, a target probe (either a letter E or F) appeared in the location previously occupied by one of the words. The probe remained on the screen until participants responded, after which the next trial started. Participants were instructed to focus their attention on the fixation cross at the start of each trial, and when a probe appeared they were to identify the probe letter (E or F) using a designated mouse button, as quickly as possible. Given the heterogeneity of trauma histories in our sample, the stimuli used were 32 trauma-related and 64 neutral words that were selected from a larger list developed by MacLeod et al (2002). Word pairs were chosen for salience to the experience of traumatic life events (eg, ‘harm’, ‘suffer’). Word pairs were matched in terms of first letter, number of letters, and frequency of usage in the English language, as suggested by MacLeod et al (2002), and were presented in random order. Stimuli presentation and data collection used E-Prime software (Psychology Software Tools, Pittsburgh, PA). To reduce the effect of anticipatory responding and outliers, response times (RTs) <200 ms and >3 SD above the mean for each trial were discarded (Salemink et al, 2007). Attentional bias to threat was calculated as the difference between average RT to targets at neutral word locations and average RT to targets at threat word locations. Negative scores indicate attentional bias away from threat, whereas positive scores indicate attentional bias toward threat.
Data Analysis
Simple descriptive statistics were computed to summarize demographic, trauma-related, and clinical variables for the sample. To reduce symptom clusters into composite measures of trauma-related threat and loss symptomatology based on prior work (Forbes et al, 2010, 2011; Grant et al, 2008), we conducted two principal components analyses (PCAs): the first contained CAPS measures of re-experiencing and hyperarousal symptoms (ie, threat symptomatology), and the second contained CAPS measure of avoidance/numbing symptoms and HAM-D and HAM-A measures of major depressive and anxiety symptoms. Pearson or Spearman correlations, as appropriate based on data distributions, were then computed to evaluate associations between [11C]OMAR VT values in the amygdala, attentional bias to threat, and composite measures of trauma-related threat and loss symptomatology. If significant associations were observed, exploratory post hoc analyses were conducted to evaluate associations between component aspects of composite measures; exploratory post hoc analyses were also conducted to evaluate associations between [11C]OMAR VT values in brain regions other than the amygdala in relation to attentional bias to threat; α was set to 0.01 for all of these analyses to reduce the likelihood of type I error. To evaluate whether attentional bias to threat mediated the relation between CB1 receptor availability in the amygdala and the phenotypic expression of trauma-related psychopathology, we conducted a bootstrapped mediation analysis with 10 000 replicates using Mplus version 7.11. Model fit was assessed using χ2, comparative fit index (CFI), and standardized root mean square residual (SRMR) fit statistics; by convention, nonsignificant χ2 values, CFI values ≥0.90, and SRMR values <0.05 indicate a good fit to the data (Kline, 2010).
RESULTS
PCAs of Trauma-Related Threat and Loss Symptomatology
Two PCAs were conducted to compute a priori composite measures of trauma-related threat and loss symptomatology (Forbes et al, 2010, 2011Grant et al, 2008). The first PCA, which included CAPS measures of re-experiencing and hyperarousal symptoms yielded a 1-factor solution (eigenvalue=1.93; 96.4% of variance explained). Factor loadings were 0.982 for both component measures. The second PCA, which included CAPS measures of avoidance/numbing symptoms, and HAM-D and HAM-A measures of major depressive and anxiety symptoms, also yielded a 1-factor solution (eigenvalue=2.91; 97.1% of variance explained). Factor loadings were very high for all component measures: 0.982 for CAPS total scores; 0.984 for HAM-A total scores; and 0.990 for HAM-D-total scores. Composite measures of trauma-related threat and loss symptomatology were correlated (r=0.86, p<0.001).
Sample Characteristics
Table 1 shows demographic, trauma-related, and clinical characteristics of the sample. On average, the sample was 33.3 years of age, had slightly more female participants (55.0%), was equally Caucasian and non-Caucasian, and had a mean 15.4 years of education. Among the 16 trauma-exposed individuals, the mean age of first trauma was 13.4, mean number of lifetime traumas was 3.8, and the most commonly endorsed index trauma (ie, worst traumatic event according to the participant) was sexual abuse (50.0%).
Correlates of Attentional Bias to Threat, and Trauma-Related Threat and Loss Psychopathology
Table 2 shows correlations between independent variables, attentional bias to threat, and scores on composite measures of trauma-related threat and loss psychopathology. Results of these analyses revealed that [11C]OMAR VT values in the amygdala and severity of trauma-related threat and loss psychopathology were significantly positively related to attentional bias to threat. Furthermore, [11C]OMAR VT values in the amygdala were significantly positively related to severity of trauma-related threat symptomatology. Exploratory post hoc analyses revealed that this association was significant at the p<0.01 level for hyperarousal symptoms (r=0.59, p=0.006); the correlation for re-experiencing symptoms was 0.52, p=0.020. Exploratory post hoc analyses of [11C]OMAR VT values in brain regions other than the amygdala revealed that [11C]OMAR VT values in the posterior cingulate cortex were associated with attentional bias to threat (r=0.56, p=0.011), but this association was not significant at the p<0.01 level. None of the other brain regions were significant (all r’s<0.36, all p’s>0.12).
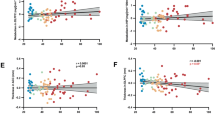
Figure 1 shows a scatterplot of the relation between [11C]OMAR VT values in amygdala and attentional bias to threat as assessed by the dot-probe task. Figure 2 shows a scatterplot of the relation between attentional bias to threat and severity of trauma-related threat symptomatology.

Scatterplot of the relation between [11C]OMAR volume of distribution (VT) values in the amygdala and performance on the dot-probe task. Note that higher attentional bias to threat scores are related to higher amygdala [11C]OMAR VT values.

Scatterplot of relation between performance on the dot-probe task and severity of threat symptomatology. Note that higher attentional bias to threat scores are related to higher threat symptoms.
Correlation of [11C]OMAR VT Values in the Amygdala, Plasma Anandamide Levels, and Attentional Bias to Threat
The [11C]OMAR VT values in the amygdala were also significantly negatively associated with plasma anandamide levels (r=−0.46, p=0.041; Figure 3), suggesting that greater CB1 receptor availability in the amygdala was associated with lower plasma levels of anandamide. Plasma anandamide levels were also significantly negatively associated with attentional bias to threat (r=−0.53, p=0.017; Figure 4).

Scatterplot of relation between [11C]OMAR volume of distribution (VT) values in the amygdala and anandamide levels.

Scatterplot of relation between anandamide levels and performance on the dot-probe task. Note that higher attentional bias to threat scores are related to lower plasma anandamide levels.
Mediation Analysis
Figure 5 shows results of a mediation analysis that evaluated the role of attentional bias to threat in mediating the relation between CB1 receptor availability in the amygdala and severity of trauma-related threat symptomatology. Although greater CB1 receptor availability in the amygdala was associated with greater severity of trauma-related threat symptomatology in a bivariate analysis (β=0.41, p=0.040), this association was no longer significant when attentional bias to threat was incorporated into the model (β=0.16, p=0.51). In the final model, which provided a good fit to the data (χ2(1)=0.51, p=0.48; CFI=1.00; SRMR=0.038), greater CB1 receptor availability was associated with increased attentional bias to threat that was in turn associated with increased severity of trauma-related threat symptomatology.

Path model of associations between CB1 receptor availability measured with [11C]OMAR and positron emission tomography in the amygdala, attentional bias to threat, and threat symptomatology. **Significant association, p<0.01. The r2 for negative attentional bias=0.28; r2 for threat symptomatology=0.31. The dashed line indicates a nonsignificant association. The 95% confidence intervals for CB1 → attentional bias to threat =0.01–0.95; for attentional bias to threat → threat symptomatology =0.16–0.85.
DISCUSSION
Using the CB1 receptor radiotracer [11C]OMAR, we found that greater CB1 receptor availability in the amygdala was associated with increased attentional bias to threat, as well as increased severity of trauma-related threat symptomatology (ie, hyperarousal) in humans presenting with a broad dimensional spectrum of trauma-related psychopathology. We also found that attentional bias to threat mediated the relation between CB1 receptor availability in the amygdala and severity of threat symptomatology. These results extend a growing body of research demonstrating an association between trauma-related disorders such as PTSD, MDD, and GAD, and attentional bias to threat (Fani et al, 2012b; Lindstrom et al, 2011; Sveen et al, 2009) by implicating the CB1 receptor system as a key neurobiological underpinning of this endophenotype and its concomitant phenotypic expression of trauma-related threat symptomatology, particularly hyperarousal symptoms. They further suggest that attentional bias to threat may mediate the association between CB1 receptor availability in the amygdala and threat symptomatology, with greater CB1 receptor availability being linked to greater attentional bias to threat that is in turn linked to greater severity of threat symptomatology.
Results of the current study build on extant neurobiological studies that have implicated the endocannabinoid system in the amygdala as an important modulator of anxiety (Mackie, 2005; Ramikie et al, 2014), as well as functional activation of the amygdala in mediating attentional bias to threat among individuals with PTSD (El Khoury-Malhame et al, 2011). Specifically, results of this study suggest that CB1 receptor availability in the amygdala may directly mediate this endophenotype and its associated phenotypic expression of trauma-related threat symptomatology. Preclinical work suggests that the activation of membrane glucocorticoid receptors appears to engage a G-protein-mediated cascade through the activation of Gs proteins (Di et al, 2003) that, in turn, increases the activity of cAMP and protein kinase A. This increase in protein kinase A appears to induce the rapid synthesis of an endocannabinoid signal through an as yet unknown mechanism that may be an increase in intracellular calcium signaling (Cadas et al, 1996; Malcher-Lopes et al, 2006; Vellani et al, 2008) that is then released from principal neurons in the amygdala and activates CB1 receptors localized on the terminals of GABAergic neurons in the amygdala. It should be noted, however, that other mechanisms than CB1 receptor stimulation by anandamide could contribute to the etiology of attentional bias to threat and threat symptomatology. First, the two endocannabinoids anandamide and 2-arachidonoylglycerol have differential roles in endocannabinoid (Ahn et al, 2008) and have distinctly different metabolic pathways (fatty acid amide hydrolase (FAAH) for anandamide and monoacylglycerollipase (MAGL) for 2-arachidonoylglycerol; (Ahn et al, 2008; Long et al, 2009). To date, the relative contribution of these two endocannabinoids and their pathways in the modulation of anxiety remains unclear. Furthermore, recent evidence suggests that CB1 receptor signaling varies across brain regions (Bosier et al, 2010), and that diverse effects of anandamide–CB1 receptor signaling mechanisms are evident even within the extended amygdala (Puente et al, 2011). Finally, the actions of anandamide are not restricted to CB1 receptors, as endocannabinoids also act on CB2 receptors (Mechoulam et al, 1995), GPR55 (Ryberg et al, 2007), transient receptor potential vanilloid type 1 channels (Melck et al, 1999; Smart et al, 2000; Zygmunt et al, 1999), and other G-protein subtypes (Glass and Felder, 1997b; Howlett, 2004).
Although additional research is needed to further evaluate how the endocannabinoid system mediates attentional bias to threat, the results of this study suggest that greater CB1 receptor availability in the amygdala, as well as lower levels of peripheral anandamide, are associated with a greater attentional bias to threat in trauma-exposed individuals. However, we acknowledge, that no human studies that we are aware of have found that anandamide concentrations directly influence CB1 receptor availability, and hence additional work is needed to ascertain how these variables are causally related. Nevertheless, the present data extend prior work linking attentional bias to threat to hyperarousal symptoms (Fani et al, 2012b) to suggest that the CB1 receptor system in the amygdala is implicated in modulating attentional bias to threat that is in turn linked to the transdiagnostic and dimensional phenotypic expression of trauma-related threat symptomatology. Further research will be useful in further elucidating molecular mechanisms that account for the observed association between CB1 receptor availability and the endophenotypic and phenotypic expression of threat processing in humans.
An important question to be addressed in future work is whether pharmacotherapies that act on catabolic enzymes for endocannabinoids may be useful in the prevention and treatment of endophenotypic and phenotypic aspects of trauma-related threat symptomatology. Emerging evidence supports the potential utility of such targets, suggesting that variation in the FAAH gene is linked to reduced expression of FAAH that consequently results in elevations in circulating levels of anadamide (Chiang et al, 2004; Sipe et al, 2010), as well as decreased amygdala response to threat (Hariri et al, 2009) and more rapid habituation of the amygdala to repeated threat (Gunduz-Cinar et al, 2013b). Notably, elevating anandamide levels via FAAH inhibition appear to provide a more circumscribed spectrum of behavioral effects than blocking MAGL (Blankman and Cravatt, 2013) that could potentially result in a more beneficial side effect profile, as anandamide is less prone to CB1 receptor desensitization and resultant behavioral tolerance (Lichtman et al, 2002; Schlosburg et al, 2010). These classes of compounds are currently being investigated for their potential efficacy in treating mood and anxiety disorders. Given that core aspects of threat symptomatology such as hyperarousal are key drivers of more disabling aspects of the trauma-related phenotype such as emotional numbing (Marshall et al, 2006; Pietrzak et al, 2013; Schell et al, 2004; Solomon et al, 2009), pharmacotherapeutic targeting of threat symptomatology in symptomatic trauma survivors may have utility in reducing the chronicity and morbidity of trauma-related psychiatric disorders such as PTSD, MDD, and GAD.
Methodological limitations of this study must be noted. First, we studied a cohort of individuals with heterogeneous trauma histories. Although this is typical for most PTSD studies and we endeavored to recruit individuals who represented a broad and representative spectrum of trauma-related psychopathology, additional studies of samples with noncivilian trauma histories will be useful in extending these results. Second, 95% confidence intervals for coefficients in the mediation analysis were markedly wide, and hence additional studies in larger samples will be useful in ascertaining magnitudes of the observed associations. Third, we observed a high correlation between threat and loss symptomatology that may call into question the extent to which these symptom clusters reflect separable components of trauma-related psychopathology that are uniquely related to CB1 receptor availability in the amygdala and attentional bias to threat. Nevertheless, high correlations among symptom clusters of trauma-related psychopathology are not uncommon, with confirmatory factor analytic studies of substantially larger samples often observing intercorrelations among symptom clusters >0.80 (Forbes et al, 2011; Grant et al, 2008; Pietrzak et al, 2010; Wang et al, 2011). Furthermore, the finding that CB1 receptor availability in the amygdala was associated only with threat, but not loss symptomatology, suggests greater specificity of association that accords with prior work (Gunduz-Cinar et al, 2013a). Fourth, it is important to recognize that our outcome measure in this study, VT, represents specific plus nondisplaceable binding. Because of the lack of a suitable reference region devoid of CB1, we and others using different CB1 receptor ligands (Ceccarini et al, 2014; Neumeister et al, 2012; 2013; Tsujikawa et al, 2014) cannot directly calculate binding potential (BPND), a measure of specific binding. Thus, an implicit assumption in the interpretation of our results is that there are no group differences in VND, the distribution volume of nondisplaceable tracer uptake. An alternative assumption would be that the magnitude of nondisplaceable binding is small compared with the total binding. To definitively address this issue would require a blocking study in humans to estimate VND. To the best of our knowledge, such data are not currently available because of the lack of suitable selective CB1 antagonist drugs approved for human use. Blocking data with the CB1 receptor antagonist rimonabant (1 mg/kg) in baboons (Horti et al, 2006), however, did show a large reduction in tracer uptake, suggesting that a substantial fraction of VT can be attributed to specific binding.
Notwithstanding these limitations, the results of this study provide the first known in vivo molecular evidence of how a candidate neuroreceptor system—CB1—relates to attentional bias to threat and the dimensional expression of trauma-related psychopathology. Results revealed that greater CB1 receptor availability in the amygdala is associated with increased attentional bias to threat, as well as the phenotypic expression of threat-related symptomatology, particularly hyperarousal symptoms. Given that these results were based on a relatively small sample, further research in larger, transdiagnostic cohorts with elevated threat symptomatology will be useful in evaluating the generalizability of these results, as well as in examining the efficacy of candidate pharmacotherapies that target the anandamide–CB1 receptor system in mitigating both the endophenotypic and phenotypic expression of threat symptomatology in symptomatic trauma survivors.
FUNDING AND DISCLOSURE
Dr Neumeister has received consulting fees from Pfizer. Dr Pietrzak is a scientific consultant to Cogstate. Dr Potenza has consulted for Ironwood, Lundbeck, Boehringer Ingelheim, Somaxon, gambling businesses and organizations, law offices, and the federal defender’s office in issues regarding impulse control disorders. He has received research support from the Mohegan Sun Casino, the National Center for Responsible Gaming, and the following pharmaceutical companies: Psyadon, Forest Laboratories, Ortho-McNeil, Oy-Control/Biotie, and GlaxoSmithKline. These activities are unrelated to the present report. The authors declare no conflict of interest.
Change history
08 September 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Ahn K, McKinney MK, Cravatt BF (2008). Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem Rev 108: 1687–1707.
Bar-Haim Y, Lamy D, Pergamin L, Bakermans-Kranenburg MJ, van IMH (2007). Threat-related attentional bias in anxious and nonanxious individuals: a meta-analytic study. Psychol Bull 133: 1–24.
Blankman JL, Cravatt BF (2013). Chemical probes of endocannabinoid metabolism. Pharmacol Rev 65: 849–871.
Bosier B, Muccioli GG, Hermans E, Lambert DM (2010). Functionally selective cannabinoid receptor signalling: therapeutic implications and opportunities. Biochem Pharmacol 80: 1–12.
Cadas H, Gaillet S, Beltramo M, Venance L, Piomelli D (1996). Biosynthesis of an endogenous cannabinoid precursor in neurons and its control by calcium and cAMP. J Neurosci 16: 3934–3942.
Carson RE, Barker WC, Liow J-S, Adler S, Johnson CA (eds) (2003). Design of a Motion-Compensation OSEM List-Mode Algorithm for Resolution-Recovery Reconstruction of the HRRT. Conf Record IEEE Nuclear Science Symposium and Medical Imaging. Portland, OR.
Ceccarini J, Hompes T, Verhaeghen A, Casteels C, Peuskens H, Bormans G et al (2014). Changes in cerebral CB1 receptor availability after acute and chronic alcohol abuse and monitored abstinence. J Neurosc 34: 2822–2831.
Chiang KP, Gerber AL, Sipe JC, Cravatt BF (2004). Reduced cellular expression and activity of the P129T mutant of human fatty acid amide hydrolase: evidence for a link between defects in the endocannabinoid system and problem drug use. Hum Mol Genet 13: 2113–2119.
Cuthbert BN (2014). The RDoC framework: facilitating transition from ICD/DSM to dimensional approaches that integrate neuroscience and psychopathology. World Psychiatry 13: 28–35.
Di S, Malcher-Lopes R, Halmos KC, Tasker JG (2003). Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci 23: 4850–4857.
El Khoury-Malhame M, Reynaud E, Soriano A, Michael K, Salgado-Pineda P, Zendjidjian X et al (2011). Amygdala activity correlates with attentional bias in PTSD. Neuropsychologia 49: 1969–1973.
Fani N, Jovanovic T, Ely TD, Bradley B, Gutman D, Tone EB et al (2012a). Neural correlates of attention bias to threat in post-traumatic stress disorder. Biol Psychol 90: 134–142.
Fani N, Tone EB, Phifer J, Norrholm SD, Bradley B, Ressler KJ et al (2012b). Attention bias toward threat is associated with exaggerated fear expression and impaired extinction in PTSD. Psychol Med 42: 533–543.
Forbes D, Lockwood E, Elhai JD, Creamer M, O'Donnell M, Bryant R et al (2011). An examination of the structure of posttraumatic stress disorder in relation to the anxiety and depressive disorders. J Affect Disord 132: 165–172.
Forbes D, Parslow R, Creamer M, O'Donnell M, Bryant R, McFarlane A et al (2010). A longitudinal analysis of posttraumatic stress disorder symptoms and their relationship with fear and anxious-misery disorders: implications for DSM-V. J Affect Disord 127: 147–152.
Glass M, Dragunow M, Faull RL (1997a). Cannabinoid receptors in the human brain: a detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience 77: 299–318.
Glass M, Felder CC (1997b). Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J Neurosci 17: 5327–5333.
Grant DM, Beck JG, Marques L, Palyo SA, Clapp JD (2008). The structure of distress following trauma: posttraumatic stress disorder, major depressive disorder, and generalized anxiety disorder. J Abnorm Psychol 117: 662–672.
Gunduz-Cinar O, Hill MN, McEwen BS, Holmes A (2013a). Amygdala FAAH and anandamide: mediating protection and recovery from stress. Trends Pharmacol Sci 34: 637–644.
Gunduz-Cinar O, MacPherson KP, Cinar R, Gamble-George J, Sugden K, Williams B et al (2013b). Convergent translational evidence of a role for anandamide in amygdala-mediated fear extinction, threat processing and stress-reactivity. Mol Psychiatry 18: 813–823.
Hariri AR, Gorka A, Hyde LW, Kimak M, Halder I, Ducci F et al (2009). Divergent effects of genetic variation in endocannabinoid signaling on human threat- and reward-related brain function. Biol Psychiatry 66: 9–16.
Herkenham M (1991). Characterization and localization of cannabinoid receptors in brain: an in vitro technique using slide-mounted tissue sections. NIDA Res Monogr 112: 129–145.
Hilton J, Yokoi F, Dannals RF, Ravert HT, Szabo Z, Wong DF (2000). Column-switching HPLC for the analysis of plasma in PET imaging studies. Nuclear Med Biol 27: 627–630.
Horti AG, Fan H, Kuwabara H, Hilton J, Ravert HT, Holt DP et al (2006). 11C-JHU75528: a radiotracer for PET imaging of CB1 cannabinoid receptors. J Nucl Med 47: 1689–1696.
Howlett AC (2004). Efficacy in CB1 receptor-mediated signal transduction. Br J Pharmacol 142: 1209–1218.
Ichise M, Toyama H, Innis RB, Carson RE (2002). Strategies to improve neuroreceptor parameter estimation by linear regression analysis. J Cereb Blood Flow Metab 22: 1271–1281.
Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN et al (2007). Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab 27: 1533–1539.
Kline RB (2010) Principles and Practice of Structural Equation Modeling 3rd edn, The Guilford Press: New York.
LeDoux JE (2000). Emotion circuits in the brain. Annu Rev Neurosci 23: 155–184.
Lichtman AH, Hawkins EG, Griffin G, Cravatt BF (2002). Pharmacological activity of fatty acid amides is regulated, but not mediated, by fatty acid amide hydrolase in vivo. J Pharmacol Exp Ther 302: 73–79.
Lindstrom KM, Mandell DJ, Musa GJ, Britton JC, Sankin LS, Mogg K et al (2011). Attention orientation in parents exposed to the 9/11 terrorist attacks and their children. Psychiatry Res 187: 261–266.
Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE et al (2009). Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol 5: 37–44.
Mackie K (2005). Distribution of cannabinoid receptors in the central and peripheral nervous system. Handb Exp Pharmacol 168: 299–325.
MacLeod C, Mathews A, Tata P (1986). Attentional bias in emotional disorders. J Abnorm Psychol 95: 15–20.
MacLeod C, Rutherford E, Campbell L, Ebsworthy G, Holker L (2002). Selective attention and emotional vulnerability: assessing the causal basis of their association through the experimental manipulation of attentional bias. J Abnorm Psychol 111: 107–123.
Malcher-Lopes R, Di S, Marcheselli VS, Weng FJ, Stuart CT, Bazan NG et al (2006). Opposing crosstalk between leptin and glucocorticoids rapidly modulates synaptic excitation via endocannabinoid release. J Neurosci 26: 6643–6650.
Marshall GN, Schell TL, Glynn SM, Shetty V (2006). The role of hyperarousal in the manifestation of posttraumatic psychological distress following injury. J Abnorm Psychol 115: 624–628.
Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR et al (1995). Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol 50: 83–90.
Melck D, Bisogno T, De Petrocellis L, Chuang H, Julius D, Bifulco M et al (1999). Unsaturated long-chain N-acyl-vanillyl-amides (N-AVAMs): vanilloid receptor ligands that inhibit anandamide-facilitated transport and bind to CB1 cannabinoid receptors. Biochem Biophys Res Commun 262: 275–284.
Neumeister A, Normandin MD, Murrough JW, Henry S, Bailey CR, Luckenbaugh DA et al (2012). Positron emission tomography shows elevated cannabinoid CB (1) receptor binding in men with alcohol dependence. Alcohol Clin Exp Res 36: 2104–2109.
Neumeister A, Normandin MD, Pietrzak RH, Piomelli D, Zheng MQ, Gujarro-Anton A et al (2013). Elevated brain cannabinoid CB1 receptor availability in post-traumatic stress disorder: a positron emission tomography study. Mol Psychiatry 18: 1034–1040.
Pietrzak RH, Feder A, Schechter CB, Singh R, Cancelmo L, Bromet EJ et al (2013). Dimensional structure and course of post-traumatic stress symptomatology in World Trade Center responders. Psycholog Med 44: 2085–2098.
Pietrzak RH, Goldstein MB, Malley JC, Rivers AJ, Southwick SM (2010). Structure of posttraumatic stress disorder symptoms and psychosocial functioning in Veterans of Operations Enduring Freedom and Iraqi Freedom. Psychiatry Res 178: 323–329.
Puente N, Cui Y, Lassalle O, Lafourcade M, Georges F, Venance L et al (2011). Polymodal activation of the endocannabinoid system in the extended amygdala. Nat Neurosci 14: 1542–1547.
Ramikie TS, Nyilas R, Bluett RJ, Gamble-George JC, Hartley ND, Mackie K et al (2014). Multiple mechanistically distinct modes of endocannabinoid mobilization at central amygdala glutamatergic synapses. Neuron 81: 1111–1125.
Rodrigues SM, Schafe GE, LeDoux JE (2004). Molecular mechanisms underlying emotional learning and memory in the lateral amygdala. Neuron 44: 75–91.
Rogan MT, Staubli UV, LeDoux JE (1997). Fear conditioning induces associative long-term potentiation in the amygdala. Nature 390: 604–607.
Ryberg E, Larsson N, Sjogren S, Hjorth S, Hermansson NO, Leonova J et al (2007). The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol 152: 1092–1101.
Salemink E, van den Hout MA, Kindt M (2007). Selective attention and threat: quick orienting versus slow disengagement and two versions of the dot probe task. Behav Res Therapy 45: 607–615.
Schell TL, Marshall GN, Jaycox LH (2004). All symptoms are not created equal: the prominent role of hyperarousal in the natural course of posttraumatic psychological distress. J Abnorm Psychol 113: 189–197.
Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG et al (2010). Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat Neurosci 13: 1113–1119.
Sipe JC, Scott TM, Murray S, Harismendy O, Simon GM, Cravatt BF et al (2010). Biomarkers of endocannabinoid system activation in severe obesity. PLoS One 5: e8792.
Smart D, Gunthorpe MJ, Jerman JC, Nasir S, Gray J, Muir AI et al (2000). The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1). Br J Pharmacol 129: 227–230.
Solomon Z, Horesh D, Ein-Dor T (2009). The longitudinal course of posttraumatic stress disorder symptom clusters among war veterans. J Clin Psychiatry 70: 837–843.
Sveen J, Dyster-Aas J, Willebrand M (2009). Attentional bias and symptoms of posttraumatic stress disorder one year after burn injury. J Nerv Ment Dis 197: 850–855.
Tsujikawa T, Zoghbi SS, Hong J, Donohue SR, Jenko KJ, Gladding RL et al (2014). In vitro and in vivo evaluation of (11)C-SD5024, a novel PET radioligand for human brain imaging of cannabinoid CB1 receptors. NeuroImage 84: 733–741.
Tzourio-Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N et al (2002). Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. NeuroImage 15: 273–289.
Vellani V, Petrosino S, De Petrocellis L, Valenti M, Prandini M, Magherini PC et al (2008). Functional lipidomics. Calcium-independent activation of endocannabinoid/endovanilloid lipid signalling in sensory neurons by protein kinases C and A and thrombin. Neuropharmacology 55: 1274–1279.
Wang L, Zhang J, Shi Z, Zhou M, Li Z, Zhang K et al (2011). Comparing alternative factor models of PTSD symptoms across earthquake victims and violent riot witnesses in China: evidence for a five-factor model proposed by Elhai et al. (2011). J Anxiety Disord 25: 771–776.
Zoellner LA, Pruitt LD, Farach FJ, Jun JJ (2014). Understanding heterogeneity in PTSD: fear, dysphoria, and distress. Depress Anxiety 31: 97–106.
Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di Marzo V et al (1999). Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400: 452–457.
Acknowledgements
We acknowledge the excellent work of the staff of the Yale PET Center and the nursing support from Brenda Breault, Cynthia D’Amico, Michelle San Pedro, Jamie Cyr, and Deborah Campbell for their contributions with patient care during the PET scans. This project was supported by the National Institutes of Health (NIH) through the following awards: R21MH096105, R21MH085627, RO1MH096876, and RO1MH102566. This publication was also made possible by CTSA Grant Number UL1 RR024139 from the National Center for Research Resources (NCRR) and the National Center for Advancing Translational Science (NCATS), components of the National Institutes of Health (NIH), NIH Roadmap for Medical Research, and Clinical Neurosciences Division of the United States Department of Veterans Affairs National Center for Posttraumatic Stress Disorder. Dr Neumeister has received grant support from Lilly. The work of Drs Neumeister, Huang, Potenza, and Carson was funded by the NIH.
The NIH or VA played no role in the design of the study, collection and analysis of the data, and decision to publish. Contents of this publication are solely the responsibility of the authors and do not necessarily represent the official view of NIH or VA.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Pietrzak, R., Huang, Y., Corsi-Travali, S. et al. RETRACTED ARTICLE: Cannabinoid Type 1 Receptor Availability in the Amygdala Mediates Threat Processing in Trauma Survivors. Neuropsychopharmacol 39, 2519–2528 (2014). https://doi.org/10.1038/npp.2014.110
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2014.110
This article is cited by
-
Increased cannabis intake during the COVID-19 pandemic is associated with worsening of depression symptoms in people with PTSD
BMC Psychiatry (2022)
-
Increased mGlu5 mRNA expression in BLA glutamate neurons facilitates resilience to the long-term effects of a single predator scent stress exposure
Brain Structure and Function (2021)
-
Sleep disturbance in PTSD and other anxiety-related disorders: an updated review of clinical features, physiological characteristics, and psychological and neurobiological mechanisms
Neuropsychopharmacology (2020)
-
Endocannabinoid signaling in psychiatric disorders: a review of positron emission tomography studies
Acta Pharmacologica Sinica (2019)
-
Cannabinoid Receptor 1 Positive Allosteric Modulators for Posttraumatic Stress Disorder
Neuropsychopharmacology (2018)
