
Abstract
The midbrain dorsal periaqueductal gray (dPAG) has an important role in orchestrating anxiety- and panic-related responses. Given the cellular and behavioral evidence suggesting opposite functions for cannabinoid type 1 receptor (CB1) and transient receptor potential vanilloid type-1 channel (TRPV1), we hypothesized that they could differentially influence panic-like reactions induced by electrical stimulation of the dPAG. Drugs were injected locally and the expression of CB1 and TRPV1 in this structure was assessed by immunofluorescence and confocal microscopy. The CB1-selective agonist, ACEA (0.01, 0.05 and 0.5 pmol) increased the threshold for the induction of panic-like responses solely at the intermediary dose, an effect prevented by the CB1-selective antagonist, AM251 (75 pmol). Panicolytic-like effects of ACEA at the higher dose were unmasked by pre-treatment with the TRPV1 antagonist capsazepine (0.1 nmol). Similarly to ACEA, capsazepine (1 and 10 nmol) raised the threshold for triggering panic-like reactions, an effect mimicked by another TRPV1 antagonist, SB366791 (1 nmol). Remarkably, the effects of both capsazepine and SB366791 were prevented by AM251 (75 pmol). These pharmacological data suggest that a common endogenous agonist may have opposite functions at a given synapse. Supporting this view, we observed that several neurons in the dPAG co-expressed CB1 and TRPV1. Thus, the present work provides evidence that an endogenous substance, possibly anandamide, may exert both panicolytic and panicogenic effects via its actions at CB1 receptors and TRPV1 channels, respectively. This tripartite set-point system might be exploited for the pharmacotherapy of panic attacks and anxiety-related disorders.
Similar content being viewed by others

INTRODUCTION
Panic disorder is a subtype of anxiety spectrum disorders characterized by recurrent episodes of panic attacks, which comprise intense feelings of fear and distress, accompanied by tachycardia, hyperventilation and increased blood pressure (Katon, 2006; Roy-Byrne et al, 2006). Although its precise neural basis has remained uncertain, evidence indicates that it might result from a malfunction in brain systems related to defense reactions (McNaughton and Gray, 2000). Accordingly, panic attacks exhibit isomorphism with flight responses regarding their autonomic, behavioral and psychological aspects (Bandler et al, 2000; McNaughton and Gray, 2000).
Among the brain regions that may participate in the elaboration of both panic and innate defense reactions is the midbrain dorsal periaqueductal gray (dPAG), comprising dorsomedial and dorsolateral columns (Del-Ben and Graeff, 2009; Schenberg et al, 2001). In humans, electrical stimulation of this structure induces an intensive feeling of aversion, fear and imminent death along with autonomic changes (Nashold et al, 1969). Moreover, neuroimaging studies detected increased activity in this region in patients suffering from panic (Del-Ben and Graeff, 2009) and in healthy volunteers exposed to a proximal threatening stimulus (Mobbs et al, 2007). In laboratory rats, local electrical or chemical dPAG stimulation induces autonomic changes along with escape responses (Beckett and Marsden, 1995; Krieger and Graeff, 1985; Schenberg et al, 2001). In addition, systemic or intradPAG injections of panicolytic drugs increase the threshold required to induce escape responses (Hogg et al, 2006; Jenck et al, 1995; Schenberg et al, 2001; Schütz et al, 1985). Due to its robust face, construct and predictive validity, the escape reaction resulting from electrical stimulation of the rat dPAG has been proposed as a model of panic attacks (Beckett and Marsden, 1995; Del-Ben and Graeff, 2009; Jenck et al, 1995; Schenberg et al, 2001).
Diverse types of receptors are expressed in the PAG, including the G-protein coupled cannabinoid type 1 receptor (CB1) and the transient receptor potential type-1 (TRPV1) channel (Cavanaugh et al, 2011; Cristino et al, 2006; Herkenham et al, 1990, 1991; Mezey et al, 2000; Tóth et al, 2005; Tsou et al, 1998). These receptors may bind common endogenous ligands, which are thus termed endocannabinoids/endovanilloids, such as arachidonoyl ethanolamide (anandamide) (for reviews, see Di Marzo, 2008; Di Marzo et al, 2001 and Starowicz et al, 2007a). Despite this similarity, electrophysiological and neurochemical pieces of evidence indicate that they may have opposite functions. In the PAG, local CB1 activation inhibits calcium channels and excitatory neuronal activity (Vaughan et al, 2000), whereas TRPV1 promotes calcium influx and glutamate release (Starowicz et al, 2007b; Xing and Li, 2007). At the behavioral level, CB1 knockout mice show deficits in extinction and habituation of conditioned fear (Kamprath et al, 2006; Marsicano et al, 2002) and exacerbated anxiety-like behavior (Haller et al, 2002, 2004; Jacob et al, 2009; Martin et al, 2002). On the contrary, TRPV1 knockout mice show reduced fear- and anxiety-like responses (Marsch et al, 2007). Finally, either local activation of CB1 or blockade of TRPV1 at level of the dorsolateral columns of the PAG-induced anxiolytic-like effects (Lisboa et al, 2008; Moreira et al, 2007, 2009; Terzian et al, 2009). The same is observed with either systemic or intraprefrontal cortex injections (Aguiar et al, 2009; Micale et al, 2009; Rubino et al, 2008).
So far, most studies have investigated the roles of CB1 and TRPV1 separately. However, there is evidence that they may act in concert to modulate behavioral responses (Maione et al 2006; Micale et al, 2009; Rubino et al, 2008). They are co-expressed in several regions related to aversion, such as prefrontal cortex, amygdaloid complex and hippocampus, where they may modulate anxiety-related responses (Cristino et al, 2006; Micale et al, 2009), as well as in the ventrolateral PAG (vlPAG), where they control nociceptive reactions (Cristino et al, 2006; Maione et al, 2006).
Considering this background, the present study tested the hypothesis that CB1 and TRPV1 exert opposite effects on panic-like responses at level of the dPAG and looked for interdependences of the two signaling systems.
MATERIALS AND METHODS
Animals
Male Wistar rats were obtained from the animal facilities at the University of São Paulo, Ribeirão Preto. The animals, weighing 300–330 g, were housed in groups of 4–6 per cage under a 12-h dark/12-h light cycle (lights on at 0700 h) at 22±1 °C, and given free access to food and water. All experiments were carried in accordance with the Brazilian Society of Neuroscience and Behavior for the care and use of laboratory animals and were approved by the Experimental Animal Ethical Committee of University of São Paulo (protocol number: 114/2007). All efforts were made to minimize animal suffering.
Surgery
Rats were anesthetized with 2,2,2 tribromoethanol (250 mg/kg i.p.) combined with local anesthesia (2% lidocaine with a vasoconstrictor; Harvey, Brazil), and fixed in a stereotaxic frame (David Kopf) for implantation of a chemitrode in the dPAG (incisor bar 3.3 mm below the interaural line, 1.7 mm lateral to lambda at an angle of 22° with the sagittal plane, 5.0 mm below the surface of the skull; according to Paxinos and Watson, 1997). The chemitrode consisted of a stainless steel guide cannula (outer diameter: 0.6 mm, length: 13 mm) glued to a brain electrode made of stainless steel wire (diameter: 250 μm) that was insulated by enamel except for the cross-section of the tip. The electrode reached 1 mm below the end of the cannula. It was connected to a male pin parallel to the outer end of the cannula that could be plugged into an amphenol socket at the end of a flexible electrical cable. The chemitrode was fixed to the skull by means of acrylic resin and two stainless steel screws. A stylet was inserted into the guide cannula in order to prevent obstruction. At the end of the surgery, animals were treated with an antibiotic (Pentabiótico, Fort Dodge, Brazil; 1.0 ml/kg, i.m.) and fluxinin meglumine (Schering-Plough, Brazil; 2.5 mg/kg, s.c.), which has analgesic, antipyretic and anti-inflammatory properties. The animals were left undisturbed for 5–7 days after the surgery.
Drugs and Treatment
The CB1 agonist, arachidonyl-2-chloro-ethylamide (ACEA; Tocris) was dissolved in Tocrisolve (vehicle). The CB1 antagonist, N-(piperidin-1yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM251; Tocris) and the TRPV1 antagonists, capsazepine (Tocris) and 4′-Chloro-3-methoxycinnamanilide (SB366791; Tocris), were dissolved in DMSO.
For local drug treatment, an injection needle (outer diameter: 0.3 mm) was inserted through the guide into the dPAG to a final depth of 6.0 mm below the surface of the skull. A volume of 0.2 μl was injected for 60 s using a 10-μl microsyringe (Hamilton) attached to a microinfusion pump (KD Scientific). The needle was left in place for another 60 s before removal.
Escape Threshold Determination
Escape behavior induced by dPAG electrical stimulation was performed in accordance with previous work (Casarotto et al, 2010). The animals were evaluated in a circular arena (40 cm in diameter) with 40 cm high walls made of transparent Plexiglas. Brain stimuli were generated by a sine-wave stimulator. The stimulation current (peak-to-peak) was monitored on the screen of an oscilloscope (Minipa, Brazil). The brain electrode was connected to the stimulator by means of an electromechanical swivel and a flexible cable, allowing ample movement of the animal inside the experimental cage. The escape threshold was determined in response to an electrical stimulus (AC, 60 Hz, 10 s) presented through the chemitrode. The interstimulus interval was 10 s. The current intensity started at 20 μA and was increased by steps of 4 μA until the rat started running around the circular arena (escape behavior). In most cases, this vigorous reaction was accompanied by vertical jumps. When these behaviors were observed, electrical stimulation to the dPAG was interrupted by the experimenter that was blind to the treatment groups. The basal escape threshold was defined as the lowest current intensity that evoked escape in three successive trials of electrical stimulation. Animals with basal thresholds >150 μA were excluded from the study. Following basal escape threshold determination, independent groups of animals were injected with the drugs or vehicle into the dPAG, and the escape threshold was re-analyzed 10 min later. The variation in escape threshold (Δ) was then calculated for each animal and refers to the difference between escape threshold values obtained post- and pre-treatment.
Experiments
Independent batches of animals were used in each experiment. In experiment 1, the rats were treated with ACEA (0.01, 0.05 and 0.5 pmol) or vehicle (n=6–7). In experiment 2, new groups of animals received AM251 (75 pmol) or its vehicle followed by ACEA (0.05 pmol) or its respective vehicle 5 min later (n=5–6). In experiment 3, they were injected with capsazepine (0.1, 1 and 10 nmol) or vehicle (n=6–9). In experiment 4, capsazepine (0.1 nmol) or vehicle injections were followed by ACEA (0.5 pmol) 5 min later (n=5 per group). In experiment 5, the rats were treated with AM251 (75 pmol) or vehicle followed by capsazepine (10 nmol) or vehicle 5 min later (n=5–7). Finally, in experiment 6, they were treated with AM251 (75 pmol) or vehicle followed by SB366791 (10 nmol) or vehicle 5 min later (n=5–7). The doses of ACEA, AM 251 and capsazepine were selected based on previous works performing injections into the dPAG (Moreira et al, 2007; Terzian et al, 2009). The dose of SB366791 was selected based on the affinity of this substance for TRPV1 as compared with other antagonists (Gawa et al, 2005; Gunthorpe et al, 2004).
Histology
After the experiments, animals were deeply anesthetized with chloral hydrate. The brain was perfused through the heart with saline solution followed by 10% formalin solution, before being removed and fixed in 10% formalin. Frozen sections of 55 μm were cut using a microtome to localize the positions of the electrode tips according to the atlas by Paxinos and Watson (1997). Only data from rats having chemitrode tips inside the dPAG were included in the statistical analysis.
Immunofluorescence
Male Wistar rats were anaesthetized with urethane and transcardially perfused with saline followed by 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (PBS, pH 7.4). The brain was removed and post-fixed over 2 h in PFA 4% and stored for at least 30 h in 30% sucrose for cryoprotection. Coronal sections (25 μm) were obtained in a cryostat and were washed in PBS 0.01 M. After this, the sections were incubated in glycine 0.1 M, washed again and incubated for 1 h in 5% bovine serum (BSA, sigma) in PBS 0.01 M, pH 7.4 containing 0.3% Triton X-100. Thereafter, the sections were incubated for 2 days at 4 °C in a mixture of anti-CB1 receptor N-terminus (1 : 250; Abcam raised in rabbit) to anti-TRPV1 receptor N-terminus (1 : 100; Santa Cruz raised in goat) diluted in PBS 0.01 M containing BSA 5% and 0.3% Triton X-100. After incubation in the primary antiserum, the tissue sections were washed in PBS and sequentially incubated in a mixture of Alexa 594 donkey anti-rabbit IgGs (1 : 1000; Molecular Probes, Eugene, OR) and Alexa 488 donkey anti-goat IgGs (1 : 1000; Molecular Probes) for 1 h. Slides were rinsed in PBS and coverslipped with Fluormount-G (Electron Microscopy Sciences, Hatfield, PA). Images were first obtained using an Olympus BX50 microscope through computerized image system (Image Pro-Plus 4.0, Media Cybernetics). For a more appropriate analysis of double-stained cells, the slides were re-examined using a modified Olympus BX61WI confocal microscope in an upright configuration, using an Argonium laser at 488 nm. Immunostained slides (Alexa Fluor 488-coupled anti-CB1 receptors; Alexa Fluor 546-coupled anti-TRPV1 receptors) were exposed for 2.71 s and green and red channels were merged digitally by Fluoview software. Images were further exported in TIFF format. The specificity of the primary antibodies has been demonstrated before by means of CB1 and TRPV1 knockout mice (Maione et al, 2006). Nevertheless, we additionally performed negative controls for non-specific labeling. The protocol was the same as described above, except for the fact that the primary antibodies for CB1 and TRPV1 were switched, so that each protocol would include a primary antibody for one receptor and a secondary for the other.
Statistical Analysis
The effects of intradPAG drug injections were analyzed by Student’s t-test or by one-way analysis of variance (ANOVA) followed by Newman-Keuls post hoc test, when appropriate. Data were expressed as mean±SEM. Statistical significance was accepted if p<0.05.
RESULTS
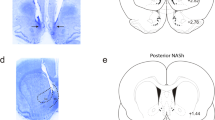
The sites of electrical stimulation and drug injection can be seen in Figure 1. Electrical stimulation of the dPAG (black dots), but not of surrounding regions (gray dots), caused an explosive panic response, which was characterized by running and jumping. Activation of CB1 receptors by local administration of ACEA modified the effects of electrical stimulation in a bell-shaped manner. It increased the threshold for inducing escape behavior at 0.05 pmol, though not at 0.01 or 0.5 pmol (F(3,23)=4.06; p<0.05; Figure 2). The panicolytic-like effect of 0.05 nmol ACEA was mediated by CB1 receptors, since it could be blocked by pre-treatment with the selective antagonist AM251 (75 pmol) (F(3,17)=13.86; p<0.01; Figure 3). Local blockade of TRPV1 channels by capsazepine also raised the escape threshold at 1 and 10 nmol (F(3,25)=16.58; p<0.05; Figure 4).

Schematic representation of coronal sections from interaural line of the rat brain (Paxions and Watson, 1997) showing the injection/electrical stimulation sites inside (dark circles) of the dPAG. Sites where electrical stimulation failed to induce escape responses are also presented (gray circles). Due to overlaps, the number of points represented is fewer than the number of rats actually employed in the experiments.

Local injection of the selective CB1-receptor agonist, ACEA, into the dPAG increases the threshold of electric current necessary for inducing panic-like response (n=7, 6, 7, 7). Following basal escape threshold determination, independent groups of animals were injected with vehicle or ACEA (0.01–0.5 pmol), and the escape threshold was re-analyzed 10 min later. The variation in escape threshold (Δ) was then calculated for each animal and refers to the difference between escape threshold values obtained post- and pre-treatment. *p<0.05 Compared with all other groups (one-way ANOVA followed by Newman-Keuls post hoc test).

Local pre-treatment (5 min) with the CB1 antagonist, AM251 (75 pmol), prevents ACEA (0.05 pmol)-induced panicolytic-like effect (n=5, 6, 5, 5). *p<0.05 Compared with all other groups (one-way ANOVA followed by Newman-Keuls post hoc test). For further details, see legend to Figure 2.

Local injection of the TRPV1 antagonist, capsazepine (CPZ; 0.1–10 nmol), into the dPAG increases the threshold of electric current required for inducing panic-like response (n=9, 7, 6, 7, 7). *p<0.05 Compared with vehicle-treated rats (one-way ANOVA followed by Newman-Keuls post hoc test). For further details, see legend to Figure 2.
These results suggest that TRPV1 has diametrically opposite functions as compared with CB1. Considering that certain cannabinoids may also bind TRPV1 at higher concentrations (Huang et al, 2002; Price et al, 2004; Silveira et al, 2010; Smart et al, 2000; Zygmunt et al, 1999), simultaneous CB1 and TRPV1 activation could explain the bell-shaped effects observed with ACEA in the first experiment (Figure 2). High-dose ACEA could activate both CB1 and TRPV1, occluding the CB1-mediated effects. Indeed, an ineffective dose of capsazepine (0.1 nmol) unmasked the panicolytic-like effect of ACEA (0.5 pmol) (t(8)=3.48; p<0.01; Figure 5).

Local pre-treatment (5 min) with the TRPV1 antagonist, capsazepine (CPZ; 0.1 nmol), unmasks the panicolytic-like effect of high-dose ACEA (0.5 pmol) (n=5 per group). *p<0.05 Compared with all other groups (one-way ANOVA followed by Newman-Keuls post hoc test). For further details, see legend to Figure 2.
Finally, we tested the hypothesis that the panicolytic-like effect of TRPV1 blockade would occur because it would shift the action of a common endogenous agonist toward CB1 activation. Supporting this hypothesis, the panicolytic-like effect of capsazepine at higher doses (10 nmol) could be prevented by pre-treatment with AM251 (75 pmol), demonstrating its dependency on CB1 signaling (F(3,20)=5.74; p<0.01; Figure 6). Remarkably, this data could be reproduced with a more selective TRPV1 antagonist, SB366791 (10 nmol), which increased the threshold for panic-like responses to the same extent as capsazepine, again in a CB1-dependent manner (75 nmol), as presented in Figure 7 (F(3,20)=16.3; p<0.01; Figure 7).

Local pre-treatment (5 min) with the CB1 antagonist, AM251 (75 pmol), prevents the panicolytic-like effect of capsazepine (CPZ; 10 nmol)-induced (n=5, 7, 5, 7). *p<0.05 Compared with all other groups (one-way ANOVA followed by Newman-Keuls post hoc test). For further details, see legend to Figure 2.

Local pre-treatment (5 min) with the CB1 antagonist, AM251 (75 pmol), prevents the panicolytic-like effect of SB366791 (10 nmol) (n=6, 6, 5, 7). *p<0.05 Compared with all other groups (one-way ANOVA followed by Newman-Keuls post hoc test). For further details, see legend to Figure 2.
At the cellular level, both CB1 and TRPV1 are expressed in neurons within the dPAG (Figure 8). Immunofluorescent labeling revealed neurons that were exclusively CB1 or TRPV1 positive. However, many neurons co-expressed both receptors in their cell bodies. This could be certified through confocal microscopy examination, which allowed us to distinguish double-stained neurons from pairs of mono-stained neurons located in adjacent plans. There was a medium degree of colocalization throughout the dorsolateral and dorsomedial aspects of the PAG. This pattern of co-expression supports the notion that they could be simultaneously activated at a given synapse. Importantly, no staining was observed in protocols in which the antibodies were switched, so that the primary for one receptor was combined with the secondary for the other (Figure 9). This re-assures the specificity of the antibodies used in the present protocols.

CB1 receptors and TRPV1channels are co-expressed in neurons in the dPAG. The photomicrographs (25 μm thick section) show image of double-label immunofluorescence of CB1 (red, Alexa Fluor 594; a, d) and TRPV1 receptors (green, Alexa Fluor 488; b, e), as revealed by fluorescence (a–c) and confocal (d–f) microscopy. Panel (c) (the composite images of (a, b)) shows neurons expressing both receptors. One representative double-stained neuron is shown in higher magnification in f (arrow). Scale bars: 100 μm (a–c) and 50 μm (d–f).

Negative controls for the double-labeled immunofluorescence. The photomicrographs (25 μm thick section) show images of the dPAG from protocols combining TRPV1 secondary (Alexa Fluor 488) with CB1 primary antibodies (a) or CB1 secondary (Alexa Fluor 594) with TRPV1 primary antibodies (b). Images resulting from protocol in which the primary antibodies were omitted as seen in the microscopy though filters for Alexa Fluor 488 (c) and Alexa Fluor 594 (d) are also shown.
DISCUSSION
The present work provides evidence for opposite functions of CB1 and TRPV1 in an animal model of panic attacks, the electrical stimulation of the dPAG. In this model, local application of antipanic drugs increases the current threshold necessary for the induction of explosive escape behaviors. We demonstrated that the selective CB1 agonist, ACEA, increased the threshold for inducing panic-like reaction in a bell-shaped manner. This panicolytic-like effect was completely prevented by the CB1 receptor antagonist, AM251. Although this effect was lost in a higher dose, it could be unmasked by previous treatment with an inactive dose of capsazepine, a TRPV1 antagonist. In addition, pharmacological blockade of TRPV1 per se also exerted panicolytic-like effects, suggesting the existence of tonic modulation by TRPV1, whose function opposed that of CB1. Remarkably, capsazepine effect could be prevented by AM251, revealing that the effect of TRPV1 blockade was completely dependent on CB1 signaling. Noteworthy, capsazepine effect could be mimicked by another TRPV1 antagonist, SB366791, whose action was also prevented by AM251. Together with the co-expression of CB1 and TRPV1 observed at level of the dPAG, these data suggest the existence of a set-point system, whereby activation of TRPV1 vs CB1 balances the induction of panic-like responses.
The dPAG model of panic has some limitations in terms of a broader interpretation in relationship to human psychopathology. It mimics only the panic attacks, which actually occur with several other symptoms of panic disorder, such as behavioral and physiological signs of intense anxiety, fear and distress (Katon, 2006; Roy-Byrne et al, 2006). Moreover, it is based on the measure of an all-or-none response to electrical stimulation of a specific brain structure, which is only part of a more complex neural circuitry generating fear, anxiety and panic responses (Del-Ben and Graeff, 2009). Despite of these drawbacks, this model presents face, construct and predictive validities (Jenck et al, 1995; Beckett and Marsden, 1995; Del-Ben and Graeff, 2009; Schenberg et al, 2001). The face validity is supported by the fact that the behavioral and autonomic responses observed in this model resemble those seen in panic attacks (Schenberg et al, 2001). As for the theoretical construct, the PAG is a midbrain structure activated during escape responses induced by natural aversive stimulus either in laboratory animals or in healthy volunteers (Dielenberg et al, 2001; Mobbs et al, 2007). Neurosurgery patients, in whom this structure has been electrically stimulated, reported feelings of fear and despair similar to those described in panic patients (Nashold et al, 1969). Finally, and more relevant for the present study, the pharmacological predictability of the model is supported by the observations that drugs used to treat panic disorder increase the threshold of escape responses induced by electrical stimulation of the dPAG (Jenck et al, 1995; Schenberg et al, 2001; Schütz et al, 1985).
In this model, ACEA induced a panicolytic-like effect in an intermediary dose, which could be prevented by AM251, in line with previous experiments in anxiety-related reactions (Moreira et al, 2007). These results support the concept of general anti-aversive effects of CB1 signaling at level of the PAG. Accordingly, local injections of other cannabinoids inhibit the escape responses induced by local injection of an excitatory amino acid (Finn et al, 2003) and promote anxiolytic-like effects in several animal models (for a review, see Moreira et al, 2009).
Contrary to CB1, TRPV1 seems to mediate pro-aversive effects, since we detected a clear panicolytic-like effect after its blockade with capsazepine or SB366791. Moreover, an ineffective dose of capsazepine was able to unmask the panicolytic-like effects of higher dose of ACEA. Therefore, the bell-shaped dose–response curve of ACEA seems to result from an activation of CB1 at lower concentrations (panicolytic), whereas higher concentration of ACEA also activates the less affine TRPV1 channels (panicogenic), counteracting CB1-mediated actions, resulting in a net null effect. This is in line with ACEA affinities profile, binding CB1 in lower dose (Gawa et al, 2005) and both CB1 and TRPV1 in higher dose (Huang et al, 2002; Price et al, 2004; Silveira et al, 2010; Smart et al, 2000; Zygmunt et al, 1999).
The panicolytic consequences of TRPV1 antagonists speak for a tonic activation of TRPV1 under basal conditions. This is in accordance with previous studies, which reported decreased anxiety upon pharmacological or genetic interruption of TRPV1 signaling. For instance, systemic injection of capsazepine reduced anxiety-like behavior in rats (Kasckow et al, 2004). Moreover, a detailed investigation on the behavior of TRPV1 knockout mice unveiled a phenotype of reduced anxiety-like behavior in the elevated plus maze and in the light-dark box, as well as reduced fear conditioning (Marsch et al, 2007). In addition, the effect of systemically administered capsazepine is mimicked by local injection into specific structures, namely the ventral hippocampus (Santos et al, 2008), the dorsolateral PAG (Terzian et al, 2009) and the prefrontal cortex (Aguiar et al, 2009; Rubino et al, 2008). This is consonant with neurochemical and electrophysiological data showing that this ion channel stimulates calcium influx and glutamate release in the PAG, increasing local neuronal activity (Starowicz et al, 2007b; Xing and Li, 2007). In addition, it has been demonstrated in other brain regions related to anxiety and emotional processing, such as the hippocampus and the nucleus accumbens, that anandamide acts upon TRPV1 modulates long-termed depression of synaptic activity (Chavez et al, 2010; Grueter et al, 2010). Although it remains unclear how these electrophysiological events would reflect behavioral responses, these studies suggest that anandamide could be the endogenous substance activating this ion channel (Di Marzo, 2010; Chavez et al, 2010; Grueter et al, 2010). Other lipids, including N-arachidonoyl dopamine, N-oleoyldopamine and some lypooxigenase products have also been among the main candidates proposed as endovanilloids (Di Marzo et al, 2001; Starowicz et al, 2007a).
Remarkably, the effects of TRPV1 blockade could be fully prevented by pre-treatment with the selective CB1 antagonist, AM251. Thus, we have shown that the effect of TRPV1 antagonists is ultimately mediated by CB1 receptor, possibly because the actions of an endogenous agonist would be entirely shifted to CB1, after TRPV1 blockade. This finding adds to previous observations about opposite roles for CB1 and TRPV1 in various behavioral responses (Maione et al, 2006; Micale et al, 2009; Rubino et al, 2008). We suggest that CB1 and TRPV1 might be simultaneously activated at a given synapse, since we observed co-expression of CB1 and TRPV1 at level of the dPAG. Similar observations were obtained before in the prefrontal cortex, amygdala complex, hippocampus and vlPAG (Cristino et al, 2006; Maione et al, 2006; Micale et al, 2009). In accordance with previous studies, we also detected dPAG neurons that were either exclusively CB1 positive (Herkenham et al, 1990, 1991; Tsou et al, 1998) or TRPV1 positive (McGaraughty et al, 2003; Mezey et al, 2000; Tóth et al, 2005).
Our pharmacological and histological experiments support a scenario in which TRPV1 and CB1 would counteract each other to balance the threshold for panic-like responses in multiple ways. For instance, the opposite effects of CB1 and TRPV1 could be mediated by different neurons that converge on the same output system. Alternatively, they could result from divergent influences on intracellular signaling, neural firing rates and neurotransmitter release within the same neurons (Kawahara et al, 2010; Starowicz et al, 2007b; Vaughan et al, 2000; Xing and Li, 2007). Regarding the physiological modulation of these mechanisms, there might be an endogenous agonist, with affinity for both, which would be part of a tripartite system to establish set points for behavioral responses. One candidate would be anandamide, which has been proposed as an endocannabinoid/endovanilloid (Zygmunt et al, 2000), with higher affinity for CB1 as compare with TRPV1 (Di Marzo et al, 2001; Starowicz et al, 2007a).
Despite of the facts that anandamide has lower affinity for TRPV1 and has its actions limited by the enzyme fatty acid amide hydrolase (FAAH; Piomelli, 2003), the local levels of this substance are significantly increased by electrical stimulation of the dPAG (Walker et al, 1999). Therefore, it is conceivable that, in the present model, anandamide levels would increase and act upon both receptors, whereby TRPV1-mediated actions may prevail over CB1 (Tognetto et al, 2001), thus facilitating panic-like behavior. Blockade of this ion channel would increase the threshold by shifting anandamide action entirely to CB1 receptors, whose selective activation is panicolytic. This hypothesis is reinforced by recent electrophysiological and behavioral experiments. In PAG slices, anandamide could either facilitate or inhibit excitatory transmission through TRPV1 and CB1, respectively, in the presence of the hydrolysis inhibitor URB597 (Kawahara et al, 2010). Accordingly, FAAH knockout mice have a phenotype of lower anxiety-like behavior due to higher CB1 activation by anandamide. However, CB1-antagonist treatment induced anxiogenic-like effects in these animals, but not in wild-type controls, possibly by shifting anandamide activity toward TRPV1 (Cassano et al, 2010).
In conclusion, we have shown opposite functions for TRPV1 and CB1 receptors in the modulation of panic-like responses at the level of the dPAG. Anandamide actions via CB1 vs TRPV1 may constitute a set-point system that represents a promising target for the pharmacotherapy of panic and other anxiety spectrum disorders.
References
Aguiar DC, Terzian AL, Guimarães FS, Moreira FA (2009). Anxiolytic-like effects induced by blockade of transient receptor potential vanilloid type 1 (TRPV1) channels in the medial prefrontal cortex of rats. Psychopharmacology (Berl) 205: 217–225.
Bandler R, Keay KA, Floyd N, Price J (2000). Central circuits mediating patterned autonomic activity during active vs. passive emotional coping. Brain Res Bull 53: 95–104.
Beckett S, Marsden CA (1995). Computer analysis and quantification of periaqueductal grey-induced defense behaviour. J Neurosci Methods 58: 157–161.
Casarotto PC, de Bortoli VC, Corrêa FM, Resstel LB, Zangrossi Jr H (2010). Panicolytic-like effect of BDNF in the rat dorsal periaqueductal grey matter: the role of 5-HT and GABA. Int J Neuropsychopharmacol 13: 573–582.
Cassano T, Gaetani S, Macheda T, Laconca L, Romano A, Morgese MG et al (2010). Evaluation of the emotional phenotype and serotonergic neurotransmission of fatty acid amide hydrolase-deficient mice. Psychopharmacology (Berl), print copy in press (originally published online 2 November 2010 at http://www.springerlink.com/content/r6351r2q7t881t25/fulltext.pdf).
Cavanaugh DJ, Chesler AT, Jackson AC, Sigal YM, Yamanaka H, Grant R et al (2011). Trpv1 reporter mice reveal highly restricted brain distribution and functional expression in arteriolar smooth muscle cells. J Neurosci 31: 5067–5077.
Chavez AE, Chiu CQ, Castillo P (2010). TRPV1 activation by endogenous anandamide triggers post synaptic long-term depression in dentate gyrus. Nat Neurosci 13: 1511–1519.
Cristino L, de Petrocellis L, Pryce G, Baker D, Guglielmotti V, Di Marzo V (2006). Immunohistochemical localization of cannabinoid type 1 and vanilloid transient receptor potential vanilloid type 1 receptors in the mouse brain. Neuroscience 139: 1405–1415.
Del-Ben CM, Graeff FG (2009). Panic disorder: is the PAG involved? Neural Plast 2009: 108135.
Di Marzo V (2008). Targeting the endocannabinoid system: to enhance or reduce? Nat Rev Drug Discov 7: 438–455.
Di Marzo V (2010). Anandamide server two masters in the brain. Nat Neurosci 13: 1446–1448.
Di Marzo V, Bisogno T, De Petrocellis L (2001). Anandamide: some like it hot. Trends Pharmacol Sci 22: 346–349.
Dielenberg RA, Hunt GE, McGregor IS (2001). ‘When a rat smells a cat’: the distribution of Fos immunoreactivity in rat brain following exposure to a predatory odor. Neuroscience 104: 1085–1097.
Finn DP, Jhaveri MD, Beckett SR, Roe CH, Kendall DA, Marsden CA et al (2003). Effects of direct periaqueductal grey administration of a cannabinoid receptor agonist on nociceptive and aversive responses in rats. Neuropharmacology 45: 594–604.
Gawa NR, Tamir R, Klionsky L, Norman MH, Louis JC, Wild KD et al (2005). Proton activation does not alter antagonist interaction with the capsaicin-binding pocket of TRPV1. Mol Pharmacol 68: 1524–1533.
Grueter BA, Brasnjo G, Malenka RC (2010). Post synaptic TRPV1 triggers cell-type specific long-term depression in the nucleus accumbens. Nat Neurosci 13: 1519–1526.
Gunthorpe MJ, Rami HK, Jerman JC, Smart D, Gill CH, Soffin EM et al (2004). Identification and characterization of SB-366791, a potent and selective vanilloid receptor (VR1/TRPV1) antagonist. Neuropharmacology 46: 133–149.
Haller J, Bakos N, Szirmay M, Ledent C, Freund TF (2002). The effects of genetic and pharmacological blockade of the CB1 cannabinoid receptor on anxiety. Eur J Neurosci 16: 1395–1398.
Haller J, Varga B, Ledent C, Freund TF (2004). CB1 cannabinoid receptors mediate anxiolytic effects: convergent genetic and pharmacological evidence with CB1-specific agents. Behav Pharmacol 15: 299–304.
Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC (1991). Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci 11: 563–583.
Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR et al (1990). Cannabinoid receptor localization in brain. Proc Natl Acad Sci USA 87: 1932–1936.
Hogg S, Michan L, Jessa M (2006). Prediction of anti-panic properties of escitalopram in the dorsal periaqueductal grey model of panic anxiety. Neuropharmacology 51: 141–145.
Huang SM, Bisogno T, Trevisani M, Al-Hayani A, De Petrocellis L, Fezza F et al (2002). An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc Natl Acad Sci USA 99: 8400–8405.
Jacob W, Yassouridis A, Marsicano G, Monory K, Lutz B, Wotjak CT (2009). Endocannabinoids render exploratory behaviour largely independent of the test aversiveness: role of glutamatergic transmission. Genes Brain Behav 8: 685–698.
Jenck F, Moreau JL, Martin JR (1995). Dorsal periaqueductal gray-induced aversion as a simulation of panic anxiety: elements of face and predictive validity. Psychiatry Res 57: 181–191.
Kamprath K, Marsicano G, Tang J, Monory K, Bisogno T, Di Marzo V et al (2006). Cannabinoid CB1 receptor mediates fear extinction via habituation-like processes. J Neurosci 26: 6677–6686.
Kasckow JW, Mulchahey JJ, Geracioti Jr TD (2004). Effects of the vanilloid agonist olvanil and antagonist capsazepine on rat behaviors. Prog Neuropsychopharmacol Biol Psychiatry 28: 291–295.
Katon WJ (2006). Clinical practice. Panic disorder. N Engl J Med 354: 2360–2367.
Kawahara H, Drew GM, Christie MJ, Vaughan CW (2010). Inhibition of fatty acid amide hydrolase unmasks CB(1) receptor and TRPV1 channel-mediated modulation of glutamatergic synaptic transmission in midbrain periaqueductal grey. Br J Pharmacol, print copy in press (originally published online 22 December 2010 at http://onlinelibrary.wiley.com/doi/10.1111/j.1476-5381.2010.01157.x/pdf).
Krieger JE, Graeff FG (1985). Defensive behavior and hypertension induced by glutamate in the midbrain central gray of the rat. Braz J Med Biol Res 18: 61–67.
Lisboa SF, Resstel LB, Aguiar DC, Guimarães FS (2008). Activation of cannabinoid CB1 receptors in the dorsolateral periaqueductal gray induces anxiolytic effects in rats submitted to the Vogel conflict test. Eur J Pharmacol 593: 73–78.
Maione S, Bisogno T, de Novellis V, Palazzo E, Cristino L, Valenti M et al (2006). Elevation of endocannabinoid levels in the ventrolateral periaqueductal grey through inhibition of fatty acid amide hydrolase affects descending nociceptive pathways via both cannabinoid receptor type 1 and transient receptor potential vanilloid type-1 receptors. J Pharmacol Exp Ther 316: 969–982.
Marsch R, Foeller E, Rammes G, Bunck M, Kössl M, Holsboer F et al (2007). Reduced anxiety, conditioned fear, and hippocampal long-term potentiation in transient receptor potential vanilloid type 1 receptor-deficient mice. J Neurosci 27: 832–939.
Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG et al (2002). The endogenous cannabinoid system controls extinction of aversive memories. Nature 418: 530–534.
Martin M, Ledent C, Parmentier M, Maldonado R, Valverde O (2002). Involvement of CB1 cannabinoid receptors in emotional behaviour. Psychopharmacology (Berl) 159: 379–387.
McGaraughty S, Chu KL, Bitner RS, Martino B, El Kouhen R, Han P et al (2003). Capsaicin infused into the PAG affects rat tail flick responses to noxious heat and alters neuronal firing in the RVM. J Neurophysiol 90: 2702–2710.
McNaughton N, Gray JA (2000). Anxiolytic action on the behavioural inhibition system implies multiple types of arousal contribute to anxiety. J Affect Disord 61: 161–176.
Mezey E, Tóth ZE, Cortright DN, Arzubi MK, Krause JE, Elde R et al (2000). Distribution of mRNA for vanilloid receptor subtype 1 (VR1), and VR1-like immunoreactivity, in the central nervous system of the rat and human. Proc Natl Acad Sci USA 97: 3655–3660.
Micale V, Cristino L, Tamburella A, Petrosino S, Leggio GM, Drago F et al (2009). Anxiolytic effects in mice of a dual blocker of fatty acid amide hydrolase and transient receptor potential vanilloid type-1 channels. Neuropsychopharmacology 34: 593–606.
Mobbs D, Petrovic P, Marchant JL, Hassabis D, Weiskopf N, Seymour B et al (2007). When fear is near: threat imminence elicits prefrontal-periaqueductal gray shifts in humans. Science 317: 1079–1083.
Moreira FA, Aguiar DC, Campos AC, Lisboa SF, Terzian AL, Resstel LB et al (2009). Antiaversive effects of cannabinoids: is the periaqueductal gray involved? Neural Plast 2009: 625469.
Moreira FA, Aguiar DC, Guimarães FS (2007). Anxiolytic-like effect of cannabinoids injected into the rat dorsolateral periaqueductal gray. Neuropharmacology 52: 958–965.
Nashold Jr BS, Wilson WP, Slaughter DG (1969). Sensations evoked by stimulation in the midbrain of man. J Neurosurg 30: 14–24.
Paxinos G, Watson C (1997). The Rat Brain in Stereotaxic Coordinates. 3rd edn. Academic Press: San Diego.
Piomelli D (2003). The molecular logic of endocannabinoid signalling. Nat Rev Neurosci 4: 873–884.
Price TJ, Patwardhan A, Akopian AN, Hargreaves KM, Flores CM (2004). Modulation of trigeminal sensory neuron activity by the dual cannabinoid-vanilloid agonists anandamide, N-arachidonoyl-dopamine and arachidonyl-2-chloroethylamide. Br J Pharmacol 141: 1118–1130.
Roy-Byrne PP, Craske MG, Stein MB (2006). Panic disorder. Lancet 368: 1023–1032.
Rubino T, Realini N, Castiglioni C, Guidali C, Viganó D, Marras E et al (2008). Role in anxiety behavior of the endocannabinoid system in the prefrontal cortex. Cereb Cortex 18: 1292–1301.
Santos CJ, Stern CA, Bertoglio LJ (2008). Attenuation of anxiety-related behaviour after the antagonism of transient receptor potential vanilloid type 1 channels in the rat ventral hippocampus. Behav Pharmacol 19: 357–360.
Schenberg LC, Bittencourt AS, Sudré EC, Vargas LC (2001). Modeling panic attacks. Neurosci Biobehav Rev 25: 647–659.
Schütz MT, de Aguiar JC, Graeff FG (1985). Anti-aversive role of serotonin in the dorsal periaqueductal grey matter. Psychopharmacology (Berl) 85: 340–345.
Silveira PE, Silveira NA, Morini Vde C, Kushmerick C, Naves LA (2010). Opposing effects of cannabinoids and vanilloids on evoked quantal release at the frog neuromuscular junction. Neurosci Lett 473: 97–101.
Smart D, Gunthorpe MJ, Jerman JC, Nasir S, Gray J, Muir AI (2000). The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1). Br J Pharmacol 129: 227–230.
Starowicz K, Maione S, Cristino L, Palazzo E, Marabese I, Rossi F et al (2007b). Tonic endovanilloid facilitation of glutamate release in brainstem descending antinociceptive pathways. J Neurosci 27: 13739–13749.
Starowicz K, Nigam S, Di Marzo V (2007a). Biochemistry and pharmacology of endovanilloids. Pharmacol Ther 114: 13–33.
Terzian AL, Aguiar DC, Guimarães FS, Moreira FA (2009). Modulation of anxiety-like behaviour by Transient Receptor Potential Vanilloid Type 1 (TRPV1) channels located in the dorsolateral periaqueductal gray. Eur Neuropsychopharmacol 19: 188–195.
Tognetto M, Amadesi S, Harrison S, Creminon C, Trevisani M, Carreras M et al (2001). Anandamide excites central terminals of dorsal root ganglion neurons via vanilloid receptor-1 activation. J Neurosci 21: 1104–1109.
Tóth A, Boczán J, Kedei N, Lizanecz E, Bagi Z, Papp Z et al (2005). Expression and distribution of vanilloid receptor 1 (TRPV1) in the adult rat brain. Brain Res Mol Brain Res 135: 162–168.
Tsou K, Brown S, Sañudo-Peña MC, Mackie K, Walker JM (1998). Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience 83: 393–411.
Vaughan CW, Connor M, Bagley EE, Christie MJ (2000). Actions of cannabinoids on membrane properties and synaptic transmission in rat periaqueductal gray neurons in vitro. Mol Pharmacol 57: 288–295.
Walker JM, Huang SM, Strangman NM, Tsou K, Sañudo-Peña MC (1999). Pain modulation by release of the endogenous cannabinoid anandamide. Proc Natl Acad Sci USA 96: 12198–11203.
Xing J, Li J (2007). TRPV1 receptor mediates glutamatergic synaptic input to dorsolateral periaqueductal gray (dl-PAG) neurons. J Neurophysiol 97: 503–511.
Zygmunt PM, Julius D, Di Marzo V, Hogestatt ED (2000). Anandamide—the other side of the coin. Trends Pharmacol Sci 21: 43–44.
Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di Marzo V et al (1999). Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400: 452–457.
Acknowledgements
We thank Dr Vincenzo, Di Marzo and Dr Luigia Cristino (Endocannabinoid Research Group, Italy) for the helpful insights with the immunofluorescence. We also thank Dr Gustavo Menezes and Dr Ana Maria de Paulo (Federal University of Minas Gerais) for the assistance with confocal microscopy. Finally, we thank Eleni T Gomes, José C de Aguiar and Afonso Padovan (University of São Paulo) for the technical assistance. ALT is a recipient of a CNPq/DAAD fellowship (290008/2009-3). FAM thanks FAPEMIG for the research grant APQ-01446-09. DCA thanks FAPEMIG for the research grant APQ-01883-09.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Casarotto, P., Terzian, A., Aguiar, D. et al. Opposing Roles for Cannabinoid Receptor Type-1 (CB1) and Transient Receptor Potential Vanilloid Type-1 Channel (TRPV1) on the Modulation of Panic-Like Responses in Rats. Neuropsychopharmacol 37, 478–486 (2012). https://doi.org/10.1038/npp.2011.207
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2011.207
Keywords
This article is cited by
-
Therapeutic Molecular Insights into the Active Engagement of Cannabinoids in the Therapy of Parkinson’s Disease: A Novel and Futuristic Approach
Neurotoxicity Research (2023)
-
Dual action of the cannabinoid receptor 1 ligand arachidonyl-2′-chloroethylamide on calcitonin gene-related peptide release
The Journal of Headache and Pain (2022)
-
The Periaqueductal Gray and Its Extended Participation in Drug Addiction Phenomena
Neuroscience Bulletin (2021)
-
The role of endocannabinoids in consolidation, retrieval, reconsolidation, and extinction of fear memory
Pharmacological Reports (2021)
-
Cannabidiol-induced panicolytic-like effects and fear-induced antinociception impairment: the role of the CB1 receptor in the ventromedial hypothalamus
Psychopharmacology (2020)
