
Abstract
Type 1 diabetes is a common metabolic disorder accompanied by an increased secretion of glucocorticoids and cognitive deficits. Chronic excess of glucocorticoids per se can evoke similar neuropathological signals linked to its major target in the brain, the hippocampus. This deleterious action exerted by excess adrenal stress hormone is mediated by glucocorticoid receptors (GRs). The aim of the present study was to assess whether excessive stimulation of GR is causal to compromised neuronal viability and cognitive performance associated with the hippocampal function of the diabetic mice. For this purpose, mice had type 1 diabetes induced by streptozotocin (STZ) administration (170 mg/kg, i.p.). After 11 days, these STZ-diabetic mice showed increased glucocorticoid secretion and hippocampal alterations characterized by: (1) increased glial fibrillary acidic protein-positive astrocytes as a marker reacting to neurodegeneration, (2) increased c-Jun expression marking neuronal activation, (3) reduced Ki-67 immunostaining indicating decreased cell proliferation. At the same time, mild cognitive deficits became obvious in the novel object-placement recognition task. After 6 days of diabetes the GR antagonist mifepristone (RU486) was administered twice daily for 4 days (200 mg/kg, p.o.). Blockade of GR during early type 1 diabetes attenuated the morphological signs of hippocampal aberrations and rescued the diabetic mice from the cognitive deficits. We conclude that hippocampal disruption and cognitive impairment at the early stage of diabetes are caused by excessive GR activation due to hypercorticism. These signs of neurodegeneration can be prevented and/or reversed by GR blockade with mifepristone.
Similar content being viewed by others


INTRODUCTION
Type 1 diabetes (T1D) is an autoimmune disease that results in the permanent destruction of insulin-producing β-cells of the pancreas. To study the disease, a commonly used rodent model is generated by injecting streptozotocin (STZ). This toxin destroys the insulin-producing cells producing diabetes (Rees and Alcolado, 2005). The impact of T1D on the central nervous system is well documented. T1D patients show impairments in explicit memory, problem solving, and intellectual development (Ryan and Williams, 1993; Ryan et al, 1993; Kramer et al, 1998; Parisi and Uccioli, 2001; McCarthy et al, 2002; Schoenle et al, 2002). In the STZ-diabetic rodent model, the severity of the cognitive deficits is related to the duration of diabetes (Kaleeswari et al, 1986; Bellush and Rowland, 1989; Flood et al, 1990; Mayer et al, 1990; Biessels et al, 1996, 1998; Popovic et al, 2001). Some studies have revealed structural and functional abnormalities, particularly in the hippocampus, such as impaired long-term potentiation, synaptic alterations (dendritic spine densities and LTP), degeneration and neuronal loss (hamsters: Luse, 1970; rats: Bestetti and Rossi, 1980; Biessels et al, 1996; Gispen and Biessels, 2000; Magarinos and McEwen, 2000; McEwen et al, 2002, mice: Saravia et al, 2002). In the hippocampus of STZ mice, we have previously demonstrated: (1) astrogliosis, as evidenced by increased number of GFAP+ cells, suggesting neuronal suffering, (2) increased immediate early gene expression such as elevated c-Jun immunoreactive neurons, a sign of neuronal activation, and (3) decreased neurogenesis in the dentate gyrus (DG), indicating that newborn cells are vulnerable to damage in diabetes (Saravia et al, 2002; Revsin et al, 2005). Overall, these alterations suggest mild hippocampal neurodegeneration in the STZ-treated mice.
In rodents, the effects of the glucocorticoid corticosterone on learning and memory processes are mediated by mineralocorticoid and glucocorticoid receptors (MR and GR), which are highly expressed in limbic brain areas. Glucocorticoids secreted after stress promote consolidation of a stressful event via transient activation of GR (Oitzl and de Kloet, 1992; Joëls, 2006). In contrast, sustained activation of GR by chronically elevated glucocorticoids impairs hippocampal function and memory processes (McEwen and Sapolsky, 1995). However, cognitive performance is improved during chronic blockade of GR with the glucocorticoid antagonist mifepristone (RU486) (Oitzl et al, 1998a, 1998b; Conrad, 2006). MR is neuroprotective and stimulates hippocampal function (Joëls et al, 2008). It mediates the role of corticosterone in the appraisal of novel situations, behavioral reactivity, and affective responses (Oitzl and de Kloet, 1992; Oitzl et al, 1994; Rozeboom et al, 2007), and enhances the performance in spatial hippocampal-dependent cognitive tasks (Ferguson and Sapolsky, 2007; Lai et al, 2007). Thus, a balanced activation of MR and GR is crucial for optimal cognitive performance.
In diabetic animals, acute cerebral dysfunction resulting from hyper- and hypoglycemic episodes was described (Biessels et al, 1994; Cryer 1994). In addition, chronic hypercorticism triggered and maintained by enhanced adrenocortical sensitivity in STZ mice, and morphological alterations of predominantly the hippocampus were reported (Fitzpatrick et al, 1992; Durant et al, 1993; Scribner et al, 1993; Saravia et al, 2002, 2004; Revsin et al, 2005, 2008). A fundamental question in the neuropathology of T1D is, therefore, whether aberrant glucose metabolism and insulin deficiency and/or glucocorticoid excess and excessive GR activation may cause the functional and morphological signs of neurodegeneration and cognitive impairment. Previous reports described the central roles of glucose and insulin in the STZ-induced animal models of T1D (Sima and Li, 2005; Inouye et al, 2005; Chan et al, 2005; McNay et al, 2006).
Recently, chronic hypercorticism was found to be responsible for the functional and morphological degeneration in hippocampus of rats and mice suffering from full-blown T1D (Stranahan et al, 2008). The study showed that impaired LTP, deficits in cognitive performance and reduced neurogenesis were normalized in diabetic rodents after adrenalectomy and corticosterone replacement at physiological concentrations (Stranahan et al, 2008). The objective of the present study is to evaluate whether these neurodegenerative effects of glucocorticoids in T1D mice were due to excessive activation of GR. We hypothesize that the STZ-induced hypercorticism of diabetic mice and the concomitant continuous activation of GR may change molecular markers of hippocampal morphology and cognitive performance. We have used the GR antagonist mifepristone (RU486) to clarify the role of continuous GR activation in the hippocampus of STZ-diabetic mice. Hippocampal integrity was based on markers for cellular proliferation and activation. For hippocampal function spatial memory tasks were used, specifically the novel object-placement recognition (NOPR) task. The data demonstrate protection of hippocampal integrity and improved cognitive performance by a brief GR antagonist treatment during the onset of diabetes in the STZ-diabetic mice.
MATERIALS AND METHODS
Animals
Twelve weeks old C57Bl/6 male mice (Janvier, NL) were group housed 3–4 per cage in humidity (55±5%) and temperature (23±2°C) controlled conditions, with 12–12 light-dark hours cycle (lights on at 0800 hours) at the animal facility of the LACDR, Leiden. Food and water was provided ad libitum. Animal experiments were performed in accordance with the European Communities Council Directive 86/609/EEC and with approval from the animal care committee of the Faculty of Medicine, Leiden University.
Experimental Schedule
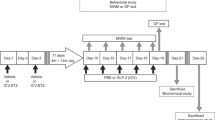
Mice received a single i.p. dose of 170 mg/kg of STZ (Sigma, St Louis, MO, USA) in 0.5 M sodium citrate buffer or vehicle; 48 h after injection glucose levels in urine (Diabur-Test 5000, Roche, Germany) indicated diabetes. Mice with blood glucose levels (Accu-Chek Compact, Roche) higher than 11 mM were classified as overtly diabetic. Six experimental groups (n=6–8 mice per group) were designed as follows: (1) control and diabetic groups with 6 days of diabetes; (2) control and diabetic groups with 11 days of diabetes, vehicle treated; (3) control and diabetic groups with 11 days of diabetes, mifepristone treated. At 6 days after diabetes onset at 0900 hours, mice received every 12 h 100 μl of mifepristone (11β-(4-dimethyl-amino)-phenyl-17β-hydroxy-17-(1-propynyl)-estra-4,9-dien-3-one, Corcept Therapeutics, CA, USA) for 4 consecutive days (200 mg/kg body weight, per o.s.) dissolved in 0.25% carboxymethylcellulose and 0.2% Tween-20 in 1 ml NaCl 0.9%, or vehicle. This dose of mifepristone ensures a complete blockade of the GR for at least 12 h (Dalm et al, 2008). Mifepristone displays antiglucocorticoid and antiprogestin activity. However, in conditions of chronic elevated glucocorticoid levels, such as occurring in T1D mice, the antiglucocorticoid activity is most prominent. Moreover, mifepristone has been used as antiglucocorticoid in animals models for stress (Krugers et al, 2007; Mayer et al, 2006; Oomen et al, 2007), drugs of abuse (Dong et al, 2006), and electroconvulsive therapy (Nagaraja et al, 2007). At day 11 of diabetes at 0800 hours, the behavioral experiment was started at a time GR blockade during the task is still ensured. Mice were killed between 1000 and 1200 hours for all the experiments (see Figure 1a).

Schematic representation of the methodology used. (a) Experimental design. (b) Handling and habituation for the novel object-placement recognition (NOPR) task before streptozotocin (STZ) or vehicle injections. (c) NOPR test trials at 11 days of diabetes.
Novel Object-Placement Recognition Task
On the basis of locomotor activity and the time exploring the objects during the last habituation trial (each object was explored approximately equally long), mice were divided into two groups that received either STZ or vehicle injection.
Apparatus and objects
Object exploration was assessed in an open field (50 × 50 × 40 cm; gray polyvinyl chloride). Mirrors were placed on the walls of the open field to improve the visual analysis of the exploratory behavior of the mice. The illumination density was approximately 50 lux at the center of the open field.
We used two identical plastic bottles filled with water as objects (0.5 l without labels). After each trial, the apparatus and objects were thoroughly cleaned with a 75% ethanol solution to remove or spread odor cues.
Procedure
A scheme of the experimental design is presented in Figure 1. Figure 1b shows the handling and habituation procedure adapted from Dere et al (2005).
At the start of each trial, mice were picked up from the base of the tail and placed into an opaque cylinder (10 cm diameter; 20 cm high) in the center of the open field. After 10 s, the cylinder was lifted and the mice were allowed to explore the environment. Depending on the experimental condition, the two bottles were present or absent. At the end of each trial, the mice were removed from the open field by a grid and returned to the home cage. This procedure prevents the mice becoming stressed by the experimenter.
On day 11 of diabetes, the NOPR test was performed (Figure 1c). In the sample trial, the two objects were present in the open field: one in the northwest zone and one in the southwest zone (SW). After 10 min exploration, the mouse was returned to its home cage for 50 min. For the consecutive 10 min test trial (T2), the SW object was relocated to the east zone.
Computerized image analysis
Exploration of an object is defined when the mouse is in the vicinity of an object at less than 2 cm and in physical contact with it. Contact was only counted as exploration when the mouse touched the object with the snout, forepaws, or vibrissae (Observer 4.1, Noldus Information Technology, Wageningen, The Netherlands). The parameters of exploration are based on Dere et al (2005).
Analysis of general locomotor activity and behavior was performed by EthoVision3.0 (Noldus Information Technology) using steps of five samples per second. The open field was divided into nine zones (Figure 1), and total distance moved (cm), mean velocity (cm/s), latency of first entry in each zone (seconds), and time per zone (percentage) were measured.
Calculations
The exploration of the two objects was calculated for the sample and test trials. The percentage of preference for exploring the relocated object was calculated as follows:

Immunocytochemistry for GFAP and Ki-67
Under pentobarbital sodium salt anesthesia (Nembutal 150 μl i.p., CEVA Sante Animale BV, The Netherlands), mice were intracardially perfused (20 ml of 0.9% NaCl followed by 40 ml of 4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS), pH 7.4), the brains were removed, kept for 2 h in the same fixative, and incubated overnight in 30% sucrose in PBS at 4°C. Thereafter, the brains were frozen on dry ice and stored at −80°C until processing.
Astrocytes and proliferative cell markers (GFAP and Ki-67) were measured in coronal brain cryosections (20 μm) from the dorsal hippocampus (bregma −1.7 to −2.06 mm; Paxinos and Frankling, 2001). Four to six sections from each brain were mounted per glass (SuperFrost Plus glasses, Menzel-Glaser, Germany) and stored at −80°C. After washes with 0.1 M Tris-buffered saline pH 7.6 (TBS), the sections were placed in plastic jars filled with citrate buffer (0.1 M for GFAP and 0.01 M for Ki-67, pH 6), microwaved for 5 min at 800, 400, and 250 W, and cooled down to room temperature (RT) for 30 min. Ki-67 immunostaining follows the previously published by Heine et al (2004). For GFAP nonspecific binding was prevented by 10% normal goat serum (NGS)/0.3% Triton X-100 in 1 M TBS for 30 min at RT. Thereafter, sections were incubated with the primary antibody rabbit polyclonal anti-GFAP (1 : 600, DakoCytomation, The Netherlands) diluted in 2% NGS/0.2% Triton X-100 in 1 M TBS (TBS+) for 1 h at RT and overnight at 4°C. For negative control, the first antibody was omitted. The next day after rinses, sections were incubated with biotinylated goat anti-rabbit immunoglobulin G (1 : 200, Jackson ImmunoResearch, The Netherlands) in TBS+ for 1 h at 37°C. After 45 min incubation with ABC kit (1 : 800, Vector Laboratory, CA, USA), 3,3′-diaminobenzidine 0.5 mg/ml, 0.05% H2O2 at RT was used for development. After dehydration with graded ethanols and xylene, the sections were mounted with Entellant.
Sections stained for Ki-67 were additionally counterstained with 0.5% cresyl violet, with unbiased stereological sampling and quantification in every 10th hippocampal section according to Saravia et al (2004) and Oomen et al (2007). The number of cells expressing GFAP per area (31 × 104 mm2) was determined in the stratum radiatum below the CA1 using computerized image analysis (Olympus Soft Imaging). Labeled cells were bilaterally counted in four to six sections per animal, using 4–8 animals per group as indicated.
Western Blot for c-Jun
Western blot was performed as described by Vreugdenhil et al (2007) in decapitated separate groups. The same amount of protein (25 μg cell lysate) from mouse hippocampal tissue (six mice per group) was used. Polyclonal anti-c-Jun primary antibody (1 : 500, Santa Cruz Biotechnology, CA, USA) and monoclonal anti-α-tubulin DM1A (1 : 1000; Sigma-Aldrich, The Netherlands), as a control for the amount of protein loaded, were applied in different membranes (Millipore). Thereafter, the secondary antibody peroxidase conjugated (1 : 5000) was added. Films were developed and the intensity of the immunoreactive labeling was analyzed (Image analysis system, Image J, NIH Bethesda, USA). The values for each sample are expressed as a percentage of optical density (o.d.) obtained using α-tubulin.
Corticosterone and ACTH Radio Immuno Assay
Trunk blood was collected individually in labeled potassium-EDTA-coated tubes (1.6 mg EDTA per ml blood, Sarstedt, Germany), kept on ice and centrifuged for 15 min at 3000 r.p.m. at 4°C. Plasma was transferred to clean tubes and stored frozen at −20°C until the determination of corticosterone or ACTH by the MP Biomedical RIA kits (ICN, Biomedicals Inc., CA).
Data Analysis and Statistics
Data are expressed as mean±SEM. Physiological and neuroendocrine parameters were analyzed by multifactorial analysis of variance (ANOVA) using SPSS software (version 7.5). Expression of molecular markers and behavioral measurements were analyzed by two-way ANOVA followed by Bonferroni-adjusted post hoc test using GraphPad Software (version 4). Differences were considered statistically significant at p<0.05.
Behavior of separate groups was tested in open field, elevated plus maze, forced swim, and Morris water maze tasks (see Supplementary data). For each behavioral task, different groups of control and diabetic animals were used.
RESULTS
Physiological and Neuroendocrine Parameters
Diabetic mice showed the characteristic clinical features of the disease after 6 days of diabetes. Multifactorial ANOVA revealed significant main effects of group (diabetic vs control: F(8, 30) 61.176, p<0.001) and treatment with mifepristone (F(8, 30) 18.485, p<0.001 and a group × treatment interaction (F(3, 30) 5.191; detailed measurements and significances are given in Table 1). As we observed before (Revsin et al, 2008), diabetic mice showed hyperglycemia (>11 mM), increased absolute adrenal weight, and adrenal/body weight ratio, and decreased body weight (BW), absolute thymus weight, and thymus/body weight ratio. Furthermore, basal plasma corticosterone levels were significantly increased, whereas ACTH concentration was significantly decreased compared to control mice. Untreated control and diabetic mice of 6 days of diabetes showed the same effects as the control and diabetic vehicle-treated groups, respectively (data not shown).
Mifepristone treatment increased adrenal weight, adrenal/BW ratio, and corticosterone levels in control as well as in diabetic mice. In addition, this mifepristone-treated diabetic group showed increased ACTH and gain in BW, but thymus weight and thymus/BW ratio remained as low as in diabetic vehicle-treated mice (Table 1).
Mifepristone Effects on the Hippocampus
Astrogliosis
The number of astrocytes (GFAP+ cells, Figure 2a) in the stratum radiatum of the hippocampus was measured in six experimental groups: control and diabetic mice killed 6 days after diabetes onset; control and diabetic mice treated with vehicle or mifepristone and killed at day 11 of diabetes. Only at 11 days of diabetes was the number of astrocytes significantly increased (Figure 2). Treating diabetic mice with mifepristone for 4 days prevented the increase in the number of GFAP+ cells, which remained comparable to nondiabetic controls. Mifepristone treatment of nondiabetic mice did not modify this parameter.

Immunocytochemistry for glial fibrillary acidic protein (GFAP). (a) Quantification of GFAP+ cell number in the stratum radiatum of the hippocampus. Values expressed mean±SEM, n=6–8, *p<0.05. (b) Microphotographs of the different experimental groups. Py=pyramidal cells of the CA1 area. Control microphotograph correspond to a control+vehicle mouse.
c-Jun protein expression
Cellular activation was studied by Western blot using a specific antibody against c-Jun (Figure 3). To normalize c-Jun protein expression between groups, the endogenous nonregulated α-tubulin was measured. The o.d. of c-Jun/α-tubulin ratio is shown in Figure 3a. The quantitative analysis reveals that hippocampal cellular activation takes place at 11, but not at 6 days after diabetes onset. Mifepristone treatment of nondiabetic mice did not alter c-Jun protein expression, but attenuated the c-Jun increase in diabetic mice. Comparable amounts of c-Jun protein expression were found in nondiabetic controls and mifepristone-treated diabetic mice.

Western blot for c-Jun. (a) Quantification of c-Jun/α-tubulin optical density (o.d.) from hippocampal homogenates. Values expressed mean±SEM, n=6, *p<0.05. (b) Autoradiograph for c-Jun and α-tubulin. 1: control day 6; 2: diabetic day 6; 3: control+vehicle; 4: diabetic+vehicle; 5: control+mifepristone; 6: diabetic+mifepristone.
Cell proliferation
Proliferative cells in the DG were detected by the endogenous proliferation marker Ki-67. The Ki-67 antigen is a protein complex present only in the G1, S, G2, and M, but not the G0 phase of the cell cycle (Gerdes et al, 1984; Endl and Gerdes, 2000). Clusters of Ki-67-positive cells were shown to be similar to BrdU after short survival time and were found almost exclusively in the subgranular zone (SGZ; Kee et al, 2002; Oomen et al, 2007). Moreover, Ki-67 is a well-accepted proliferation marker (Gerdes et al, 1991; Heine et al, 2004; Krugers et al, 2007; Veenema et al, 2007).
After 6 days of diabetes, a significantly decreased cell proliferation in the SGZ of the DG was observed (diabetic day 6 vs control day 6, p<0.05, Figure 4). This reduction in the number of proliferative cells was maintained at 11 days of the disease: Ki-67-positive cell number of diabetic+vehicle group is significantly lower as compared to the control+vehicle (p<0.001). After mifepristone administration, the number of Ki-67-positive cells is significantly increased in diabetic mice (diabetic+mifepristone vs diabetic+vehicle, p<0.05). Mifepristone treatment of control mice did not modify the Ki-67-positive cell number in comparison to the control vehicle-treated group.

Cell proliferation in the subgranular zone (SGZ) of the dentate gyrus. (a) Quantitative analysis of the immunocytochemistry for Ki-67 in the SGZ. Values expressed mean±SEM, n=6–8, *p<0.05. (b) Microphotographs of the different experimental groups. Insert A: four times magnification of Ki-67+ cells. Control microphotograph correspond to a control+vehicle mouse.
Diabetes Effect on Cognition
Only the more subtle hippocampal-dependent task, the NOPR, revealed significant impairments in cognitive performance of diabetic mice at 11 days of the disease. In the sample trial, the percentage of time that both objects are explored is similar in both groups (control: 50.51±5.34, diabetic: 57.84±3.84, expressed as percentage of preference for the object that will be relocated in the test trial, n=7–8). In contrast to mice of the control group, diabetic mice had no preference for the relocated object in the test trial (control: 61.37±5.54, diabetic: 44.41±4.98%, of time exploring the relocated object, p<0.05).
Mifepristone Effects on Cognition: the NOPR Task
Locomotor activity
Locomotor activity expressed as distance moved, duration of the movement, and mean velocity of the movement is presented in Table 2. During the sample trial of the NOPR task, both vehicle- and mifepristone-treated diabetic mice walked slower and shorter distances, with shorter duration of movement when compared to control groups. At 50 min later, during the test trial, the diabetic vehicle-treated group was still less active. However, diabetic mifepristone-treated mice significantly increased their locomotor activity. Locomotor activity of control groups remained comparable in sample and test trials.
The latency to first exploration of the objects and to the zones, as well as the percentage of time in each zone did not differ between groups (data not shown). The number of mice at the beginning of the behavioral experiment was 12 per group. However, some diabetic and control+mifepristone mice died before the end of the experimental procedure. Moreover, two mice were discarded from the behavioral analysis of the diabetic+mifepristone group due to their inability to walk as a consequence of the metabolic deficits originated by the disease. The number of mice used was: control+vehicle, n=12; diabetic+vehicle, n=10; control+mifepristone, n=8; diabetic+mifepristone, n=6.
Spatial memory: exploration of the relocated object
During the sample trial, all groups spent a comparable percentage of time exploring both objects, expressed as percentage of preference for the object that will be relocated in the test trial (control+vehicle=42.67±2.07, diabetic+vehicle=49.47±5.17, control+mifepristone=44.59±4.32, diabetic+mifepristone=51.03±2.41). Figure 5 shows the percentage of preference for the object placed in the new location (ie the relocated object) during the test trial. As previously reported (Dere et al, 2005), the control vehicle-treated group preferred to explore the relocated object. The same effect was found in mifepristone-treated control mice. Conversely, diabetic mice explored both objects equally (50% of the exploration time for each object). The preference for the relocated object is restored when diabetic mice are treated with mifepristone. In addition, the difference in preference in this group is higher compared to its respective control (p<0.05, Bonferroni-adjusted post hoc test).

Mifepristone effect on the novel object-recognition task. Percentage of preference for the relocated object during the test trial is shown for control and 11 days diabetic mice vehicle- and mifepristone treated. Values expressed mean±SEM, *p<0.05 Bonferroni-adjusted post hoc test.
DISCUSSION
Our results show that: (1) hippocampal integrity is threatened in STZ-treated diabetic mice as revealed by astrogliosis, increased cellular activation, and decreased neuronal proliferation at 11 days of disease progression; (2) diabetic mice show spatial memory deficits as is indicated by the impaired performance in a specific hippocampal-dependent task (the NOPR task); and (3) 4 days of treatment with the GR antagonist mifepristone in the early phase of diabetes prevents the change in markers for hippocampal integrity and improves cognitive performance in the face of corticosterone hypersecretion.
Mifepristone Effects on Hippocampal Alterations of STZ-Diabetic Mice
Prevention of astrogliosis
Increased GFAP immunoreactivity representing astrogliosis in the hippocampus was previously reported at day 28 of STZ diabetes in mice (Saravia et al, 2002). We found similar results already in 11-day-diabetic mice, whereas GFAP immunoreactivity was not affected at 6 days of diabetes. Treatment with the GR antagonist mifepristone from days 6 to 10, prevented the GFAP increase at day 11, indicating that blockade of excess glucocorticoid activation of the GR can prevent hippocampal astrogliosis.
Reactive, GFAP-expressing astrocytes provide neuroprotection during metabolic insults, stress, or injury to nearby neurons by secreting growth factors, substrate-bound neurite-promoting factors, and the removal of neurotoxins and excess glutamate (Kiessling et al, 1986; Liedtke et al, 1996; Lambert et al, 2000). Astrocytes may also protect neurons by increasing glucose uptake, metabolism, and transport (Magistretti and Pellerin, 1999; Vesce et al, 1999), in addition to being necessary for the preservation of myelin and normal white matter architecture (Louw et al, 1998). Moreover, the close connection of astrocytes to the blood–brain barrier makes them early sensors of variations of glucose homeostasis, which can be communicated to neurons (Magistretti and Pellerin, 1999). The astrogliosis therefore can be considered to be a neuroprotective response to brain damage inflicted by excessive GR activation, which is prevented by the GR antagonist.
Prevention of cellular activation
We previously reported that T1D also resulted in cellular activation, as shown by an increase of c-Jun- and c-fos-positive cells in neuronal populations of the CA1, CA3, and DG hippocampal areas (Revsin et al, 2005), and this is further supported by the current findings. Although some authors imply a functional role of early gene products in neuronal cell death (Eilers et al, 2001; Barone et al, 2008), others suggested that they might rather contribute to cellular repair and/or regenerating processes (Herzog and Morgan, 1996; Herdegen et al, 1997; Waetzig et al, 2006). As c-Jun was increased at day 11 but not at day 6 of STZ diabetes, we conclude that either corticosterone caused the hippocampal cellular activation or that increased c-Jun-positive cells are a response to an emerging corticosterone-induced neuronal derangement in the hippocampus of diabetic animals, which is prevented by the GR antagonist.
Restoration of cell proliferation
Previously, we demonstrated a strong reduction in cell proliferation rate in STZ-diabetic mice (Saravia et al, 2004). The results shown in the present study confirm these previous observations and demonstrate that significant suppression of cell proliferation already occurs at early stages of diabetes, at days 6 and 11, suggesting that newborn cells are particularly vulnerable to the detrimental effects of glucocorticoid excess. These data are in support of Stranahan et al (2008) showing that adrenalectomy and low-dose corticosterone replacement prevent the decreased proliferation and survival of newborn DG neurons in long-term STZ-induced diabetes.
The data also show that GR blockade in diabetic mice restores the number of Ki-67-positive cells in the DG toward control levels. Therefore, the suppression of proliferation in diabetes is mediated directly and/or indirectly by glucocorticoids via the GR. In other studies, neurogenesis was suppressed in high stress and corticosterone environments (Wong and Herbert, 2005; Mayer et al, 2006; Oomen et al, 2007), but not the Ki-67 marker observed in this study and by Stranahan et al (2008). Therefore, the proliferative response marker might be sensitive to a high corticosterone environment during hyperglycemia and this effect is reversible by blockade of the GR. Nevertheless, the action of glucocorticoids on proliferation is likely to be indirect as newly formed cells in the DG have been found not to express corticosteroid receptors (MR and GR; Cameron et al, 1993; Garcia et al, 2004; Wong and Herbert, 2005). Therefore, mature receptor-expressing dentate granule cells or glia surrounding the progenitors might determine proliferation and survival of these cells (Song et al, 2002; Hastings and Gould, 2003; Seki, 2003). However, a study from Garcia et al (2004) showed that a small proportion of newly formed cells (15%) does in fact express GR 24 h after division.
In summary, the molecular changes described above indicate that mifepristone treatment interferes with the disruption of hippocampal integrity at early stages of STZ diabetes. Whether mifepristone's prevention and/or reversal of the disturbances caused by diabetes is directly on hippocampal astrocytes, neurons, and proliferative cells, or by changing its surroundings (ie their inputs) requires further investigation. It is worth noting that 4 days mifepristone treatment can affect not only the functioning of these cells (present study, Karst and Joëls, 2007; Wu et al, 2007; Mayer et al, 2006; Wong and Herbert, 2005), but also the innervating projections from other brain regions (Witter, 2007). Moreover, in the present study, we reported that reversion of hippocampal alterations takes place in the face of hypercorticism. Recently, Oomen et al (2007) and Mayer et al (2006) reported similar beneficial effects of mifepristone in rats subjected to chronic unpredictable stressors and to high doses of exogenous corticosterone, respectively.
Cognitive Performance
The previously observed HPA axis dysregulation in the type 1 diabetic mice (Revsin et al, 2008) suggested that hypercorticism and excessive GR activation might impair cognitive performance. Therefore, we assessed whether the learning and memory capabilities of STZ-induced diabetes mice were altered at different early states of the disease in a series of behavioral tasks (open field, forced swim test, Morris water maze, and elevated plus maze). Surprisingly, diabetic and nondiabetic control mice performed equally well in these early stages of diabetes (Supplementary Figures S1–S3 and Supplementary Table S1), although during a more prolonged state of diabetes behavioral deficits were established (Stranahan et al, 2008). It is possible that in our study these tasks are insensitive to the moderate hippocampal disturbances observed at early diabetes. For this reason, we decided to adopt another behavioral task for hippocampus-specific memory, the NOPR task (Dere et al, 2005).
The NOPR task uses the spontaneous exploration of objects in novel locations (Li et al, 2004; Dere et al, 2005), and thus allows the study of mild hippocampal alterations. In accordance with the literature, nondiabetic control mice preferred exploring the object placed in the novel location. The diabetic mice, however, exhibit less exploration of the object in the novel location compared to controls (Figure 5). This reduced exploration displayed by the diabetic mouse indicates impaired spatial object-placement memory. It is noteworthy that although diabetic mice showed a significantly decreased locomotor activity and exploration time of the objects than nondiabetic controls, the influence of these parameters on the preference for the objects was prevented by the method used to calculate the percentage of preference for the relocated object.
Mifepristone restores spatial memory deficits of STZ-diabetic mice
Mifepristone treatment of diabetic mice restored the preference of these animals to explore an object in a novel location. This effect exerted by the GR antagonist occurred the day after termination of the treatment. In the studies of Stranahan et al (2008), the reinstatement in long-term adrenalectomized T1D animals with exogenous corticosterone in physiological concentrations levels was a prerequisite to prevent deterioration of cognitive functions and impairment of LTP. The current study shows that similar results are achieved during the early stages of diabetes when the mild cognitive impairments are ameliorated by pharmacological blockade of the GR in an environment of extremely high-circulating corticosterone. This observation raises a number of interesting issues:
First, the GR-mediated action of corticosterone on hippocampal function overrides the influence of insulin and glycemia (Stranahan et al, 2008). The current study also shows that chronic mifepristone causes extreme increases in corticosterone, and reverses some parameters (body weight, hippocampal function) but not others (thymus weight; Table 1; van Haarst et al, 1996). The treatment schedule was based on the efficacy of mifepristone in the treatment of psychosis (van der Lely et al, 1991) and psychotic depression (Flores et al, 2006), and previous animal studies using corticosterone injections and chronic stress (Mayer et al, 2006; Oomen et al, 2007). How precisely mifepristone exerts its lasting antagonistic central action through GR requires further investigation.
Second, the blockade with the antagonist becomes only effective when the GRs are fully occupied with high levels of corticosterone. At low levels of corticosterone GR is hardly occupied, and GR blockade would be less effective (Ratka et al, 1989), and hence mifepristone is not effective in the nondiabetic controls.
Third, during GR blockade the MR is still freely accessible for corticosterone. In the NOPR test, we specifically address MR-mediated functions, ie behavioral reactivity in the acquisition and retrieval of memory (Oitzl and de Kloet, 1992; Oitzl et al, 1994). Overexpression of MR could block some of the impairing behavioral effects mediated by GR (Ferguson and Sapolsky, 2007) and perhaps this explains why the GR antagonism in our diabetic mice not only rescued cognitive performance, but also even enhanced it. GR antagonism also allows a more prominent function of the MR-mediated action in preserving integrity and stability of the hippocampus (Joëls et al, 2008). MR is neuroprotective (Lai et al, 2007), promotes neuronal survival, and facilitates hippocampus function (Joëls et al, 2008). Hence this would predict that during GR blockade in the face of high-circulating glucocorticoids, the maintenance of hippocampal integrity is a necessary condition for improved performance in the NOPR task. Further experiments with administration of GR and MR antagonists are needed to test this possibility.
Fourth, understanding the mechanism would require the identification of the functional recovery of plasticity related genes downregulated by the excess of glucocorticoids. Such genes are related to synaptic plasticity and glutamate transmission, alterations in dendrite and spine morphology, neurogenesis in the DG, and to the rapid functional responses underlying information processing in the NOPR test (Joëls et al, 2007; Morsink et al, 2007). Obviously, future research should be focused on the question how the molecular and cellular changes during diabetes and anti-glucocorticoid treatment are linked to behavior.
In conclusion, in the present study, we have provided evidence that glucocorticoid excess and continuous activation of the GR compromise hippocampal integrity and function in T1D regardless of the hyperglycemic state. This deleterious effect of excess glucocorticoid can be abolished by a brief treatment with the GR antagonist mifepristone. The beneficial effect of mifepristone on molecular markers of hippocampal plasticity and spatial recognition is exerted in a high-corticosterone environment and probably depends on blockade of excess GR activation, perhaps, facilitated by more prominent MR-mediated actions preserving hippocampal integrity.
References
Barone MC, Desouza LA, Freeman RS (2008). Pin1 promotes cell death in NGF-dependent neurons through a mechanism requiring c-Jun activity. J Neurochem. (originally published online May 9, PMID: 18419764) 106: 734–745.
Bellush LL, Rowland NE (1989). Stress and behavior in streptozotocin diabetic rats: biochemical correlates of passive avoidance learning. Behav Neurosci 103: 144–150.
Bestetti G, Rossi GL (1980). Hypothalamic lesions in rats with long-term streptozotocin-induced diabetes mellitus. A semiquantitative light- and electron-microscopic study. Acta Neuropathol (Berl) 52: 119–127.
Biessels GJ, Kamal A, Ramakers GM, Urban IJ, Spruijt BM, Erkelens DW et al (1996). Place learning and hippocampal synaptic plasticity in streptozotocin-induced diabetic rats. Diabetes 45: 1259–1266.
Biessels GJ, Kamal A, Urban IJ, Spruijt BM, Erkelens DW, Gispen WH (1998). Water maze learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: effects of insulin treatment. Brain Res 800: 125–135.
Biessels GJ, Kappelle AC, Bravenboer B, Erkelens DW, Gispen WH (1994). Cerebral function in diabetes mellitus. Diabetologia 37: 643–650.
Cameron HA, Woolley CS, Gould E (1993). Adrenal steroid receptor immunoreactivity in cells born in the adult rat dentate gyrus. Brain Res 611: 342–346.
Chan O, Inouye K, Akirav EM, Park E, Riddell MC, Matthews SG et al (2005). Hyperglycemia does not increase basal hypothalamo–pituitary–adrenal activity in diabetes but it does impair the HPA response to insulin-induced hypoglycemia. Am J Physiol Regul Integr Comp Physiol 289: R235–R246.
Conrad CD (2006). What is the functional significance of chronic stress-induced CA3 dendritic retraction within the hippocampus? Behav Cogn Neurosci Rev 5: 41–60.
Cryer PE (1994). Banting lecture. Hypoglycemia: the limiting factor in the management of IDDM. Diabetes 43: 1378–1389.
Dalm S, Brinks V, van der Mark MH, de Kloet ER, Oitzl MS (2008). Non-invasive stress-free application of glucocorticoid ligands in mice. J Neurosci Methods 170: 77–84.
Dere E, Huston JP, Souza Silva MA (2005). Episodic-like memory in mice: simultaneous assessment of object, place and temporal order memory. Brain Res Brain Res Protoc 16: 10–19.
Dong Z, Han H, Wang M, Xu L, Hao W, Cao J (2006). Morphine conditioned place preference depends on glucocorticoid receptors in both hippocampus and nucleus accumbens. Hippocampus 16: 809–813.
Durant S, Coulaud J, Amrani A, el Hasnaoui A, Dardenne M, Homo-Delarche F (1993). Effects of various environmental stress paradigms and adrenalectomy on the expression of autoimmune type 1 diabetes in the non-obese diabetic (NOD) mouse. J Autoimmun 6: 735–751.
Eilers A, Whitfield J, Shah B, Spadoni C, Desmond H, Ham J (2001). Direct inhibition of c-Jun N-terminal kinase in sympathetic neurones prevents c-jun promoter activation and NGF withdrawal-induced death. J Neurochem 76: 1439–1454.
Endl E, Gerdes J (2000). Posttranslational modifications of the KI-67 protein coincide with two major checkpoints during mitosis. J Cell Physiol 182: 371–380.
Ferguson D, Sapolsky R (2007). Mineralocorticoid receptor overexpression differentially modulates specific phases of spatial and nonspatial memory. J Neurosci 27: 8046–8052.
Fitzpatrick F, Christeff N, Durant S, Dardenne M, Nunez EA, Homo-Delarche F (1992). Glucocorticoids in the nonobese diabetic (NOD) mouse: basal serum levels, effect of endocrine manipulation and immobilization stress. Life Sci 50: 1063–1069.
Flood JF, Mooradian AD, Morley JE (1990). Characteristics of learning and memory in streptozocin-induced diabetic mice. Diabetes 39: 1391–1398.
Flores BH, Kenna H, Keller J, Solvason HB, Schatzberg AF (2006). Clinical and biological effects of mifepristone treatment for psychotic depression. Neuropsychopharmacology 31: 628–636.
Garcia A, Steiner B, Kronenberg G, Bick-Sander A, Kempermann G (2004). Age-dependent expression of glucocorticoid- and mineralocorticoid receptors on neural precursor cell populations in the adult murine hippocampus. Aging Cell 3: 363–371.
Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H (1984). Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol 133: 1710–1715.
Gerdes J, Li L, Schlueter C, Duchrow M, Wohlenberg C, Gerlach C et al (1991). Immunobiochemical and molecular biologic characterization of the cell proliferation-associated nuclear antigen that is defined by monoclonal antibody Ki-67. Am J Pathol 138: 867–873.
Gispen WH, Biessels GJ (2000). Cognition and synaptic plasticity in diabetes mellitus. Trends Neurosci 23: 542–549.
Hastings NB, Gould E (2003). Neurons inhibit neurogenesis. Nat Med 9: 264–266.
Heine VM, Maslam S, Zareno J, Joëls M, Lucassen PJ (2004). Suppressed proliferation and apoptotic changes in the rat dentate gyrus after acute and chronic stress are reversible. Eur J Neurosci 19: 131–144.
Herdegen T, Skene P, Bähr M (1997). The c-Jun transcription factor—bipotential mediator of neuronal death, survival and regeneration. Trends Neurosci 20: 227–231.
Herzog KH, Morgan JI (1996). Cellular immediate-early genes and cell death in the nervous system. Neuropathol Appl Neurobiol 22: 484–488.
Inouye KE, Chan O, Yue JT, Matthews SG, Vranic M (2005). Effects of diabetes and recurrent hypoglycemia on the regulation of the sympathoadrenal system and hypothalamo–pituitary–adrenal axis. Am J Physiol Endocrinol Metab 288: E422–E429.
Joëls M (2006). Corticosteroid effects in the brain: U-shape it. Trends Pharmacol Sci 27: 244–250.
Joëls M, Karst H, DeRijk R, de Kloet ER (2008). The coming out of the brain mineralocorticoid receptor. Trends Neurosci 31: 1–7.
Joëls M, Karst H, Krugers HJ, Lucassen PJ (2007). Chronic stress: implications for neuronal morphology, function and neurogenesis. Front Neuroendocrinol 28: 72–96.
Kaleeswari R, Thombre DP, Chakrabarty AS (1986). Acute effect of streptozotocin induced diabetes on bar pressing for food reward in albino rats. Indian J Physiol Pharmacol 30: 319–321.
Karst H, Joëls M (2007). Brief RU 38486 treatment normalizes the effects of chronic stress on calcium currents in rat hippocampal CA1 neurons. Neuropsychopharmacology 32: 1830–1839.
Kee N, Sivalingam S, Boonstra R, Wojtowicz JM (2002). The utility of Ki-67 and BrdU as proliferative markers of adult neurogenesis. J Neurosci Methods 115: 97–105.
Kiessling M, Auer RN, Kleihues P, Siesjo BK (1986). Cerebral protein synthesis during long-term recovery from severe hypoglycemia. J Cereb Blood Flow Metab 6: 42–51.
Kramer L, Fasching P, Madl C, Schneider B, Damjancic P, Waldhausl W et al (1998). Previous episodes of hypoglycemic coma are not associated with permanent cognitive brain dysfunction in IDDM patients on intensive insulin treatment. Diabetes 47: 1909–1914.
Krugers HJ, van der LS, van Olst E, Alfarez DN, Maslam S, Lucassen PJ et al (2007). Dissociation between apoptosis, neurogenesis, and synaptic potentiation in the dentate gyrus of adrenalectomized rats. Synapse 61: 221–230.
Lai M, Horsburgh K, Bae SE, Carter RN, Stenvers DJ, Fowler JH et al (2007). Forebrain mineralocorticoid receptor overexpression enhances memory, reduces anxiety and attenuates neuronal loss in cerebral ischaemia. Eur J Neurosci 25: 1832–1842.
Lambert KG, Gerecke KM, Quadros PS, Doudera E, Jasnow AM, Kinsley CH (2000). Activity-stress increases density of GFAP-immunoreactive astrocytes in the rat hippocampus. Stress 3: 275–284.
Li C, Brake WG, Romeo RD, Dunlop JC, Gordon M, Buzescu R et al (2004). Estrogen alters hippocampal dendritic spine shape and enhances synaptic protein immunoreactivity and spatial memory in female mice. Proc Natl Acad Sci USA 101: 2185–2190.
Liedtke W, Edelmann W, Bieri PL, Chiu FC, Cowan NJ, Kucherlapati R et al (1996). GFAP is necessary for the integrity of CNS white matter architecture and long-term maintenance of myelination. Neuron 17: 607–615.
Louw DF, Masada T, Sutherland GR (1998). Ischemic neuronal injury is ameliorated by astrocyte activation. Can J Neurol Sci 25: 102–107.
Luse SA (1970). The ultrastructure of the brain in the diabetic Chinese hamster with special reference to synaptic abnormalities. Electroencephalogr Clin Neurophysiol 29: 410.
Magarinos AM, McEwen BS (2000). Experimental diabetes in rats causes hippocampal dendritic and synaptic reorganization and increased glucocorticoid reactivity to stress. Proc Natl Acad Sci USA 97: 11056–11061.
Magistretti PJ, Pellerin L (1999). Astrocytes couple synaptic activity to glucose utilization in the brain. News Physiol Sci 14: 177–182.
Mayer G, Nitsch R, Hoyer S (1990). Effects of changes in peripheral and cerebral glucose metabolism on locomotor activity, learning and memory in adult male rats. Brain Res 532: 95–100.
Mayer JL, Klumpers L, Maslam S, de Kloet ER, Joëls M, Lucassen PJ (2006). Brief treatment with the glucocorticoid receptor antagonist mifepristone normalises the corticosterone-induced reduction of adult hippocampal neurogenesis. J Neuroendocrinol 18: 629–631.
McCarthy AM, Lindgren S, Mengeling MA, Tsalikian E, Engvall JC (2002). Effects of diabetes on learning in children. Pediatrics 109: E9.
McEwen BS, Magarinos AM, Reagan LP (2002). Studies of hormone action in the hippocampal formation: possible relevance to depression and diabetes. J Psychosom Res 53: 883–890.
McEwen BS, Sapolsky RM (1995). Stress and cognitive function. Curr Opin Neurobiol 5: 205–216.
McNay EC, Williamson A, McCrimmon RJ, Sherwin RS (2006). Cognitive and neural hippocampal effects of long-term moderate recurrent hypoglycemia. Diabetes 55: 1088–1095.
Morsink MC, Van Gemert NG, Steenbergen PJ, Joels M, de Kloet ER, Datson NA (2007). Rapid glucocorticoid effects on the expression of hippocampal neurotransmission-related genes. Brain Res 1150: 14–20.
Nagaraja N, Andrade C, Sudha S, Madan Singh N, Chandra JS, Venkataraman BV (2007). Glucocorticoid mechanisms may contribute to ECT-induced retrograde amnesia. Psychopharmacology (Berl) 190: 73–80.
Oitzl MS, de Kloet ER (1992). Selective corticosteroid antagonists modulate specific aspects of spatial orientation learning. Behav Neurosci 106: 62–71.
Oitzl MS, Fluttert M, de Kloet ER (1994). The effect of corticosterone on reactivity to spatial novelty is mediated by central mineralocorticosteroid receptors. Eur J Neurosci 6: 1072–1079.
Oitzl MS, Fluttert M, de Kloet ER (1998a). Acute blockade of hippocampal glucocorticoid receptors facilitates spatial learning in rats. Brain Res 797: 159–162.
Oitzl MS, Fluttert M, Sutanto W, de Kloet ER (1998b). Continuous blockade of brain glucocorticoid receptors facilitates spatial learning and memory in rats. Eur J Neurosci 10: 3759–3766.
Oomen CA, Mayer JL, de Kloet ER, Joëls M, Lucassen PJ (2007). Brief treatment with the glucocorticoid receptor antagonist mifepristone normalizes the reduction in neurogenesis after chronic stress. Eur J Neurosci 26: 3395–3401.
Parisi V, Uccioli L (2001). Visual electrophysiological responses in persons with type 1 diabetes. Diabetes Metab Res Rev 17: 12–18.
Paxinos G, Frankling KBJ (2001). The Mouse Brain in Stereotaxic Coordinates. Academic Press: San Diego, USA.
Popovic M, Biessels GJ, Isaacson RL, Gispen WH (2001). Learning and memory in streptozotocin-induced diabetic rats in a novel spatial/object discrimination task. Behav Brain Res 122: 201–207.
Ratka A, Sutanto W, Bloemers M, de Kloet ER (1989). On the role of brain mineralocorticoid (type I) and glucocorticoid (type II) receptors in neuroendocrine regulation. Neuroendocrinology 50: 117–123.
Rees DA, Alcolado JC (2005). Animal models of diabetes mellitus. Diabet Med 22: 359–370.
Revsin Y, Saravia F, Roig P, Lima A, de Kloet ER, Homo-Delarche F et al (2005). Neuronal and astroglial alterations in the hippocampus of a mouse model for type 1 diabetes. Brain Res 1038: 22–31.
Revsin Y, van Wijk D, Saravia FE, Oitzl MS, De Nicola AF, de Kloet ER (2008). Adrenal hypersensitivity precedes chronic hypercorticism in Streptozotocin-induced diabetes mice. Endocrinology 149: 3531–3539.
Rozeboom AM, Akil H, Seasholtz AF (2007). Mineralocorticoid receptor overexpression in forebrain decreases anxiety-like behavior and alters the stress response in mice. Proc Natl Acad Sci USA 104: 4688–4693.
Ryan CM, Williams TM (1993). Effects of insulin-dependent diabetes on learning and memory efficiency in adults. J Clin Exp Neuropsychol 15: 685–700.
Ryan CM, Williams TM, Finegold DN, Orchard TJ (1993). Cognitive dysfunction in adults with type 1 (insulin-dependent) diabetes mellitus of long duration: effects of recurrent hypoglycaemia and other chronic complications. Diabetologia 36: 329–334.
Saravia F, Revsin Y, Lux-Lantos V, Beauquis J, Homo-Delarche F, De Nicola AF (2004). Oestradiol restores cell proliferation in dentate gyrus and subventricular zone of streptozotocin-diabetic mice. J Neuroendocrinol 16: 704–710.
Saravia FE, Revsin Y, Gonzalez Deniselle MC, Gonzalez SL, Roig P, Lima A et al (2002). Increased astrocyte reactivity in the hippocampus of murine models of type 1 diabetes: the nonobese diabetic (NOD) and streptozotocin-treated mice. Brain Res 957: 345–353.
Schoenle EJ, Schoenle D, Molinari L, Largo RH (2002). Impaired intellectual development in children with Type I diabetes: association with HbA(1c), age at diagnosis and sex. Diabetologia 45: 108–114.
Scribner KA, Akana SF, Walker CD, Dallman MF (1993). Streptozotocin-diabetic rats exhibit facilitated adrenocorticotropin responses to acute stress, but normal sensitivity to feedback by corticosteroids. Endocrinology 133: 2667–2674.
Seki T (2003). Microenvironmental elements supporting adult hippocampal neurogenesis. Anat Sci Int 78: 69–78.
Sima AA, Li ZG (2005). The effect of C-peptide on cognitive dysfunction and hippocampal apoptosis in type 1 diabetic rats. Diabetes 54: 1497–1505.
Song H, Stevens CF, Gage FH (2002). Astroglia induce neurogenesis from adult neural stem cells. Nature 417: 39–44.
Stranahan AM, Arumugam TV, Cutler RG, Lee K, Egan JM, Mattson MP (2008). Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat Neurosci 11: 309–317.
van der Lely AJ, Foeken K, van der Mast RC, Lamberts SW (1991). Rapid reversal of acute psychosis in the Cushing syndrome with the cortisol-receptor antagonist mifepristone (RU486). Ann Intern Med 114: 143–144.
van Haarst AD, Oitzl MS, Workel JO, de Kloet ER (1996). Chronic brain glucocorticoid receptor blockade enhances the rise in circadian and stress-induced pituitary-adrenal activity. Endocrinology 137: 4935–4943.
Veenema AH, de Kloet ER, de Wilde MC, Roelofs AJ, Kawata M, Buwalda B et al (2007). Differential effects of stress on adult hippocampal cell proliferation in low and high aggressive mice. J Neuroendocrinol 19: 489–498.
Vesce S, Bezzi P, Volterra A (1999). The active role of astrocytes in synaptic transmission. Cell Mol Life Sci 56: 991–1000.
Vreugdenhil E, Kolk SM, Boekhoorn K, Fitzsimons CP, Schaaf M, Schouten T et al (2007). Doublecortin-like, a microtubule-associated protein expressed in radial glia, is crucial for neuronal precursor division and radial process stability. Eur J Neurosci 25: 635–648.
Waetzig V, Zhao Y, Herdegen T (2006). The bright side of JNKs-Multitalented mediators in neuronal sprouting, brain development and nerve fiber regeneration. Prog Neurobiol 80: 84–97.
Witter MP (2007). The perforant path: projections from the entorhinal cortex to the dentate gyrus. Prog Brain Res 163: 43–61.
Wong EY, Herbert J (2005). Roles of mineralocorticoid and glucocorticoid receptors in the regulation of progenitor proliferation in the adult hippocampus. Eur J Neurosci 22: 785–792.
Wu LM, Han H, Wang QN, Hou HL, Tong H, Yan XB et al (2007). Mifepristone repairs region-dependent alteration of synapsin I in hippocampus in rat model of depression. Neuropsychopharmacology 32: 2500–2510.
Acknowledgements
We thank Dr Danielle Champagne for critically reading the paper and Wendy Rodger for editorial assistance. The support by The Netherlands Science Foundation-WOTRO (NWO-WOTRO) project no. 88-252, Leiden-Trier International Research Training Group of the Deutsche Forschungs Gemeinschaft, and NWO (project no. NWO-DN-95-420) and The Royal Netherlands Academy of Arts and Sciences (KNAW) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
DISCLOSURE/CONFLICT OF INTEREST
The authors Yanina Revsin, Niels V Rekers, Mieke C Louwe, Flavia E Saravia, Alejandro F De Nicola, and Melly S Oitzl have nothing to declare. E Ronald de Kloet has been a member of the Scientific Advisory Board of Corcept Therapeutics Inc., the manufacturer of mifepristone over the past 3 years.
Supplementary Information accompanies the paper on the Neuropsychopharmacology website (http://www.nature.com/npp)
Rights and permissions
About this article
Cite this article
Revsin, Y., Rekers, N., Louwe, M. et al. Glucocorticoid Receptor Blockade Normalizes Hippocampal Alterations and Cognitive Impairment in Streptozotocin-Induced Type 1 Diabetes Mice. Neuropsychopharmacol 34, 747–758 (2009). https://doi.org/10.1038/npp.2008.136
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2008.136
Keywords
This article is cited by
-
Memory deficits in a juvenile rat model of type 1 diabetes are due to excess 11β-HSD1 activity, which is upregulated by high glucose concentrations rather than insulin deficiency
Diabetologia (2023)
-
Spinal Cord Injury Leads to Hippocampal Glial Alterations and Neural Stem Cell Inactivation
Cellular and Molecular Neurobiology (2022)
-
Activation of mineralocorticoid receptors facilitate the acquisition of fear memory extinction and impair the generalization of fear memory in diabetic animals
Psychopharmacology (2020)
-
Keap1/Nrf2/HO-1 signaling pathway contributes to p-chlorodiphenyl diselenide antidepressant-like action in diabetic mice
Psychopharmacology (2020)
-
Time-dependent impairments in learning and memory in Streptozotocin-induced hyperglycemic rats
Metabolic Brain Disease (2019)

{kind=link}
{kind=link}
{kind=link}