
Abstract
The antiepileptic drug levetiracetam (LEV) is a potential treatment for alcohol use disorders, yet few preclinical studies exist on its effects in animal models relevant to drug or alcohol abuse. We investigated the effects of LEV on locomotor stimulation following acute and repeated administration of alcohol or cocaine and on alcohol- and cocaine-mediated changes in responding for brain stimulation reward (BSR) in C57BL/6J mice. LEV alone (10.0–100.0 mg/kg intraperitoneally) had no significant effect on locomotor activity or intracranial self-stimulation. Pretreatment with LEV reduced acute locomotor stimulation by 2.0 g/kg alcohol, attenuated the development of locomotor sensitization to alcohol with repeated exposure, and produced a shift in the dose-response curve for alcohol on BSR threshold without affecting maximum operant response rate (MAX). Conversely, LEV pretreatment enhanced both acute locomotor stimulation by 15 mg/kg cocaine and development of locomotor sensitization following repeated exposure and produced a leftward shift in the dose-response curve for cocaine on BSR threshold without affecting MAX. Electrophysiological recordings in vitro showed that LEV reduced excitatory currents in both ventral tegmental area (VTA) dopamine neurons and nucleus accumbens (NAc) medium spiny neurons, consistent with a presynaptic effect. The opposite effects of LEV pretreatment on alcohol- and cocaine-related behaviors may predict its clinical utility in the treatment of patients with alcohol, but not psychostimulant abuse disorders.
Similar content being viewed by others


INTRODUCTION
The mood-elevating effects of drugs and alcohol are important contributors to substance abuse, and reducing their acute rewarding effects represents a viable strategy for limiting consumption. One example of this clinical approach is the use of the opioid receptor antagonist naltrexone, which blunts the alcohol ‘high’ reported by users and can reduce alcohol intake (Anton et al, 2006). As new pharmacotherapeutic candidates are identified, it is important to determine how they alter the acute rewarding effects of drugs and alcohol. Preclinical studies in laboratory animals provide the advantages of precise control over drug administration and the individual subject’s history of drug exposure.
In laboratory animals, drugs such as psychostimulants that increase locomotor activity commonly also enhance intracranial self-stimulation (ICSS) through activation of the mesocorticolimbic dopamine system (Wise, 1996). ICSS is an operant behavioral method that measures the effects of a drug on responding for rewarding electrical brain stimulation by detecting changes in the minimum amount of stimulation required to sustain responding, the brain stimulation reward (BSR) threshold (θ0, Miliaressis et al, 1986). Acutely administered alcohol or cocaine lowers BSR threshold in mice, as well as stimulates locomotor activity (Fish et al, 2010). Identifying candidate drugs that block alcohol- or cocaine-mediated changes in ICSS or locomotor activity may be useful in the development of new treatments for substance abuse.
While studies examining acute drug effects are useful in determining mechanisms of action, they are unable to assess long-term neural adaptations to repeated drug exposure. Behavioral sensitization is the progressive enhancement of locomotor responses elicited by repeated drug administration and is associated with persistent hypersensitivity of mesocorticolimbic reward circuits to acute drug challenge (reviewed in Vanderschuren and Kalivas, 2000). The induction of behavioral sensitization has consistently been shown to be separable from effects on ICSS, as the potentiation of BSR does not change with repeated administration of alcohol and other drugs of abuse (Esposito and Kornetsky, 1977; Bauco and Wise, 1997; Riday et al, 2012). This phenomenon allows researchers to measure the acute rewarding effects of a drug independent from the neural adaptations that occur following prolonged exposure.
Drug effects on acute locomotor stimulation, sensitized locomotor activity, and ICSS share a common neural substrate, the mesencephalic dopaminergic projections to the nucleus accumbens (NAc), dorsal striatum (STR), prefrontal cortex (PFC), and amygdala (Wise, 2005). The NAc also receives convergent glutamatergic input from PFC, hippocampus, and basolateral amygdala (Britt et al, 2012), which interacts with dopaminergic signaling to generate adaptive responses to behaviorally relevant stimuli (Vanderschuren and Kalivas, 2000). Electrical stimulation of this circuitry is potently reinforcing (Olds and Milner, 1954), and animals will perform operant tasks for stimulation of the medial forebrain bundle, which carries ascending dopaminergic projections and descending glutamatergic and GABAergic fibers to and from the midbrain (Wise, 2005). Glutamatergic neurotransmission is particularly relevant to drug-mediated behaviors, as both ionotropic and metabotropic glutamate receptor antagonists alter drug and alcohol seeking and prevent the induction of behavioral sensitization (reviewed in Gass and Olive, 2008).
One approach to block the acute and adaptive effects of drug exposure, therefore, is to interfere with glutamatergic neurotransmission. Levetiracetam (LEV) (S-α-ethyl-2-oxo-pyrrolidine acetamide) is an antiepileptic drug that decreases excitatory transmission preferentially at more active synapses in vitro (Yang et al, 2007; Yang and Rothman, 2009; Meehan et al, 2011). LEV selectively binds to synaptic vesicle glycoprotein 2A (SV2A) in presynaptic terminals throughout the brain (Lynch et al, 2004; Bragina et al, 2011). SV2A interacts with the calcium-sensor synaptotagmin through its N-terminal (cytosolic) domain (Schivell et al, 2005) and appears to have a modulatory rather than a necessary role in vesicular neurotransmitter release, as SV2A knockout mice show reduction but not elimination of excitatory neurotransmission (Custer et al, 2006).
Despite clinical interest in LEV, there have been few studies examining its effects on alcohol- or drug-influenced behaviors in laboratory animals. We investigated the effects of LEV on locomotor activation by alcohol or cocaine in C57BL/6J mice to determine if it affects acute or sensitized responses. We also assessed the effects of LEV on alcohol- or cocaine-potentiated reward using ICSS and the effects of LEV on excitatory neurotransmission in both ventral tegmental area (VTA) dopaminergic neurons and NAc medium spiny neurons (MSNs) in vitro. Given its ability to alter glutamatergic neurotransmission, we hypothesized that LEV would attenuate the acute rewarding and locomotor-stimulating effects of alcohol and cocaine, as well as interfere with behavioral sensitization following repeated administration.
MATERIALS AND METHODS
Mice
Adult male C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME) were group-housed, four mice per cage for locomotor activity (n=85) and in vitro electrophysiology experiments (n=18), or housed individually for ICSS experiments (n=11), and had free access to food and water. The vivarium was at 21 °C with a 12-h light cycle (lights on at 2000 h). All procedures were conducted between 0830 and 12:30 h, approved by The Institutional Animal Care and Use Committee (IACUC) of the University of North Carolina at Chapel Hill, and conducted according to the Guide for the Care and Use of Laboratory Animals (NIH publication 85-23, revised 2011).
Locomotor Activity
Locomotion was measured in 28 × 28 cm plexiglass chambers containing two sets of 16 infrared photobeams (MedAssociates, St. Albans, VT). Data were collected with software (MED-PC v4.1; MedAssociates) that calculated the total distance traveled (cm) by measuring the position of the mouse every 100 ms. During test sessions, mice were placed into the center of the chamber and locomotion was measured for 15 min. Mice were removed from the chambers, injected intraperitoneally (i.p.) with saline or LEV (100 mg/kg), and returned to the chamber for 30 min. Mice were removed again and injected i.p. with saline, alcohol (2.0 g/kg), or cocaine (15.0 mg/kg) and returned to the chamber for 15 min.
Locomotor Sensitization
Mice were habituated to i.p. saline injections on Days 1 and 2 for alcohol and cocaine sensitization experiments. Alcohol sensitization experiments were adapted from Lessov et al (2001) and Melón and Boehm (2011). On Day 3, mice were randomly assigned and tested for acute effects of the following treatments on locomotor activity: saline/saline (SAL/SAL; n=10), LEV/saline (LEV/SAL; n=12), saline/alcohol (SAL/ALC; n=12), or LEV/alcohol (LEV/ALC; n=15). On Days 4–13, mice received treatments in their home cages with SAL/SAL, LEV/SAL, SAL/alcohol (3.0 g/kg), or LEV/alcohol (3.0 g/kg). On Day 14, all mice received saline followed by alcohol 2.0 g/kg i.p. On Day 15, all mice received LEV 100 mg/kg i.p. followed by alcohol 2.0 g/kg i.p.
Cocaine locomotor sensitization experiments were adapted from Thomas et al (2001). On Day 3, mice were randomly assigned to the following treatment groups: saline/saline (SAL/SAL; n=9), LEV/saline (LEV/SAL; n=9), saline/cocaine (SAL/COC; n=9), or LEV/cocaine (LEV/COC; n=9), and locomotion before and after injection with each drug was measured on Days 3–7 as described above. Ten days after the fifth cocaine injection, all mice were retested after injection of saline followed by saline on Day 18, saline followed by 15 mg/kg i.p. cocaine on Day 19, and LEV 100 mg/kg i.p. followed by cocaine 15 mg/kg i.p. on Day 20.
Electrode Implantation
ICSS experiments were conducted in a separate group of mice (n=11) that were anesthetized (120 mg/kg ketamine and 18 mg/kg xylazine; Sigma, St Louis, MO) and stereotaxically implanted in the right medial forebrain bundle at the level of the lateral hypothalamus (A/P −1.3, M/L −1.0, D/V −5.0; Paxinos and Franklin, 1996) with insulated monopolar stainless steel electrodes (0.28 mm diameter; Plastics One, Roanoke, VA). The electrode was grounded to the skull with a stainless steel screw and secured to the skull using dental cement. Following surgery, mice were returned to their cages for 1 week of recovery.
Intracranial Self-Stimulation
ICSS experiments were performed as previously described (Fish et al, 2010) in operant conditioning chambers with a grid floor, wheel manipulandum, and house light (MedAssociates). Delivery of electrical stimulation was controlled by MED-PC software (v4.1; MedAssociates) and a stimulator (PHM-150B/2; MedAssociates) connected to a swivel commutator and insulated wire (Plastics One) attached to the stimulating electrode. A computer interface recorded responses (1 response=¼ turn of the wheel), activated the house light, and issued electrical current (BSR). Each stimulation was a 500-ms train of unipolar cathodal square-wave current pulses (100 μs) delivered at a trial-dependent frequency. During the 500 ms stimulation period, responses were recorded but did not earn additional stimulation. Each response was accompanied by illumination of the house light for 500 ms.
During each testing session, mice responded during three consecutive series of 15 descending stimulus frequencies. The first series served as a warm-up and was discarded; daily baseline BSR thresholds (θ0) were calculated from the second and third series. In initial LEV experiments, mice were removed from the chambers after baseline determinations, injected (i.p.) with saline or LEV (10.0–100.0 mg/kg), and returned to the chambers for 30 min (ie, two 15-min response series). In drug pretreatment studies, mice were removed from the chambers after baseline determinations, injected with saline or LEV (100.0 mg/kg, i.p.), and placed in their home cages for 30 min. Mice were then either injected (i.p.) with saline or cocaine (1.0, 5.0, or 15.0 mg/kg) or gavaged (p.o.) with water or alcohol (0.6, 1.7, or 2.4 g/kg) and immediately returned to the chambers for one 15-min response series. Maximum response rate (MAX) and θ0 were calculated with custom-designed software.
Brain Slice Preparations
Adult male C57BL/6J mice (n=18) were anesthetized (100 mg/kg ketamine and 10 mg/kg xylazine, i.p.), transcardially perfused with ice-cold (4 °C) sucrose cutting solution (75 mM sucrose, 87 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 7 mM MgCl2, 0.5 mM CaCl2, 10 mM glucose, 26 mM NaHCO3, and 1 mM ascorbic acid) and decapitated. The brain was removed and sectioned in the same sucrose cutting solution at 4 °C with a vibratome. Coronal slices (250 μm) were used for NAc (n=6) and horizontal slices (200 μm) were used for VTA recordings (n=12). Slices were recovered in a submerged holding chamber with oxygenated (95% O2/5% CO2) artificial cerebrospinal fluid (aCSF: 126 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 2.5 mM CaCl2, 1.2 mM MgCl2, 26 mM NaHCO3, and 11 mM glucose) at 32 °C for at least 30 min before recording.
In Vitro Electrophysiology
After recovery, slices were placed in the recording chamber and continuously perfused with oxygenated aCSF (2 ml/min). All recordings were performed at 32 °C in the presence of picrotoxin (50 μM) to block inhibitory currents. Whole cell recordings were made under voltage clamp at a holding potential of −70 mV using a MultiClamp 700B amplifier and DigiData 1322A interface (Molecular Devices, Sunnyvale, CA). Excitatory postsynaptic currents (EPSCs) were evoked with a bipolar stainless-steel electrode through an isolated stimulator (Digitimer, Letchworth Garden City, England). Signals were sampled at 10 KHz and filtered at 2 KHz. Recordings were performed on NAc MSNs with the stimulation electrode at the NAc core/anterior commissure border. Recording pipettes had a resistance of 3–5 MΩ and were filled with a cesium methanesulfonate-based internal solution (120 mM CsMeSO4, 3.7 mM NaCl, 10 mM TEA-Cl, 20 mM HEPES, 0.3 mM EGTA, 4 mM MgATP, 0.3 mM Na3GTP). The VTA region was identified by the interpeduncular nucleus and medial terminal nucleus of accessory optic tract, and recordings were obtained from neurons identified by the presence of a hyperpolarization-activated cation current (Ih) with hyperpolarizing steps from −60 mV to −120 mV in −10 mV increments. Stimulation electrodes were placed rostral to the recording site. Recording pipettes had a resistance of 2–4 MΩ and were filled with a potassium gluconate-based internal solution (135 mM K-Gluconate, 4 mM KCl, 10 mM NaCl, 10 mM HEPES, 0.3 mM EGTA, 2.5 mg/ml MgATP, and 0.25 mg/ml Na3GTP).
Drugs
Cocaine HCL (doses calculated as salt) and LEV were purchased from Sigma, dissolved in 0.9% saline, and injected intraperitoneally in a volume of 1 ml/100 g body weight. For locomotion experiments, alcohol was diluted in 0.9% saline to a 15% v-v solution and injected intraperitoneally to deliver either 2.0 or 3.0 g/kg. For ICSS experiments, alcohol solutions were prepared in tap water and administered via oral gavage in a volume of 1 ml/100 g body weight. For ICSS experiments, drug doses and vehicle injections were given in a random order using a Latin square design, each dose was given twice and the results averaged; and no drug was given 2 days in a row. For in vitro electrophysiology experiments, all drugs were bath-applied.
Data Analysis
All statistical analyses were performed with SPSS (IBM, Armonk, NY). Comparisons of acute effects of LEV were performed using a t-test for locomotor activity and one-way analysis of variance (ANOVA) for ICSS responses. Comparisons of pretreatment effects on locomotor activity or ICSS responses were made using one- or two-way ANOVAs. Bonferroni-corrected post hocs were performed when p<0.05. Electrophysiological data were analyzed using Clampfit 9.2 software (Molecular Devices). Three sweeps were averaged, and the baseline was adjusted manually by subtracting a line from the first stimulus to 100 ms after the last stimulus. EPSC amplitudes were adjusted by subtracting the baseline amplitude and normalized to the amplitude of the first EPSC in each train. Comparison of acute effects of LEV was made using ANOVA for repeated measures. For all comparisons, p<0.05 was considered significant.
RESULTS
For ICSS experiments, electrode tip locations ranged between −1.34 and −1.82 mm relative to bregma (Figure 1a and b) and the average baseline BSR threshold (θ0) expressed as charge delivery when mice met training criteria (⩽±10% variation of θ0 × 3 consecutive days) was 5.49±0.39 μC (n=11). Throughout all experiments, mice responded in a frequency-dependent manner (Figure 1c and d). In ICSS experiments, after initial dose-response determinations to LEV alone, alcohol and cocaine doses were administered in a random order following LEV pretreatment.

ICSS electrode tip locations and representative rate frequency curves. (a) Photomicrograph (4 × ) displaying the electrode tip location in a Nissl-stained coronal section from a representative mouse. (b) Ventral electrode tip placements (mm posterior to bregma) for each mouse (black circles, n=11). (c) Representative rate-frequency curves from one C57BL/6J mouse displaying the effects of treatment with saline i.p./water p.o. (VEH/H2O; white circles), 100.0 mg/kg levetiracetam i.p./water p.o. (LEV/H2O; gray circles), saline i.p./0.6 g/kg alcohol p.o. (SAL/ALC0.6; gray triangles), or 100.0 mg/kg levetiracetam i.p./0.6 g/kg alcohol p.o. (LEV/ALC0.6; black triangles). (d) Representative rate-frequency curves from one C57BL/6J mouse displaying the effects of intraperitoneal injection with saline/saline (SAL/SAL; white circles), 100.0 mg/kg levetiracetam/saline (LEV/SAL; gray circles), saline/5.0 mg/kg cocaine (SAL/COC5.0; gray diamonds), or 100.0 mg/kg levetiracetam/5.0 mg/kg cocaine (LEV/COC5.0; black diamonds).
Behavioral Effects of LEV on Locomotor Activity and ICSS
In initial experiments, the effects of LEV on both locomotor activity and ICSS were determined before pretreatment studies. When given alone, 100 mg/kg LEV had no significant effect on locomotion during the 30-min period following injection when compared with saline (1757.1±202.2 cm vs 1543.6±164.9 cm; t80=1.1, p=0.30). LEV alone (10.0, 30.0, or 100.0 mg/kg) had no effect on BSR threshold (θ0) or MAX during the first or second 15-min post injection response series (Table 1), suggesting that these doses of LEV do not possess rewarding potency. While LEV appeared to elevate MAX during the first response series, this effect was not statistically significant (F3,30=0.66, p=0.59).
Behavioral Effects of LEV and Alcohol
Locomotor activity
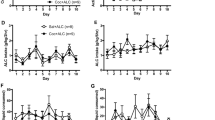
There was no significant effect of acute drug treatment combination on the total distance traveled during the first 15-min testing period (Figure 2b—Day 3; F3,45=1.2, p=0.33); however, there was a significant interaction between drug combination and time after alcohol administration (Figure 2a; F3,90=5.2, p<0.001). Post hoc analyses revealed that alcohol (2.0 g/kg, i.p.) administered after saline pretreatment significantly enhanced locomotor activity during the first 5 min of testing, but not in mice pretreated with LEV. During the last 5 min of testing, locomotor activity after alcohol was significantly lower in the LEV pretreatment group (Figure 2a).

Effects of levetiracetam (LEV) pretreatment on alcohol-affected behaviors in C57BL/6J mice. (a) Locomotor activity (total distance traveled, in cm) during each 5-min interval of the 15-min testing period on Day 3 (±SEM, vertical lines). Mice were randomized to receive intraperitoneal injections of the following treatments: saline/saline (SAL/SAL, white circles, n=10), 100.0 mg/kg levetiracetam/saline (LEV/SAL, light gray circles, n=12), saline/2.0 g/kg alcohol (SAL/ALC, dark gray circles, n=12), or 100.0 mg/kg levetiracetam/2.0 g/kg alcohol (LEV/ALC, black circles, n=15). (b) Locomotor activity during the 15-min post injection test on each experimental day (±SEM, vertical lines). On Days 1–2, all mice received saline/saline. On Day 3, mice received the following treatments, as in (a): SAL/SAL (white bars), LEV/SAL (light gray bars), SAL/ALC (dark gray bars), or LEV/ALC (black bars). On Day 14, all mice received saline/2.0 g/kg alcohol. On Day 15, all mice received 100.0 mg/kg levetiracetam/2.0 g/kg alcohol. (c) Comparison of locomotor activity during the 15-min post injection test on Days 14 (white bars) and 15 (gray bars;±SEM, vertical lines). Chronic daily treatment groups are abbreviated on the x axis (S/S=SAL/SAL, L/S=LEV/SAL, S/A=SAL/ALC, L/A=LEV/ALC). Number signs (#) indicate significant differences (p<0.05) between days. (d) Effect of levetiracetam pretreatment on ICSS after alcohol in C57BL/6J mice. Changes in BSR threshold (θ0) and maximum response rate (MAX) are shown as mean percentages (±SEM, vertical lines) of preinjection baseline after intraperitoneal injection with saline (+SAL, white circles) or 100.0 mg/kg levetiracetam (+LEV, gray circles) followed by oral gavage with water (H2O) or alcohol (n=11). Asterisks (*) indicate significance (p<0.05) vs SAL/SAL (SAL/H2O in d). Daggers (†) indicate significance (p<0.05) vs SAL/ALC.
On Day 14, following repeated treatments, the effect of 2.0 g/kg alcohol (i.p.) depended on repeated drug treatment (Figure 2b—Day 14; F3,45=6.1, p=0.001). Post hoc analyses revealed that locomotion was significantly higher in mice that received daily alcohol injections after saline but not LEV pretreatment on Days 4–13. On Day 15, when all mice received 100 mg/kg LEV before 2.0 g/kg alcohol (i.p.), there was no significant effect of repeated drug treatments, and alcohol did not affect locomotor activity in any of the treatment groups (Figure 2b—Day 15). When locomotor activity on Day 14 was compared with Day 15, the behavioral effects of 2.0 g/kg alcohol were dependent on 100.0 mg/kg LEV (F3,45=4.6, p=0.007; Figure 2c). Post hoc analyses revealed that the effects of alcohol on locomotor activity were decreased following LEV 100.0 mg/kg (Day 15) when compared with saline pretreatment (Day 14) in mice that received saline pretreatment on Days 3–7.
Intracranial self-stimulation
As we have previously shown (Fish et al, 2010), alcohol (0.6, 1.7, and 2.4 g/kg, p.o.) had biphasic, dose-dependent effects on BSR threshold (θ0) in C57BL/6J mice (Figure 2d). These effects depended on LEV or saline pretreatment (F3,27=3.2, p<0.05). Post hoc analyses revealed that 0.6 g/kg alcohol (p.o.) significantly lowered θ0 when mice were pretreated with saline, but not with LEV 100.0 mg/kg (i.p.). The 1.7 g/kg alcohol dose (p.o.) significantly elevated θ0 when mice were pretreated with LEV, but not with saline. The 2.4 g/kg alcohol dose significantly elevated θ0 when mice were pretreated with either saline or LEV. Pretreatment with LEV 100.0 mg/kg appeared to increase maximum response rate, but there was no interaction between LEV and alcohol dose (F3,27=0.18, p=0.91; Figure 2d).
Behavioral Effects of LEV and Cocaine
Locomotor activity
To determine if the effects of LEV on locomotor sensitization were specific to alcohol, similar experiments were conducted with cocaine. There was no significant effect of drug treatment combination on the total distance traveled during the first 15-min testing period (Figure 3b—Day 3); however, there was a significant interaction between drug combination and time after cocaine administration (Figure 3a; F3,64=6.2, p<0.001). Post hoc analyses revealed that cocaine (15.0 mg/kg, i.p.) increased total distance traveled during the final 10 min of testing when compared with saline, and this effect was greater in mice that received LEV pretreatment.

Effects of levetiracetam (LEV) pretreatment on cocaine-affected behaviors in C57BL/6J mice. (a) Locomotor activity (total distance traveled, in cm) during each 5-min interval of the 15-min testing period on Day 3 (±SEM, vertical lines). Mice were randomized to receive intraperitoneal injections of the following treatments: saline/saline (SAL/SAL, white circles, n=9), 100.0 mg/kg levetiracetam/saline (LEV/SAL, gray circles, n=9), saline/15.0 mg/kg cocaine (SAL/COC, gray diamonds, n=9), 100.0 mg/kg levetiracetam/15.0 mg/kg cocaine (LEV/COC, black diamonds, n=9). (b) Locomotor activity during the 15-min post injection test on each experimental day (±SEM, vertical lines). On Days 1–2, all mice received saline/saline. On Days 3–7, mice received the following treatments, as in (a): SAL/SAL (white circles), LEV/SAL (gray circles), SAL/COC (gray diamonds), or LEV/COC (black diamonds). On Day 18, all mice received saline/saline. On Day 19, all mice received saline/15.0 mg/kg cocaine. On Day 20, all mice received 100 mg/kg levetiracetam/15.0 mg/kg cocaine. (c) Comparison of locomotor activity during the 15-min post injection test on Days 14 (white bars) and 15 (gray bars;±SEM, vertical lines). Chronic daily treatment groups are abbreviated on the x axis (S/S=SAL/SAL, L/S=LEV/SAL, S/C=SAL/COC, L/A=LEV/COC). Number signs (#) indicate significant differences (p<0.05) between days. (d) Effect of levetiracetam pretreatment on ICSS after cocaine in C57BL/6J mice. Changes in BSR threshold (θ0) and maximum response rate (MAX) are shown as mean percentages (±SEM, vertical lines) of preinjection baseline after intraperitoneal injection with saline (+SAL, white circles) or 100.0 mg/kg levetiracetam (+LEV, gray circles) followed by injection with saline (V) or cocaine (n=11). Asterisks (*) indicate significance (p<0.05) vs SAL/SAL (SAL/V in d). Daggers (†) indicate significance (p<0.05) vs SAL/COC.
On Days 3–7, there was a significant interaction between repeated drug treatments and day (F3,128=18.2, p<0.001). Post hoc analyses revealed that locomotor activity was higher in the mice receiving cocaine than in saline controls. Within the mice receiving cocaine, locomotor activity was greater on Days 4–7 than on Day 3. LEV pretreatment was associated with a greater increase in locomotor activity on Days 6 and 7 than saline pretreatment (Figure 3b).
Following 10 days of withdrawal, there was no significant difference between treatment groups when all mice were treated with saline injections (Figure 3b—Day 18). On Day 19, when all mice received saline followed by 15.0 mg/kg cocaine, locomotor activity depended on prior repeated drug treatment (Figure 3b—Day 19; F3,32=30.0, p<0.001). Post hoc analyses revealed that locomotion during the 15-min post injection testing period was significantly greater in groups with previous exposure to cocaine. On Day 20, when all groups received 100.0 mg/kg LEV followed by 15.0 mg/kg cocaine, there was no effect of prior repeated drug treatment on total distance traveled (Figure 3b—Day 20). When total distance traveled on Day 19 was compared with Day 20, the locomotor effects of cocaine depended on LEV administration (F3,32=12.3, p<0.001; Figure 3c). Post hoc analyses revealed that the effects of 15.0 mg/kg cocaine on locomotor activity were increased following 100.0 mg/kg LEV on Day 20 when compared with saline pretreatment on Day 19 in mice that received saline on Days 3–7.
Intracranial self-stimulation
To determine if the effects of LEV on potentiation of BSR were specific to alcohol, similar experiments were performed using cocaine. Cocaine (1.0, 5.0, and 15.0 mg/kg, i.p.) dose dependently lowered θ0, and the magnitude of this effect depended on pretreatment with 100.0 mg/kg LEV (Figure 3d; F3,27=6.2, p<0.01). Post hoc analyses revealed that all cocaine doses lowered θ0, and 1.0 and 5.0 mg/kg cocaine produced a greater reduction in BSR threshold following LEV pretreatment. Pretreatment with LEV appeared to increase maximum response rate, but there was no interaction between LEV and cocaine dose (F3,27=0.41, p=0.75; Figure 3d).
Effects of LEV on Excitatory Neurotransmission in VTA and NAc Neurons
To further explore the possible mechanisms underlying the behavioral effects of LEV, we examined the effect of LEV on excitatory synaptic transmission in NAc MSNs and VTA dopaminergic neurons. Acute brain slices for LEV experiments were exposed to LEV (100 μM) for at least 1.5 h before and continuously throughout all recordings. Stimulus intensity was adjusted with single pulses 20 s apart to elicit stable evoked EPSCs of 200–500 pA and stimulus intensity was adjusted to match the amplitude of the first EPSC in control and LEV-exposed cells (−281.7±26.8 pA vs −272.2±23.5 pA, p=0.80 in VTA; −318.6±38.1 pA vs −394.9±46.0 pA, p=0.22 in NAc). Trains of 20 stimuli at 20 and 40 Hz were used for LEV experiments (Yang and Rothman, 2009; Meehan et al, 2011). The amplitude of EPSCs decreased significantly across stimulus trains in both control and LEV-exposed cells; however, in both NAc and VTA, LEV-exposed cells showed more reduction in EPSC amplitude than controls (Figure 4). Repeated-measures ANOVA of the 2nd to 20th EPSC normalized to the first EPSC in each train revealed significant reduction in EPSC amplitude at 20 Hz (F1, 28=4.70, p=0.04) but not at 40 Hz (F1, 23=0.71, p=0.41) in NAc MSNs (Figure 4a). In recordings of VTA dopaminergic neurons, a significant effect of LEV on EPSC amplitude was observed at 40 Hz (F1,26=6.6, p=0.02), while depression of EPSCs by LEV at 20 Hz was observed but did not reach significance (F1,27=4.01, p=0.055; Figure 4b).

Effect of bath-applied levetiracetam (LEV) on excitatory neurotransmission in nucleus accumbens medium spiny neurons (NAc) and ventral tegmental area dopaminergic neurons (VTA) in vitro. In both cell types, stimulus trains at 20 and 40 Hz were repeated three times each at a 1.5 min interval, 2.5 min apart. LEV (+LEV 100 μM, red traces) progressively reduced mean excitatory postsynaptic current (EPSC) amplitudes in averaged raw current traces (right panels) of recordings from both (a) NAc (n=15 control, 17 LEV cells) and (b) VTA (n=19 control, 16 LEV cells). Amplitudes of each successive EPSC normalized to the first EPSC in each stimulus train (EPSCn/EPSC1±SEM, left panels) show a greater progressive reduction in EPSC amplitude in LEV-exposed (red circles) than control neurons (black circles) in both NAc and VTA. At the end of each stimulus train (EPSC16–20/EPSC1±SEM) this inhibitory effect of LEV (insets, red bars) was significant at 20 Hz stimulation in NAc and at 40 Hz in VTA. Asterisks (*) indicate significance (p<0.05) of LEV exposed vs controls.
DISCUSSION
Although the antiepileptic drug LEV is a possible novel therapeutic agent in the treatment of alcohol and other substance abuse disorders, surprisingly little is known about its behavioral effects in preclinical animal models. In these studies, we found that LEV differentially affected responses to alcohol and cocaine in two behaviors relevant to drug and alcohol abuse, locomotor stimulation and ICSS. LEV pretreatment reduced alcohol-stimulated motor activity after both acute and repeated administration and caused a vertical shift in the alcohol dose-response curve on BSR threshold, biasing alcohol toward more aversive than rewarding effects. Conversely, LEV pretreatment increased the locomotor stimulant effects of acute and repeated cocaine and produced a leftward shift in the dose-response curve of cocaine on BSR threshold. We also found that LEV reduced excitatory neurotransmission in the VTA and NAc, neural substrates that are critical to the behavioral effects of alcohol and other drugs of abuse.
The exact cellular mechanism of action of LEV is not completely understood, although it is known that that LEV binds selectively to SV2A (Lynch et al, 2004; Gillard et al, 2006). SV2A is expressed throughout the brain and localizes to both glutamatergic and GABAergic presynaptic terminals. The other two members of the SV2 protein family, SV2B and SV2C, which have no apparent affinity for LEV, are enriched in glutamatergic and GABAergic vesicles, respectively (Bragina et al, 2011). SV2A has been shown to reduce excitatory (Yang and Rothman, 2009) and possibly inhibitory (Meehan et al, 2012; but see also Margineanu and Klitgaard, 2003) neurotransmission by affecting vesicular release mechanisms in response to intracellular calcium (Schivell et al, 2005; Chang and Sudhof, 2009; Wan et al, 2010). The molecular mechanisms by which SV2 proteins function in the synaptic vesicle cycle remain unknown, but may relate to a role in vesicle priming (Chang and Sudhof, 2009; Nowack et al, 2010). However, it is evident that binding of LEV to SV2A inhibits excitatory neurotransmission, and that this effect occurs preferentially at more active synapses (Yang et al, 2007; Yang and Rothman, 2009). Therefore, we hypothesized that LEV administration would affect behaviors that are strongly regulated by glutamatergic neurotransmission.
Levetiracetam (Keppra) is approved by the US Food and Drug Administration for the treatment of epilepsy, and is virtually unaffected by hepatic metabolism, making it extremely unlikely that changes in alcohol or cocaine pharmacokinetics can account for our behavioral findings in mice. We chose the 100 mg/kg LEV dose because it approximates the weight-based dose used for seizure prevention in patients with epilepsy, typically 40–100 mg/kg/day, and had no independent effects on mouse behavior. LEV crosses the blood–brain barrier freely, with peak serum concentrations achieved within 30 min after intraperitoneal administration and a serum half-life between 1 and 3 h in rats and mice. In the rat, cerebrospinal fluid concentrations of LEV reached concentrations of 300 μM by 30 min after a dose of 80 mg/kg i.p. (Doheny et al, 1999).
When given alone, LEV (100 mg/kg i.p.) did not significantly affect locomotion, although there was a trend toward motor activation. Across a range of doses (10–100 mg/kg i.p.), LEV did not affect BSR threshold or the maximum operant response rate, although there was a trend toward increased operant responding. The absence of effects on motor activity and ICSS was unexpected, as electrical stimulation of the medial forebrain bundle in the lateral hypothalamus affects both descending glutamatergic fibers (You et al, 2001) and firing of GABAergic MSNs in the NAc (Cheer et al, 2005). However, in vitro studies in the hippocampus have demonstrated that entry of LEV into presynaptic vesicles is highly activity dependent (Yang et al, 2007; Yang and Rothman, 2009; Meehan et al, 2011), and our electrophysiological data suggest that LEV may have a similar effect in NAc and VTA neurons; that is, the amplitude of stimulated EPSCs with repetitive firing is progressively decreased by LEV. These in vitro data are consistent with, but do not conclusively prove, that LEV may have a presynaptic effect on excitatory neurotransmission in the VTA and NAc, and in the absence of a stimulus that induces sustained cell firing (eg, following exposure to drugs of abuse) LEV may not fully enter the necessary cells to affect neurotransmission and thereby alter behavior. Nevertheless, when LEV was administered before alcohol or cocaine injection, pronounced effects on alcohol or cocaine-induced behaviors were observed.
Acutely administered alcohol exerts dose- and time-dependent effects on motor activity (Crabbe et al, 1982). In C57BL/6J mice, alcohol (2.0 g/kg, i.p.) produced an initial stimulant response in the first 5 min of testing followed by a return to baseline activity. Although it did not affect activity on its own, LEV pretreatment prevented this initial stimulation and appeared to depress activity during the final 5 min of testing, suggesting that LEV either directly antagonized motor stimulation by alcohol or enhanced its sedative or ataxic effects. Acute alcohol administration stimulates glutamate release in the NAc (Moghaddam and Bolinao, 1994; Lominac et al, 2006) and excites dopaminergic VTA neurons (Gessa et al, 1985; Verbanck et al, 1990), both directly (Brodie et al, 1990; Brodie and Appel, 1998) and through glutamate-dependent mechanisms (Deng et al, 2009; Xiao et al, 2009). Overall, the effects of LEV on motor activity following acute alcohol exposure are consistent with attenuation of excitatory neurotransmission, as both AMPA and NMDA antagonists also reduce the locomotor stimulant effects of alcohol (Broadbent et al, 2003). Our electrophysiological data support this interpretation and demonstrate for the first time that LEV attenuates excitatory neurotransmission in both NAc and VTA neurons in vitro.
Repeated administration of alcohol can induce behavioral sensitization in mice. Consistent with previous results using this procedure in C57BL/6J mice (Lessov et al, 2001; Melón and Boehm, 2011), we observed that 10 days of repeated alcohol treatments (3.0 g/kg) increased motor activity upon exposure to the 2.0 g/kg test dose of alcohol. This sensitized response did not occur in mice that received LEV before each repeated alcohol treatment, suggesting that LEV is capable of preventing the development of alcohol sensitization. Moreover, acute LEV administration can attenuate the expression of behavioral sensitization, as seen in the reduction of the locomotor response to alcohol on Day 15 in sensitized mice. NMDA receptor antagonists prevent the induction of behavioral sensitization to alcohol in mice (Broadbent and Weitemier, 1999; Camarini et al, 2000), while both NMDA and AMPA receptor antagonists block the expression of alcohol sensitization (Broadbent et al, 2003). These previous findings support the hypothesis that LEV may decrease the development of alcohol sensitization by decreasing glutamatergic neurotransmission in vivo. Dopamine receptor antagonists also attenuate both acute and sensitized responses to alcohol (Harrison and Nobrega, 2009). It is therefore possible that LEV affects these behaviors by decreasing excitatory neurotransmission to VTA dopaminergic neurons, which we have now demonstrated LEV can do in vitro, thereby decreasing dopamine release in forebrain targets, including the NAc. The reduction in acute alcohol-stimulated activity in the LEV/SAL mice on Day 14 replicates our finding that LEV can attenuate acute stimulation by alcohol. Interestingly, repeated LEV exposure appeared to reduce its efficacy to attenuate acute alcohol-mediated locomotor stimulation, as the reduction in the acute effects of a first alcohol exposure by LEV was greater in the chronically saline pretreated (SAL/SAL) than LEV pretreated (LEV/SAL) mice (ie, Day 14 vs Day 15 in Figure 2c). Further study with repeated LEV administration is necessary to determine if tolerance to these effects of LEV develops over time.
Previous studies in our laboratory have shown that alcohol administered by oral gavage has biphasic dose effects on BSR threshold in C57BL/6J mice (Fish et al, 2010). We found that LEV pretreatment resulted in a vertical shift in the alcohol dose-response curve, antagonizing the rewarding effects of 0.6 g/kg and enhancing the aversive-like effects of 1.7 g/kg alcohol compared with the saline-pretreated group. Alcohol-stimulated excitation of dopaminergic VTA neurons may contribute to the potentiation of BSR by the 0.6 g/kg alcohol dose. It is therefore possible that the effects of LEV on changes in ICSS by alcohol were due to inhibition of glutamatergic neurotransmission in the VTA, consistent with our in vitro results. This hypothesis is supported by the finding that NMDA receptor antagonists microinjected into the VTA suppress NAc dopamine transients evoked by MFB stimulation (Sombers et al, 2009). In addition, one in vivo microdialysis study (Fukuyama et al, 2012) has shown that local infusion of LEV decreases stimulated but not basal neurotransmitter release in a concentration-dependent manner in the medial PFC. Further studies measuring the effects of systemic LEV on neurotransmitter release using microdialysis or microvoltammetry in the NAc and other reward-relevant targets will be informative in this regard.
Similarly to alcohol, cocaine produces a dose-dependent increase in acute locomotor stimulation in mice that sensitizes with repeated treatment. In contrast to its effects on alcohol-mediated behaviors, LEV pretreatment enhanced locomotor stimulation by cocaine (15.0 mg/kg, i.p.) during the last 10 min of testing when compared with saline-pretreated mice. Repeated cocaine dosing (Days 3–7) produced behavioral sensitization in both the LEV and saline pretreatment groups, and the magnitude of sensitization was greater following LEV pretreatment. After 10 days without drug exposure, mice demonstrated a persistent sensitized response to cocaine challenge that did not depend on repeated pretreatment (Days 19 and 20), indicating that LEV did not affect the expression of behavioral sensitization. Since both cocaine-treated groups showed behavioral sensitization, it is unclear whether LEV altered cellular adaptations to repeated cocaine administration on Days 3–7, and it is possible that LEV only has an acute effect on the locomotor response to cocaine that becomes more apparent as behavioral sensitization develops. However, the absence of an acute LEV effect when comparing cocaine challenge days (Days 19 and 20) in mice from repeated treatment groups suggests that LEV may have altered the neuroadaptive response to cocaine. Since glutamate release in the NAc sensitizes with repeated cocaine administration (Pierce et al, 1996; Bell et al, 2000), further investigation using neurochemical and electrophysiological methods will be necessary to test this hypothesis.
Binding of SV2A by LEV enhances the effects of acute cocaine on locomotor activity and the development of behavioral sensitization, possibly due to events up- or downstream from glutamatergic signaling. While acute cocaine and alcohol administration both stimulate glutamate release in the NAc (Smith et al, 1995), cocaine only appears to stimulate significant glutamate release in the VTA after cocaine sensitization (Kalivas and Duffy, 1998) or a history of cocaine self-administration (You et al, 2007). Although LEV decreases excitatory neurotransmission both in vitro and in vivo, its behavioral effects are opposite to those of systemically administered glutamate receptor antagonists on the locomotor response to acute or repeated cocaine. Both AMPA (Li et al, 1997; Jackson et al, 1998) and NMDA (Karler and Calder, 1992; Kim et al, 1996) receptor antagonists attenuate acute locomotor stimulation and the development of behavioral sensitization to cocaine. Interestingly, relatively few drugs increase cocaine-induced locomotor stimulation, including mGluR2 antagonists (O'Neill et al, 2003) and low dose benzodiazepines, although GABAA receptor antagonists do not affect this behavior (Thiebot et al, 1981). Future studies will be necessary to identify the specific neural substrates of cocaine that are altered by presynaptic SV2A antagonism.
The mechanism underlying the ability of LEV to enhance the acute and sensitized locomotor response to cocaine and inhibit the response to alcohol is unknown, but may involve changes in glutamatergic neurotransmission in mesocorticolimbic circuitry during drug exposure. Although both alcohol and cocaine exert effects throughout brain reward circuitry, in general the rewarding effects of alcohol are thought mainly to involve its actions in the VTA while those of cocaine are thought mainly to involve its actions in the NAc (McBride et al, 1999). Acute cocaine administration does not stimulate significant glutamate release in VTA (reviewed in Wise, 2009) and depresses spontaneous VTA firing but not excitatory synaptic responses from descending cortical projections onto dopaminergic neurons (Almodovar-Fabregas et al, 2002). In contrast, acute alcohol increases the firing rate of dopaminergic VTA neurons in vivo both directly and through synaptic mechanisms (Morikawa and Morrisett, 2010). These differences in responses of VTA neurons to acute drug administration may explain in part why LEV blocks the development of locomotor sensitization to alcohol but not to cocaine, despite observations that increased glutamatergic sensitivity of dopaminergic VTA neurons is an early triggering event in sensitization to both cocaine and alcohol (Stuber et al, 2010).
Several investigations have demonstrated that cocaine dose dependently decreases BSR threshold in mice (Riday et al, 2012), and we found that LEV produces a leftward shift in the cocaine dose-response curve in C57BL/6J mice. These results may reflect an effect of LEV on glutamatergic neurotransmission, as the NMDA receptor antagonists MK-801 (Ranaldi et al, 1997) and memantine (Tzchentke and Schmidt, 2000) also increase the potentiation of BSR by cocaine. Cocaine, which blocks monoamine reuptake, elevates dopamine levels and decreases excitatory neurotransmission in the NAc. Since decreased NAc MSN firing and output are thought to be one common pathway by which drugs of abuse may be perceived as rewarding (Thomas et al, 2001; Beurrier and Malenka, 2002; Cheer et al, 2005; Taha and Fields, 2006), LEV may have enhanced the effects of cocaine on BSR by attenuating activity-dependent glutamate release in the NAc. In this context, it is also important to reiterate that, unlike their effects on locomotor behavior, the ability of cocaine (Bauco and Wise, 1997; Riday et al, 2012) and alcohol (our laboratory, unpublished observations) to potentiate BSR does not sensitize with repeated exposure.
In these studies, we observed that LEV oppositely affected the psychomotor and rewarding effects of alcohol and cocaine. While LEV diminished the effects of alcohol on locomotor activity, behavioral sensitization, and BSR, it augmented the effects of cocaine on these same behaviors. LEV reduced excitatory neurotransmission in vitro in both the VTA, where alcohol is thought to exert much of its rewarding effect, and the NAc, where cocaine may exert its primary rewarding effect. Our results suggest that LEV may have clinical utility in the treatment of alcohol use disorders by decreasing alcohol reward, by interfering with the development of neuroadaptations to chronic alcohol, or both. While results from open-label trials of LEV for maintenance of sobriety in alcohol-abusing patients were initially positive (Mariani and Levin, 2008; Sarid-Segal et al, 2008; Müller et al, 2010), recent double-blind placebo-controlled clinical trials have been less convincing (Fertig et al, 2012; Richter et al, 2012). However, the use of LEV as an add-on agent to other pharmacotherapy regimens has not been extensively explored (Müller et al, 2011), but may provide benefit for patients with alcohol abuse disorders. In contrast, LEV enhanced both the acute and sensitized effects of cocaine, suggesting that it may increase the abuse potential of psychostimulants. Taken together, these findings suggest that further studies in preclinical models relevant to drug and alcohol abuse in addition to carefully designed clinical trials are necessary to determine how LEV alters the behavioral effects of abused drugs.
References
Almodovar-Fabregas LJ, Segarra O, Colon N, Dones JG, Mercado M, Mejias-Aponte CA et al (2002). Effects of cocaine administration on VTA cell activity in response to prefrontal cortex stimulation. Ann NY Acad Sci 965: 157–171.
Anton RF, O'Malley SS, Ciraulo DA, Cisler RA, Couper D, Donovan DM et al (2006). Combined pharmacotherapies and behavioral interventions for alcohol dependence: the COMBINE study: a randomized controlled trial. JAMA 295: 2003–2017.
Bauco P, Wise RA (1997). Synergistic effects of cocaine with lateral hypothalamic brain stimulation reward: Lack of tolerance or sensitization. J Pharmacol Exp Ther 283: 1160–1167.
Bell K, Duffy P, Kalivas PW (2000). Context-specific enhancement of glutamate transmission by cocaine. Neuropsychopharmacology 23: 335–344.
Beurrier C, Malenka RC (2002). Enhanced inhibition of synaptic transmission by dopamine in the nucleus accumbens during behavioral sensitization to cocaine. J Neurosci 22: 5817–5822.
Bragina L, Fattorini G, Giovedi S, Melone M, Bosco F, Benfenati F et al (2011). Analysis of synaptotagmin, SV2, and Rab3 expression in cortical glutamatergic and GABAergic axon terminals. Front Cell Neurosci 5: 32.
Britt JP, Benaliouad F, McDevitt RA, Stuber GD, Wise RA, Bonci A (2012). Synaptic and behavioral profile of multiple glutamatergic inputs to the nucleus accumbens. Neuron 76: 790–803.
Broadbent J, Kampmueller KM, Koonse SA (2003). Expression of behavioral sensitization to ethanol by DBA/2J mice: The role of NMDA and non-NMDA glutamate receptors. Psychopharmacology (Berl) 167: 225–234.
Broadbent J, Weitemier AZ (1999). Dizocilpine (MK-801) prevents the development of sensitization to ethanol in DBA/2J mice. Alcohol Alcohol 34: 283–288.
Brodie MS, Appel SB (1998). The effects of ethanol on dopaminergic neurons of the ventral tegmental area studied with intracellular recording in brain slices. Alcohol Clin Exp Res 22: 236–244.
Brodie MS, Shefner SA, Dunwiddie TV (1990). Ethanol increases the firing rate of dopamine neurons of the rat ventral tegmental area in vitro. Brain Res 508: 65–69.
Camarini R, Frussa-Filho R, Monteiro MG, Calil HM (2000). MK-801 blocks the development of behavioral sensitization to the ethanol. Alcohol Clin Exp Res 24: 285–290.
Chang WP, Sudhof TC (2009). SV2 renders primed synaptic vesicles competent for Ca2+ -induced exocytosis. J Neurosci 29: 883–897.
Cheer JF, Heien ML, Garris PA, Carelli RM, Wightman RM (2005). Simultaneous dopamine and single-unit recordings reveal accumbens GABAergic responses: Implications for intracranial self-stimulation. Proc Natl Acad Sci USA 102: 19150–19155.
Crabbe JC, Johnson NA, Gray DK, Kosobud A, Young ER (1982). Biphasic effects of ethanol on open-field activity: sensitivity and tolerance in C57BL/6N and DBA/2J mice. J Comp Physiol Psychol 96: 440–451.
Custer KL, Austin NS, Sullivan JM, Bajjalieh SM (2006). Synaptic vesicle protein 2 enhances release probability at quiescent synapses. J Neurosci 26: 1303–1313.
Deng C, Li KY, Zhou C, Ye JH (2009). Ethanol enhances glutamate transmission by retrograde dopamine signaling in a postsynaptic neuron/synaptic bouton preparation from the ventral tegmental area. Neuropsychopharmacology 34: 1233–1244.
Doheny HC, Ratnaraj N, Whittington MA, Jefferys JG, Patsalos PN (1999). Blood and cerebrospinal fluid pharmacokinetics of the novel anticonvulsant levetiracetam (UCB 1059) in the rat. Epilepsy Res 34: 161–168.
Esposito R, Kornetsky C (1977). Morphine lowering of self-stimulation thresholds: Lack of tolerance with long-term administration. Science 195: 189–191.
Fertig JB, Ryan ML, Falk DE, Litten RZ, Mattson ME, Ransom J et al (2012). A double-blind, placebo-controlled trial assessing the efficacy of levetiracetam extended-release in very heavy drinking alcohol-dependent patients. Alcohol Clin Exp Res 36: 1421–1430.
Fish EW, Riday TT, McGuigan MM, Faccidomo S, Hodge CW, Malanga CJ (2010). Alcohol, cocaine, and brain stimulation-reward in C57BL6/J and DBA2/J mice. Alcohol Clin Exp Res 34: 81–89.
Fukuyama K, Tanahashi S, Nakagawa M, Yamamura S, Motomura E, Shiroyama T et al (2012). Levetiracetam inhibits neurotransmitter release associated with CICR. Neurosci Lett 518: 69–74.
Gass JT, Olive MF (2008). Glutamatergic substrates of drug addiction and alcoholism. Biochem Pharmacol 75: 218–265.
Gessa GL, Muntoni F, Collu M, Vargiu L, Mereu G (1985). Low doses of ethanol activate dopaminergic neurons in the ventral tegmental area. Brain Res 348: 201–203.
Gillard M, Chatelain P, Fuks B (2006). Binding characteristics of levetiracetam to synaptic vesicle protein 2A (SV2A) in human brain and in CHO cells expressing the human recombinant protein. Eur J Pharmacol 536: 102–108.
Harrison SJ, Nobrega JN (2009). A functional role for the dopamine D3 receptor in the induction and expression of behavioural sensitization to ethanol in mice. Psychopharmacology (Berl) 207: 47–56.
Jackson A, Mead AN, Rocha BA, Stephens DN (1998). AMPA receptors and motivation for drug: Effect of the selective antagonist NBQX on behavioural sensitization and on self-administration in mice. Behav Pharmacol 9: 457–467.
Kalivas PW, Duffy P (1998). Repeated cocaine administration alters extracellular glutamate in the ventral tegmental area. J Neurochem 70: 1497–1502.
Karler R, Calder LD (1992). Excitatory amino acids and the actions of cocaine. Brain Res 582: 143–146.
Kim HS, Park WK, Jang CG, Oh S (1996). Inhibition by MK-801 of cocaine-induced sensitization, conditioned place preference, and dopamine-receptor supersensitivity in mice. Brain Res Bull 40: 201–207.
Lessov CN, Palmer AA, Quick EA, Phillips TJ (2001). Voluntary ethanol drinking in C57BL/6J and DBA/2J mice before and after sensitization to the locomotor stimulant effects of ethanol. Psychopharmacology (Berl) 155: 91–99.
Li Y, Vartanian AJ, White FJ, Xue CJ, Wolf ME (1997). Effects of the AMPA receptor antagonist NBQX on the development and expression of behavioral sensitization to cocaine and amphetamine. Psychopharmacology (Berl) 134: 266–276.
Lominac KD, Kapasova Z, Hannun RA, Patterson C, Middaugh LD, Szumlinski KK (2006). Behavioral and neurochemical interactions between group 1 mGluR antagonists and ethanol: Potential insight into their anti-addictive properties. Drug Alcohol Depend 85: 142–156.
Lynch BA, Lambeng N, Nocka K, Kensel-Hammes P, Bajjalieh SM, Matagne A et al (2004). The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Natl Acad Sci USA 101: 9861–9866.
Margineanu DG, Klitgaard H (2003). Levetiracetam has no significant gamma-aminobutyric acid-related effect on paired-pulse interaction in the dentate gyrus of rats. Eur J Pharmacol 466: 255–261.
Mariani JJ, Levin FR (2008). Levetiracetam for the treatment of co-occurring alcohol dependence and anxiety: Case series and review. Am J Drug Alcohol Abuse 34: 683–691.
McBride WJ, Murphy JM, Ikemoto S (1999). Localization of brain reinforcement mechanisms: Intracranial self-administration and intracranial place-conditioning studies. Behav Brain Res 101: 129–152.
Meehan AL, Yang X, McAdams BD, Yuan L, Rothman SM (2011). A new mechanism for antiepileptic drug action: Vesicular entry may mediate the effects of levetiracetam. J Neurophysiol 106: 1227–1239.
Meehan AL, Yang X, Yuan LL, Rothman SM (2012). Levetiracetam has an activity-dependent effect on inhibitory transmission. Epilepsia 53: 469–476.
Melón LC, Boehm SL (2011). Role of genotype in the development of locomotor sensitization to alcohol in adult and adolescent mice: Comparison of the DBA/2J and C57BL/6J inbred mouse strains. Alcohol Clin Exp Res 35: 1351–1360.
Miliaressis E, Rompre PP, Laviolette P, Philippe L, Coulombe D (1986). The curve-shift paradigm in self-stimulation. Physiol Behav 37: 85–91.
Moghaddam B, Bolinao ML (1994). Biphasic effect of ethanol on extracellular accumulation of glutamate in the hippocampus and the nucleus accumbens. Neurosci Lett 178: 99–102.
Morikawa H, Morrisett RA (2010). Ethanol action on dopaminergic neurons in the ventral tegmental area: Interaction with intrinsic ion channels and neurotransmitter inputs. Int Rev Neurobiol 91: 235–288.
Müller CA, Schafer M, Banas R, Heimann HM, Volkmar K, Forg A et al (2011). A combination of levetiracetam and tiapride for outpatient alcohol detoxification: a case series. J Addict Med 5: 153–156.
Müller CA, Schafer M, Schneider S, Heimann HM, Hinzpeter A, Volkmar K et al (2010). Efficacy and safety of levetiracetam for outpatient alcohol detoxification. Pharmacopsychiatry 43: 184–189.
Nowack A, Yao J, Custer KL, Bajjalieh SM (2010). SV2 regulates neurotransmitter release via multiple mechanisms. Am J Physiol Cell Physiol 299: C960–C967.
O'Neill MF, Heron-Maxwell C, Conway MW, Monn JA, Ornstein P (2003). Group II metabotropic glutamate receptor antagonists LY341495 and LY366457 increase locomotor activity in mice. Neuropharmacology 45: 565–574.
Olds J, Milner P (1954). Positive reinforcement produced by electrical stimulation of septal area and other regions of rat brain. J Comp Physiol Psychol 47: 419–427.
Paxinos GT, Franklin KB (1996) The Mouse Brain in Stereotaxic Coordinates. Academic Press: San Diego, USA.
Pierce RC, Bell K, Duffy P, Kalivas PW (1996). Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J Neurosci 16: 1550–1560.
Ranaldi R, Bauco P, Wise RA (1997). Synergistic effects of cocaine and dizocilpine (MK-801) on brain stimulation reward. Brain Res 760: 231–237.
Richter C, Effenberger S, Bschor T, Bonnet U, Haasen C, Preuss UW et al (2012). Efficacy and safety of levetiracetam for the prevention of alcohol relapse in recently detoxified alcohol-dependent patients: a randomized trial. J Clin Psychopharmacol 32: 558–562.
Riday TT, Kosofsky BE, Malanga CJ (2012). The rewarding and locomotor-sensitizing effects of repeated cocaine administration are distinct and separable in mice. Neuropharmacology 62: 1858–1866.
Sarid-Segal O, Piechniczek-Buczek J, Knapp C, Afshar M, Devine E, Sickles L et al (2008). The effects of levetiracetam on alcohol consumption in alcohol-dependent subjects: an open label study. Am J Drug Alcohol Abuse 34: 441–447.
Schivell AE, Mochida S, Kensel-Hammes P, Custer KL, Bajjalieh SM (2005). SV2A and SV2C contain a unique synaptotagmin-binding site. Mol Cell Neurosci 29: 56–64.
Smith JA, Mo Q, Guo H, Kunko PM, Robinson SE (1995). Cocaine increases extraneuronal levels of aspartate and glutamate in the nucleus accumbens. Brain Res 683: 264–269.
Sombers LA, Beyene M, Carelli RM, Wightman RM (2009). Synaptic overflow of dopamine in the nucleus accumbens arises from neuronal activity in the ventral tegmental area. J Neurosci 29: 1735–1742.
Stuber GD, Hopf FW, Tye KM, Chen BT, Bonci A (2010). Neuroplastic alterations in the limbic system following cocaine or alcohol exposure. Curr Top Behav Neurosci 3: 3–27.
Taha SA, Fields HL (2006). Inhibitions of nucleus accumbens neurons encode a gating signal for reward-directed behavior. J Neurosci 26: 217–222.
Thiebot MH, Kloczko J, Chermat R, Puech AJ, Soubrie P, Simon P (1981). Enhancement of cocaine-induced hyperactivity in mice by benzodiazepines: evidence for an interaction of GABAergic processes with catecholaminergic neurons? Eur J Pharmacol 76: 335–343.
Thomas MJ, Beurrier C, Bonci A, Malenka RC (2001). Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci 4: 1217–1223.
Tzchentke TM, Schmidt WJ (2000). Effects of the non-competitive NMDA-receptor antagonist memantine on morphine- and cocaine-induced potentiation of lateral hypothalamic brain stimulation reward. Psychopharmacology (Berl) 149: 225–234.
Vanderschuren LJ, Kalivas PW (2000). Alterations in dopaminergic and glutamatergic transmission in the induction and expression of behavioral sensitization: a critical review of preclinical studies. Psychopharmacology (Berl) 151: 99–120.
Verbanck P, Seutin V, Dresse A, Scuvee J, Massotte L, Giesbers I et al (1990). Electrophysiological effects of ethanol on monoaminergic neurons: An in vivo and in vitro study. Alcohol Clin Exp Res 14: 728–735.
Wan QF, Zhou ZY, Thakur P, Vila A, Sherry DM, Janz R et al (2010). SV2 acts via presynaptic calcium to regulate neurotransmitter release. Neuron 66: 884–895.
Wise RA (1996). Addictive drugs and brain stimulation reward. Annu Rev Neurosci 19: 319–340.
Wise RA (2005). Forebrain substrates of reward and motivation. J Comp Neurol 493: 115–121.
Wise RA (2009). Ventral tegmental glutamate: A role in stress-, cue-, and cocaine-induced reinstatement of cocaine-seeking. Neuropharmacology 56 (Suppl 1): 174–176.
Xiao C, Shao XM, Olive MF, Griffin WC, Li KY, Krnjevic K et al (2009). Ethanol facilitates glutamatergic transmission to dopamine neurons in the ventral tegmental area. Neuropsychopharmacology 34: 307–318.
Yang XF, Rothman SM (2009). Levetiracetam has a time- and stimulation-dependent effect on synaptic transmission. Seizure 18: 615–619.
Yang XF, Weisenfeld A, Rothman SM (2007). Prolonged exposure to levetiracetam reveals a presynaptic effect on neurotransmission. Epilepsia 48: 1861–1869.
You ZB, Chen YQ, Wise RA (2001). Dopamine and glutamate release in the nucleus accumbens and ventral tegmental area of rat following lateral hypothalamic self-stimulation. Neuroscience 107: 629–639.
You ZB, Wang B, Zitzman D, Azari S, Wise RA (2007). A role for conditioned ventral tegmental glutamate release in cocaine seeking. J Neurosci 27: 10546–10555.
Acknowledgements
We are very grateful to Dr Joyce Besheer and Dr Garret Stuber for their advice and helpful comments in the preparation of this manuscript. This work was funded by grants from the National Institute on Alcohol Abuse and Alcoholism to CJM (AA 018335), CWH (AA 014983 and AA 011605), and JER (AA 021312).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that, except for income from primary employers, no financial support or compensation has been received from any institutional or corporate entity over the past three years for research or professional service, and there are no personal holdings that could be perceived as constituting a potential conflict of interest.
Rights and permissions
About this article
Cite this article
Robinson, J., Chen, M., Stamatakis, A. et al. Levetiracetam Has Opposite Effects on Alcohol- and Cocaine-Related Behaviors in C57BL/6J Mice. Neuropsychopharmacol 38, 1322–1333 (2013). https://doi.org/10.1038/npp.2013.30
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2013.30
Keywords
This article is cited by
-
Receptor Reserve Moderates Mesolimbic Responses to Opioids in a Humanized Mouse Model of the OPRM1 A118G Polymorphism
Neuropsychopharmacology (2015)
-
Effects of the neuroactive steroid allopregnanolone on intracranial self-stimulation in C57BL/6J Mice
Psychopharmacology (2014)
