
Abstract
Despite management with opioids and other pain modifying therapies, neuropathic pain continues to reduce the quality of life and daily functioning in HIV-infected individuals. Cannabinoid receptors in the central and peripheral nervous systems have been shown to modulate pain perception. We conducted a clinical trial to assess the impact of smoked cannabis on neuropathic pain in HIV. This was a phase II, double-blind, placebo-controlled, crossover trial of analgesia with smoked cannabis in HIV-associated distal sensory predominant polyneuropathy (DSPN). Eligible subjects had neuropathic pain refractory to at least two previous analgesic classes; they continued on their prestudy analgesic regimens throughout the trial. Regulatory considerations dictated that subjects smoke under direct observation in a hospital setting. Treatments were placebo and active cannabis ranging in potency between 1 and 8% Δ-9-tetrahydrocannabinol, four times daily for 5 consecutive days during each of 2 treatment weeks, separated by a 2-week washout. The primary outcome was change in pain intensity as measured by the Descriptor Differential Scale (DDS) from a pretreatment baseline to the end of each treatment week. Secondary measures included assessments of mood and daily functioning. Of 127 volunteers screened, 34 eligible subjects enrolled and 28 completed both cannabis and placebo treatments. Among the completers, pain relief was greater with cannabis than placebo (median difference in DDS pain intensity change, 3.3 points, effect size=0.60; p=0.016). The proportions of subjects achieving at least 30% pain relief with cannabis versus placebo were 0.46 (95%CI 0.28, 0.65) and 0.18 (0.03, 0.32). Mood and daily functioning improved to a similar extent during both treatment periods. Although most side effects were mild and self-limited, two subjects experienced treatment-limiting toxicities. Smoked cannabis was generally well tolerated and effective when added to concomitant analgesic therapy in patients with medically refractory pain due to HIV DSPN.
Similar content being viewed by others

INTRODUCTION
In 1999, a report of the United States Institute of Medicine (Watson et al, 2000) recommended further investigations of the possible benefits of cannabis (marijuana) as a medicinal agent for a variety of conditions, including neuropathic pain due to HIV distal sensory polyneuropathy (DSPN). The most abundant active ingredient in cannabis, tetrahydrocannabinol (THC), and its synthetic derivatives, produce effective analgesia in most animal models of pain (Mao et al, 2000; Martin and Lichtman, 1998). The antinociceptive effects of THC are mediated through cannabinoid receptors (CB1, CB2) in the central and peripheral nervous systems (Calignano et al, 1998), which in turn interact with noradrenergic and κ-opioid systems in the spinal cord to modulate the perception of painful stimuli. The endogenous ligand of CB1, anandamide, itself is an effective antinociceptive agent (Calignano et al, 1998). In open-label clinical trials and one recent controlled trial (Abrams et al, 2007), medicinal cannabis has shown preliminary efficacy in relieving neuropathic pain.
Neuropathic pain in HIV is an important and persisting clinical problem, affecting 30% or more of HIV-infected individuals. Although combination antiretroviral (ARV) therapy has improved immunity and survival in HIV, it does not significantly benefit neuropathic pain. In fact, certain nucleoside-analogue HIV reverse transcriptase inhibitors, such as didanosine and stavudine, contribute to the frequent occurrence of painful DSPN, possibly through mitochondrial toxicity. Existing analgesic and adjunctive treatments are inadequate; neuropathic pain in DSPN persists in many cases despite attempts at management with opioids, nonsteroidal anti-inflammatory agents, and adjunctive pain modifying therapies, and patients suffer unfavorable side effects, reducing life quality and socioeconomic productivity.
Cannabis also may have adverse effects, including cognitive and motor dysfunction. Yet the extent to which these effects are treatment limiting has received little study. Given the paucity of rigorous scientific assessment of the potential medicinal value of cannabis, the State of California in 2001 commissioned research addressing this topic. As at that time alternative cannabis delivery systems had not been developed to provide the rapid tissue distribution afforded by smoking, the State specifically solicited research using smoked cannabis. We therefore conducted a clinical trial to ascertain a safe, clinically useful, and efficacious dosing range for smoked medicinal cannabis as a short-term analgesic in the treatment of refractory neuropathic pain in HIV DSPN. We evaluated the magnitude and clinical significance of side effects.
METHODS
Design
This was a phase II, single group, double-blind, placebo-controlled, crossover trial of smoked cannabis for the short-term treatment of neuropathic pain associated with HIV infection. Each subject participated in five study phases over 7 weeks as schematized in Figure 1: (1) a 1-week wash-in phase to obtain baseline measurements of pain and neuropsychological (NP) functioning; (2) 5 days of smoked active or placebo cannabis; (3) 2 weeks wash-out to allow for drug clearance and to assess possible extended benefits or rebound worsening of pain after treatment is withdrawn; (4) 5 days smoked active or placebo cannabis; and (5) 2 weeks final wash-out.

Study Schema. After screening, eligible subjects were randomized to receive cannabis or placebo first (treatment week 1; Rx 1), followed by the alternative treatment (treatment week 2; Rx 2). The principal measure of pain, the Descriptor Differential Scale (DDS), was measured at five time points (DDS1–5; arrowheads). The primary outcome was the difference in DDS change from baseline (DDS1) to the end of each treatment (active or placebo) week (DDS2/4). Remaining DDS assessments (3, 5) were used in secondary analyses. During each day of the 5-day treatment week, subjects smoked cannabis or placebo cigarettes four times daily. On day 1 of each week, cannabis dose was titrated to efficacy and tolerability as described in the text. On the remaining days (2–4), subjects smoked the maximum tolerated dose achieved on day 1.
Participants
Study participants were adults with documented HIV infection, neuropathic pain refractory to a least two previous analgesics, and an average score of 5 or higher on the pain intensity subscale of the Descriptor Differential Scale (DDS), described below. HIV DSPN was diagnosed by a board-certified clinical neurologist (RJE). The association of DSPN with HIV disease and ARV treatment was established according to the previously published research diagnostic criteria and included the presence of abnormal bilateral physical findings (reduced distal tendon reflexes, distal sensory loss) or electrophysiological abnormalities (distal leg sensory nerve conduction studies), plus symptoms of pain and paresthesias, acquired in the setting of HIV infection (AAN, 1991). Exclusion criteria were (1) current DSM-IV substance use disorders; (2) lifetime history of dependence on cannabis; (3) previous psychosis with or intolerance to cannabinoids; (4) concurrent use of approved cannabinoid medications (ie Marinol); (5) positive urine toxicology screen for cannabinoids during the wash-in week before initiating study treatment; and (6) serious medical conditions that might affect participant safety or the conduct of the trial. Individuals with a previous history of alcohol or other drug dependence were eligible provided that criteria for dependence had not been met within the last 12 months. Subjects were excluded if urine toxicology demonstrated ongoing use of nonprescribed, recreational drugs such as methamphetamine and cocaine.
Screening and baseline evaluations
Before administering study treatments, all subjects underwent comprehensive clinical and laboratory evaluations. Plasma HIV RNA (viral load; VL) was quantified by reverse transcriptase-polymerase chain reaction (Amplicor, Roche Diagnostic Systems, Indianapolis, IN) using the ultrasensitive assay (nominal lower limit of quantitation, 50 copies per ml). Blood CD4+ lymphocyte counts were measured by flow cytometry. Standard blood chemistry and hematology panels were performed. The overall severity of DSPN was characterized using the Total Neuropathy Score (TNS) (Cornblath et al, 1999). TNS is a validated measure, which combines information obtained from assessment of reported symptoms, physical signs, nerve conduction studies, and quantitative sensory testing. To evaluate potential cardiovascular, pulmonary, and other medical risks, we performed electrocardiogram (ECG) and chest radiography, and assessed past medical history, medication history, and conducted a focused general physical and neurological examination. Also performed were a drug use history, NP testing and an abridged Composite International Diagnostic Interview to assess for bipolar disorder, schizophrenia, recent drug or alcohol addiction, and other psychiatric exclusion criteria. Participants watched a video demonstrating the standardized smoking technique (Foltin et al, 1988), and each participant was observed practicing the smoking technique with a placebo cigarette.
Regulatory Issues and Study Medication
This trial was performed as an outpatient study at the General Clinical Research Center at the University of California, San Diego (UCSD) Medical Center. This study was approved and monitored by the UCSD Institutional Review Board, the Research Advisory Panel of California, the US Food and Drug Administration, the US Drug Enforcement Administration, the US Department of Health and Human Services, and the University of California Center for Medicinal Cannabis Research. Confidentiality of research participants was protected by a federal Certificate of Confidentiality. All participants gave written informed consent to participate in this study.
All cannabis and placebo cigarettes were provided by the National Institute on Drug Abuse and were constructed of the same base material. Active strengths ranged from 1% to 8% Δ-9-THC concentration by weight. Placebo cigarettes were made from whole plant material with cannabinoids removed and were identical in appearance to active cigarettes. Cannabis was placed in an airtight container and stored in a locked, alarmed freezer at the UCSD Medical Center Investigational Drug Service Pharmacy. Cannabis was humidified at room temperature within a dessicator using a saturated sodium chloride solution for 12–24 h before use. Periodic assays for THC content were performed to confirm stability of material over time in storage. Nurses weighed material before and after smoking and returned all used and unused medication to the pharmacy Investigational Drug Service for appropriate disposal. Randomization was performed by a research pharmacist using a random number generator, and the key to study assignment was withheld from investigators until completion statistical analyses.
Cannabis Administration
On study days, participants smoked randomly assigned active or placebo cannabis under the observation of the study nurse who provided smoking cues (‘inhale’, ‘hold’, ‘exhale’) from an adjacent room. On day 1 of each intervention week, a dose escalation/titration protocol was employed to accommodate individual differences in sensitivity to the analgesic and adverse effects of cannabis (Figure 2). Over four smoking sessions, each participant titrated to the dose (‘target dose’) affording the best achievable pain relief without unacceptable adverse effects. Titration was started at 4% THC or placebo and adjusted incrementally downwards (to 2 or 1%) if side effects were intolerable, or upwards (to 6 or 8%) if pain relief was incomplete. The target dose was that providing the best analgesia whereas maintaining side effects, if any, at a tolerable level. Treatment was discontinued if side effects were intolerable despite adjusting to the minimum study dose (1%). The target dose was administered for the remaining 4 days, except that downward titration or dose withholding was available if adverse effects became intolerable.

Dose escalation schedule for day 1 of the study treatment weeks. See text for details. The objective of the dose escalation was to find, for each study subject, a dose of smoked cannabis that optimized pain relief, while minimizing unwanted adverse effects.
To provide near-continuous drug effect for the duration of the 8-h study day, treatments were administered in four daily smoking sessions separated by intervals of 90–120 min. This interval was chosen based on previous studies demonstrating that the subjective ‘high’ after varying doses of cannabis declined to 50% of maximal effect after an average of 100 min (Harder and Rietbrock, 1997). Although the effect-time course for analgesia with cannabis may differ from the effect-time course for subjective ‘highness’, no formal studies of cannabis-related analgesia were available on which to base estimates of effect duration.
Outcome Measures
Outcome measures selected for this study were standardized, validated measures of multiple pain-associated constructs, including analgesia, improvement in function, and relief of pain-associated emotional distress. Details of these measures are provided below and the schedule of their administration is provided in Table 1.
Pain quality and impact
Descriptor Differential Scale. The principal evaluation of treatment efficacy was change in self-reported pain magnitude assessed by the DDS. The DDS is a ratio scale containing 24 words describing pain intensity and unpleasantness. Ratings are aggregated to provide a summary score on a 0- to 20-point scale. Participants rated their ‘current’ pain magnitude (at the time of assessment) relative to these descriptors. Pain intensity changes were compared from baseline to the end of each treatment week as shown in Figure 1. The DDS demonstrates good internal consistency, reliability, objective correlation with experimentally induced pain (Gracely et al, 1978a, 1978b), and sensitivity to analgesic effects on clinical pain syndromes (Gracely and Kwilosz, 1988). Participants also rated the quality and intensity of their pain experience on the McGill Pain Questionnaire (Chapman et al, 1985; Melzack, 1975). This included the Visual Analog Scale (VAS), a 10-cm line anchored at one end by the descriptor ‘No Pain’ and at the other by the words ‘Worst Pain Imaginable.’
Additional clinical assessments
Table 1 specifies the schedule for additional clinical assessments. Disability, mood, and quality of life in study subjects were assessed using the Sickness Impact Profile (SIP; Gilson et al, 1975), the Profile of Mood States (POMS; McNair et al, 1992) and the Brief Symptom Inventory (BSI; Derogatis and Melisaratos, 1983). Treatment emergent effects of cannabis were assessed by clinician interview and self-report of physical and psychological symptoms as captured using a standardized inventory, the UKU Side Effect Rating Scale (Lingjaerde et al, 1987a, 1987b). Also, a subjective Highness/Sedation Scale adapted from Block et al (1998) was administered to assess the intensity of psychological effects commonly associated with the inhalation of cannabis. Subjects were asked to ‘guess’ the treatment to which they had been assigned using established procedures (Moscucci et al, 1987).
Safety assessements
Participants were monitored carefully before, during and after study treatments to detect clinically significant changes in blood pressure, heart rate, respiration, temperature, and HIV disease parameters including plasma VL and blood CD4+ lymphocyte counts. Additional evaluations included blood hematology and chemistry, urine dipstick toxicology for drugs of abuse, chest radiography, and ECG. Participants were instructed not to drive while on study and were provided with taxi transportation if unable to make other arrangements. Adverse drug effects were graded according to the Division of AIDS Table for Grading Severity of Adult Adverse Experiences (AACTG, 1992). For events rated Grade 2 or 1, study treatment was temporarily suspended until the event resolved. For events rated Grade 3 or 4, study treatment was permanently discontinued. In the event that treatment suspension was required more than once, the next lower dosing level was used for the remaining smoking sessions.
Concomitant nonstudy analgesics
As intractable pain was a criterion for study inclusion, subjects were permitted to continue taking concomitant analgesics such as opioids, nonsteroidal anti-inflammatory agents, and adjunctive pain medications. They were asked to maintain regular dosing during the study. However, to monitor compliance with these instructions, we recorded the average daily dose of these agents at each visit. For analytic purposes, these data were expressed as aspirin or morphine equivalents using standard conversions (AHCRP, 1992).
Statistical Analyses
Primary analyses
Baseline DDS values between the two arms were compared using the Wilcoxon's rank sum test. Prestudy power analyses indicated that a sample size of 30 individuals would yield an 80% chance (α=0.05) of detecting at least a 1.8 point difference between placebo- and active treatment-related changes in pain intensity as measured by DDS. The principal evaluation of treatment efficacy/tolerability in this study was the change in DDS pain intensity scores from baseline to the end of each treatment week (Figure 1) used completers only, as randomized. A conservative ITT analysis was also performed, using multiple imputation (MI) for the six subjects with incomplete data. For MI, the missing Δ values were imputed from the most unfavorable (highest) 50% of the observed (completers) values. These comparisons used the t-test with the MI adjustment (Little and Rubin, 2002).
Secondary analyses were performed for study completers, except for the adverse event (AE) analysis, which included all randomized subjects. Change in average weekly VAS values between the placebo and active treatment weeks was analyzed using Wilcoxon's signed rank test (WSRT). The association between baseline pain and titrated dosing used the F-test of linear regression. The change in use of analgesics during the study was compared between placebo and active cannabis weeks using WSRT. The proportion of subjects guessing their treatment allocation was compared to a chance guess (50% correct guess) using the χ2 one-sample test for proportions.
The proportions of subjects with moderate or severe UKU symptoms possibly or probably attributable to study treatment were compared for the placebo and active cannabis weeks using the McNemar test, for each UKU side effect. Similarly, the proportions of subjects with clinically significant changes in heart rate, blood pressure, VL, and CD4 counts were compared between the two arms using the McNemar test. This test considers pairs of outcomes for the two treatment weeks and is appropriate for a crossover trial. The number of AEs (including the six dropouts) was compared between the two treatment weeks using WSRT.
RESULTS
Recruitment, Screening, and Completion of Assigned Treatments
Screening and subject disposition are summarized in the CONSORT diagram (Figure 3). Between February 2002 and November 2006, 127 subjects were screened, 34 met inclusion/ exclusion criteria and 28 completed treatment with both active and placebo cannabis. Six randomized subjects failed to complete the study. As demonstrated in Table 2, completers did not differ significantly from the ITT population on demographics, medical variables, and cannabis experience. Two subjects were withdrawn for safety reasons. One cannabis-naive subject had an acute, cannabis-induced psychosis at the start of the second smoking week; unblinding revealed that he had received placebo during the first week and active cannabis during the second. A second subject developed an intractable, smoking-related cough during cannabis treatment; symptoms resolved spontaneously after smoking cessation. A third subject experienced intractable diarrhea deemed unlikely to be related to study treatments. A fourth elected to discontinue the protocol in order to fulfill an unanticipated personal commitment, and a fifth was lost to follow-up. The sixth was dropped because of a protocol-defined exclusion when urine toxicology was positive for methamphetamine.

CONSORT Flow Diagram. Disposition of subjects screened, randomized, and completing both treatment periods. Placebo 1, subjects randomized to receive placebo cannabis during the first treatment week; Active 1, subjects randomized to receive active cannabis during the first treatment week. DSPN, distal sensory polyneuropathy; +Utox, positive urine toxicology for substances of abuse, including cannabis.
Baseline Characteristics
Study participants were typically white (75%), high-school educated (mean 13.6, SD±2.0 years) men (100%) in their late 40s (48.8±6.8 years), who had been HIV infected for more than 5 years, and who were prescribed combination ARV therapy (93%) for advanced HIV disease. Most (72%) had been exposed to potentially neurotoxic dideoxynucleoside reverse transcriptase inhibitors (d-drugs). Almost all (96%) had previous exposure to cannabis, generally remote (>1 year; 63%). The mean baseline TNS, reflecting symptoms, disability, neurological exam findings, and quantitative measures of peripheral nerve function, was 16 points (range, 9–34), corresponding to mild-to-moderately severe neuropathy as described previously (Cornblath et al, 1999). Of the 28 participants, 18 (64%) took opioid pain medications, 10 (36%) used concurrent NSAIDS, 8 (29%) used tricyclic antidepressants, and 18 (64%) used anticonvulsants. All participants continued to take concomitant analgesics and adjunctive pain-modifying medications throughout the trial.
Treatment Effects
The median (range) baseline pain as measured by DDS pain intensity scale was 11.1. (9.1, 13.7) points. During the placebo treatment week, 26 subjects (93%) titrated to a maximum nominal dose of 8% THC; the remaining two chose 6%. In comparison, during the cannabis treatment week, most subjects titrated to the 2% (N=9) or 4% (N=10) dose; the remainder titrated to 1% (N=1), 6% (N=4), and 8% (N=4). Subjects with greater pain at baseline as measured by DDS chose higher nominal doses, although this association was statistically modest (linear regression p=0.052, R2=0.14).
Primary analysis
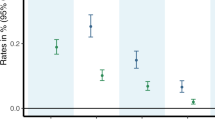
Pain reduction was significantly greater with cannabis compared to placebo (median difference in pain reduction=3.3 DDS points; effect size=0.60; p=0.016, all completers included; Figure 4). The results were consistent for the ITT analysis (p=0.020), and for the comparison based on the first week of treatment alone (median change in DDS pain =−4.1 and 0.1 for the cannabis and placebo arms, p=0.029). There were no evident sequence effects: the degree of pain relief achieved with active cannabis did not differ significantly according to whether it was administered during the first or the second treatment week (mean reduction in DDS points, 4.1 vs 0.96; p=0.13).

Plot of treatment effect. DDS pain severity scores (mean, 95% CI) for participants in the cannabis (CNB) and placebo (PCB) arms before study treatment (W/I), during each of the 2 treatment weeks (1, 2) and during the Washout (W/O) between treatment weeks. Cannabis was superior to placebo in this crossover trial whether subjects were treated with cannabis during the first or second treatment week. The median difference in DDS pain severity change was 3.3 points (p = 0.016, WRT).
Additional analyses
The proportion of subjects achieving pain reduction of 30% or more was greater for the active cannabis than for the placebo cannabis week (0.46 (95%CI 0.28, 0.65) vs 0.18 (0.03, 0.32), p=0.043). The number needed to treat (NNT) to achieve 30% pain reduction (active vs placebo cannabis) was 3.5 (95% CI 1.9, 20.8). In a secondary analysis of changes in reported pain as measured by the VAS, the median (range) change in pain scores from baseline was −17 (−58, 52) for cannabis as compared to −4 (−56, 28) for placebo (p<0.001). As measured by the POMS, SIP, and BSI, there were similar improvements in total mood disturbance, physical disability, and quality of life for the cannabis and placebo treatments (data not shown).
Concomitant Analgesic Use
As intractable pain was a criterion for study inclusion, subjects were permitted to continue taking concomitant analgesics such as opioids, nonsteroidal anti-inflammatories and adjunctive pain medications. They were asked to maintain regular dosing during the study; however, to monitor compliance with these instructions, average daily doses of these agents were collected according to the schedule in Table 1. Concomitant opioids were used by 18 of the 28 subjects (64%). Changes from baseline in morphine equivalent doses were minimal and did not differ significantly for placebo (+5.8%) as compared to cannabis (+0.1%). Changes in DDS pain severity did not differ for those who did and did not use opioids (mean difference 0.21, 95%CI (−3.7, 4.1)). Of the 28 subjects 10 (36%) used nonopioid analgesics such as acetaminophen and NSAIDS. Changes in aspirin equivalents were minimal: 7.4% for placebo and 0.7% for active cannabis.
Preservation of Blinding
To assess preservation of the blind, subjects were asked to guess the treatment to which they were assigned at the end of dose titration (day 1) and at the end of each treatment week. After dose titration, subjects receiving placebo guessed no better than chance (5/13 (38%) incorrect vs 50% chance guessing), whereas those receiving cannabis rarely guessed incorrectly (1/15 (93%)). At the end of the first treatment week, subjects receiving placebo still guessed no better than chance (4/13 (31%) incorrect guesses). At the end of the first treatment week, DDS pain reduction was larger for the cannabis than placebo (median change=−4.08 vs 0.08, respectively). Most of the subjects crossing over to active cannabis during their second treatment week correctly guessed their treatment assignment (12/13, 92%).
Treatment Safety and Adverse Events
Dose- and treatment-limiting AEs occurred in two subjects as described above. As assessed by the UKU and AE reports, the frequency of some nontreatment-limiting side effects was greater for cannabis than placebo. These included concentration difficulties, fatigue, sleepiness or sedation, increased duration of sleep, reduced salivation, and thirst. The combined UKU and DAIDS side effects frequency was greater with cannabis than placebo and there was a trend for moderate or severe AEs to be more frequent during active than during placebo administration. Changes in heart rate and blood pressure were asymptomatic and resolved spontaneously; none resulted in unblinding of the investigators. Increases in heart rate of 30 points or more within 30 min of a smoking session were more frequent with cannabis (13/28; 46%) than placebo (1/28; 4%). Blood pressure alterations and changes in VL and CD4 counts did not differ for cannabis and placebo.
DISCUSSION
In this randomized clinical trial, smoked cannabis at maximum tolerable dose (1–8% THC), significantly reduced neuropathic pain intensity in HIV-associated DSPN compared to placebo, when added to stable concomitant analgesics. Using verbal descriptors of pain magnitude from DDS, cannabis was associated with an average reduction of pain intensity from ‘strong’ to ‘mild to moderate’. Also, cannabis was associated with a sizeable (46%) and significantly greater (vs 18% for placebo) proportion of patients who achieved what is generally considered clinically meaningful pain relief (eg ⩾30% reduction in pain; Farrar et al, 2001). Mood disturbance, physical disability, and quality of life all improved significantly for subjects during study treatments, regardless of treatment order.
A recently published, influential review concluded that the potential medicinal benefits of cannabis, including analgesia for neuropathic pain, warranted further high quality research (Watson et al, 2000). We employed methodological criteria generally regarded as essential for establishing the validity of treatment outcome research in chronic pain syndromes, including rigorous specification of neurologic diagnosis, randomization and placebo control, assessment of study blinding, tracking of cointerventions, and an individualized dosing strategy designed to optimize outcomes (Deyo, 1983). The study sample is arguably representative of clinic populations of painful HIV DSPN, given the duration and stage of HIV disease, use of concurrent analgesics, as well as history of exposure to ARV agents known to be associated with painful DSPN.
This study's findings are consistent with and extend other recent research supporting the short-term efficacy of cannabis for neuropathic pain. Thus one recent, inpatient randomized clinical trial of painful DSPN noted that inhaled cannabis, in doses comparable to those in the present report, significantly reduced pain intensity (34%) compared to placebo (17%; Abrams et al, 2007). Our findings extend the efficacy of cannabis to individuals with intractable pain, as our cohort had substantially greater number of subjects taking concomitant analgesics (100%) than did Abrams et al (22%). Most of our subjects took concomitant opioid therapy and almost all took at least one other concurrent pain-modifying medication. This afforded us the opportunity to evaluate potential pharmacodynamic interactions, such as synergy with opioids, as suggested by previous investigators. We observed no interaction (positive or negative synergism) between opioids and cannabis. Two other placebo-controlled studies of neuropathic pain associated with multiple sclerosis indicated that both sublingual Δ-9-THC alone or with cannabidiol (Rog et al, 2005), and oral synthetic Δ-9-THC (Svendsen et al, 2004) significantly outperformed placebo. As regards the pain benefits of cannabis compared to other available therapies for painful DSPN, as assessed by NNT: our results (NNT=3.5) are equivalent to those achieved by Abrams et al (2007) (NNT=3.6), are in the range of the leading anticonvulsants (lamotrigine, NNT=5.4; gabapentin, NNT=3.8) (Simpson et al, 2003; Backonja, 2002) and are superior to null results obtained for amitriptyline (Kieburtz et al, 1998; Shlay et al, 1998) and mexiletine (Kieburtz et al, 1998).
Blinding in this study was performed using conventional measures, which included randomization of subjects to treatment assignments known only to the study pharmacist. We expected that because the prominent psychoactive effects of cannabis would distinguish it from placebo—as is true for other potent analgesic agents such as opioids—some subjects would correctly ‘guess’ their treatment assignment. To evaluate preservation of the blind, we asked each subject to report his or her impression of what treatment they received at several time points during the study as previously described. Blinding was considered to be preserved when the accuracy of treatment guesses was no different from random guessing (50%). Correct guessing was related to two factors: first, whether the subject received placebo or cannabis first; and second, when during the study they were asked to make their guess. Thus among subjects randomized to receive placebo first, guessing was no better than chance through the end of the first treatment week, whereas among subjects randomized to receive cannabis first, the majority correctly guessed their treatment assignment at all time points. Furthermore, by the second treatment week, when all subjects had been given the opportunity to compare the cannabis placebo and treatments, even those randomized to receive placebo first correctly guessed their treatment assignment. These findings raise the possibility that some of the DDS pain reduction was placebo driven. To assess whether correct treatment guessing influenced treatment responses, we performed secondary analyses showing that in the placebo group during the first treatment week, when guessing was no better than chance, cannabis still provided pain relief superior to that of placebo. This finding suggests that although placebo effects were present, treatment effects were independent.
Several other potential limitations were addressed. Attrition, approximately 18%, was somewhat higher than projected, but was within the range of other trials of HIV-associated and other painful neuropathic syndromes (Kieburtz et al, 1998; Max et al, 1992; Shlay et al, 1998; Simpson et al, 2003). However, an ITT sensitivity analysis demonstrated that the superiority of cannabis was robust to reasonable assumptions about the treatment responses of the dropouts. We included subjects with DSPN related either to HIV itself or to nucleoside ARV drug exposure; a more homogeneous sample may have had a different outcome. Finally, durability of analgesia, which is of paramount concern in chronic pain syndromes, could not be assessed in this short-term study. Because DDS is a relatively complex instrument for capturing pain reports, its validity and reliability might be limited by confusion and sedation during cannabis treatment. We therefore considered a simpler pain assessment tool, VAS, which is less susceptible to confounding by neurocognitive side effects. Similar to DDS, VAS also showed superior analgesia with cannabis.
The therapeutic application of cannabis depends on palatability and safety concerns as well as efficacy. Smoking is not an optimal delivery system. Long-term use of smoked cannabis is associated with symptoms suggestive of obstructive lung disease, and although short-term use is not (Tetrault et al, 2007), many individuals cannot tolerate smoking. Alternative administration routes for cannabinoids, including vaporization and mucosal sprays, are currently approved for clinical use in Great Britain and Canada and are under evaluation in the United States. Cannabis has potent psychotropic effects including ‘paradoxical’ effects (eg depersonalization, hallucination, suspiciousness) in an important minority of individuals (Hall and Solowij, 1998). A recent meta-analysis suggested an increased risk of psychotic illness in individuals who had ever used cannabis (Moore et al, 2007), although it was acknowledged that vulnerability to psychotic disorder and use of cannabis may be confounded.
Our findings suggest that cannabinoid therapy may be an effective option for pain relief in patients with medically intractable pain due to HIV-associated DSPN. As with all analgesics, dose limiting side effects should be carefully monitored, particularly during the initial trials of therapy.
References
AACTG (1992). Table for Grading Severity of Adult Adverse Experiences. Division of AIDS, National Institute of Allergy and Infectious Diseases: Rockville, Maryland.
AAN (1991). Nomenclature and research case definitions for neurologic manifestations of human immunodeficiency virus-type 1 (HIV-1) infection. Report of a Working Group of the American Academy of Neurology AIDS Task Force. Neurology 41: 778–785.
Abrams DI, Jay CA, Shade SB, Vizoso H, Reda H, Press S et al (2007). Cannabis in painful HIV-associated sensory neuropathy: a randomized placebo-controlled trial. Neurology 68: 515–521.
Agency for Health Care Policy and Research (1992). Acute Pain Management in Adults: Operative Procedures. Quick Reference Guide for Clinicians AHCPR Pub No. 92-0019. Rockville, MD: Agency for Health Care Policy and Research, Public Health Service, U.S. Department of Health and Human Services.
Backonja MM (2002). Use of anticonvulsants for treatment of neuropathic pain. Neurology 59: S14–S17.
Block RI, Erwin WJ, Farinpour R, Braverman K (1998). Sedative, stimulant, and other subjective effects of marijuana: relationships to smoking techniques. Pharmacol Biochem Behav 59: 405–412.
Calignano A, La Rana G, Giuffrida A, Piomelli D (1998). Control of pain initiation by endogenous cannabinoids. Nature 394: 277–281.
Chapman CR, Syrjala K, Sargur M (1985). Pain as a manifestation of cancer treatment. Semin Oncol Nurs 1: 100–108.
Cornblath DR, Chaudhry V, Carter K, Lee D, Seysedadr M, Miernicki M et al (1999). Total neuropathy score: validation and reliability study. Neurology 53: 1660–1664.
Derogatis LR, Melisaratos N (1983). The Brief Symptom Inventory: an introductory report. Psychol Med 13: 595–605.
Deyo RA (1983). Conservative therapy for low back pain. Distinguishing useful from useless therapy. JAMAama 250: 1057–1062.
Farrar JT, Young Jr JP, LaMoreaux L, Werth JL, Poole RM (2001). Clinical importance of changes in chronic pain intensity measured on an 11-point numerical pain rating scale. Pain 94: 149–158.
Foltin RW, Fischman MW, Byrne MF (1988). Effects of smoked marijuana on food intake and body weight of humans living in a residential laboratory. Appetite 11: 1–14.
Gilson BS, Gilson JS, Bergner M, Bobbit RA, Kressel S, Pollard WE et al (1975). The sickness impact profile. Development of an outcome measure of health care. Am J Public Health 65: 1304–1310.
Gracely RH, Kwilosz DM (1988). The Descriptor Differential Scale: applying psychophysical principles to clinical pain assessment. Pain 35: 279–288.
Gracely RH, McGrath F, Dubner R (1978a). Ratio scales of sensory and affective verbal pain descriptors. Pain 5: 5–18.
Gracely RH, McGrath P, Dubner R (1978b). Validity and sensitivity of ratio scales of sensory and affective verbal pain descriptors: manipulation of affect by diazepam. Pain 5: 19–29.
Hall W, Solowij N (1998). Adverse effects of cannabis. Lancet 352: 1611–1616.
Harder S, Rietbrock S (1997). Concentration-effect relationship of delta-9-tetrahydrocannabiol and prediction of psychotropic effects after smoking marijuana. Int J Clin Pharmacol Ther 35: 155–159.
Kieburtz K, Simpson D, Yiannoutsos C, Max MB, Hall CD, Ellis RJ et al (1998). A randomized trial of amitriptyline and mexiletine for painful neuropathy in HIV infection. AIDS Clinical Trial Group 242 Protocol Team. Neurology 51: 1682–1688.
Lingjaerde O, Ahlfors U, Bech P, Dencker S, Elgen K (1987a). The UKU side effects rating scale. Acta Psychiatr Scand 76: 11–79.
Lingjaerde O, Ahlfors UG, Bech P, Dencker SJ, Elgen K (1987b). The UKU side effect rating scale. A new comprehensive rating scale for psychotropic drugs and a cross-sectional study of side effects in neuroleptic-treated patients. Acta Psychiatr Scand Suppl 334: 1–100.
Little R, Rubin D (2002). Statistical Analysis with Missing Data. Wiley-Interscience: Hoboken, New Jersey.
Mao J, Price DD, Lu J, Keniston L, Mayer DJ (2000). Two distinctive antinociceptive systems in rats with pathological pain. Neurosci Lett 280: 13–16.
Martin BR, Lichtman AH (1998). Cannabinoid transmission and pain perception. Neurobiol Dis 5: 447–461.
Max MB, Lynch SA, Muir J, Shoaf SE, Smoller B, Dubner R (1992). Effects of desipramine, amitriptyline, and fluoxetine on pain in diabetic neuropathy. N Engl J Med 326: 1250–1256.
McNair D, Lorr M, Droppleman L (1992). Profile of Mood States (POMS) Manual. Educational and Industrial Testing Services: San Diego.
Melzack R (1975). The McGill Pain Questionnaire: major properties and scoring methods. Pain 1: 277–299.
Moore TH, Zammit S, Lingford-Hughes A, Barnes TR, Jones PB, Burke M et al (2007). Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet 370: 319–328.
Moscucci M, Byrne L, Weintraub M, Cox C (1987). Blinding, unblinding, and the placebo effect: an analysis of patients′ guesses of treatment assignment in a double-blind clinical trial. Clin Pharmacol Ther 41: 259–265.
Rog DJ, Nurmikko TJ, Friede T, Young CA (2005). Randomized, controlled trial of cannabis-based medicine in central pain in multiple sclerosis. Neurology 65: 812–819.
Shlay JC, Chaloner K, Max MB, Flaws B, Reichelderfer P, Wentworth D et al (1998). Acupuncture and amitriptyline for pain due to HIV-related peripheral neuropathy: a randomized controlled trial. Terry Beirn Community Programs for Clinical Research on AIDS. JAMA 280: 1590–1595.
Simpson DM, McArthur JC, Olney R, Clifford D, So Y, Ross D et al (2003). Lamotrigine for HIV-associated painful sensory neuropathies: a placebo-controlled trial. Neurology 60: 1508–1514.
Svendsen KB, Jensen TS, Bach FW (2004). Does the cannabinoid dronabinol reduce central pain in multiple sclerosis? Randomised double blind placebo controlled crossover trial. BMJmj 329: 253.
Tetrault JM, Crothers K, Moore BA, Mehra R, Concato J, Fiellin DA (2007). Effects of marijuana smoking on pulmonary function and respiratory complications: a systematic review. Arch Intern Med 167: 221–228.
Watson SJ, Benson Jr JA, Joy JE (2000). Marijuana and medicine: assessing the science base: a summary of the 1999 Institute of Medicine report. Arch Gen Psychiatry 57: 547–552.
Acknowledgements
This project was supported by Grant C00-SD-104 from the University of California, Center for Medicinal Cannabis Research. We thank Dr Geoffrey Sheean and the staff of the General Clinical Research Center at the University of California, San Diego Medical Center for supporting this project. Statistical analyses were conducted by Florin Vaida.
Author information
Authors and Affiliations
Corresponding author
Additional information
DISCLOSURE
Heather Bentley and Ben Gouaux are employees of the Center for Medicinal Cannabis Research at the University of California, San Diego, the study sponsor. Ms Bentley is Project Manager for the CMCR and assisted the investigator with regulatory issues, oversight/monitoring, and preparation of the manuscript. Mr. Gouaux is a Research Associate with the CMCR and assisted the investigator with regulatory issues, oversight/monitoring, data preparation and analysis, and preparation and submission of the article. The authors declare that over the past 3 years Dr. Atkinson has received compensation from Eli Lilly Pharmaceuticals. The authors declare that they have not received other financial support or compensation in the past 3 years or have any personal financial holdings that could be perceived as constituting a conflict of interest.
Rights and permissions
About this article
Cite this article
Ellis, R., Toperoff, W., Vaida, F. et al. Smoked Medicinal Cannabis for Neuropathic Pain in HIV: A Randomized, Crossover Clinical Trial. Neuropsychopharmacol 34, 672–680 (2009). https://doi.org/10.1038/npp.2008.120
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2008.120
Keywords
This article is cited by
-
An answered call for aid? Cannabinoid clinical framework for the opioid epidemic
Harm Reduction Journal (2023)
-
The Role of Cannabis, Cannabidiol and Other Cannabinoids in Chronic Pain. The Perspective of Physicians
Journal of Neuroimmune Pharmacology (2022)
-
Interactive Effects of HIV Infection and Cannabis Use on Insula Subregion Functional Connectivity
Journal of Neuroimmune Pharmacology (2022)
-
Cannabinoide zur Therapie chronischer Nervenschmerzen und Spastik
MMW - Fortschritte der Medizin (2022)
-
Pharmacologic and Non-Pharmacologic Treatments for Chronic Pain Used by Patients with Pain, HIV, and Depression
AIDS and Behavior (2022)
