
Abstract
Finding causal relationships between genotypic and phenotypic variation is a key focus of evolutionary biology, human genetics and plant breeding. To identify genome-wide patterns underlying trait diversity, we assembled a high-quality reference genome of Cardamine hirsuta, a close relative of the model plant Arabidopsis thaliana. We combined comparative genome and transcriptome analyses with the experimental tools available in C. hirsuta to investigate gene function and phenotypic diversification. Our findings highlight the prevalent role of transcription factors and tandem gene duplications in morphological evolution. We identified a specific role for the transcriptional regulators PLETHORA5/7 in shaping leaf diversity and link tandem gene duplication with differential gene expression in the explosive seed pod of C. hirsuta. Our work highlights the value of comparative approaches in genetically tractable species to understand the genetic basis for evolutionary change.
Similar content being viewed by others

Parallel genetic studies in C. hirsuta and the related model A. thaliana have provided a powerful platform to identify the molecular causes of trait diversity between these species at a gene-by-gene level1–4. In particular, leaf shape differences have provided an attractive model to investigate the genetic basis for morphological evolution1–5. To extend this approach to a genome-wide level and broaden its scope, we constructed a high-quality reference genome of the C. hirsuta strain ‘Oxford’2 for comparison with A. thaliana6. C. hirsuta and A. thaliana belong to lineage I in the Brassicaceae family7,8, together with A. lyrata9 and Capsella rubella10, all of which have fully assembled genomes (Fig. 1a). Complete or partial genome sequences are available for a number of other Brassicaceae species6,9–14, including Aethionema arabicum11 in the earliest diverging lineage of Brassicaceae (Fig. 1a). This allows comparisons between C. hirsuta and A. thaliana to gain additional context from comparative analyses within lineage I and the Brassicaceae family as a whole.

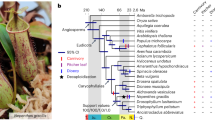
a, Phylogenetic tree for A. thaliana, A. lyrata, C. rubella, C. hirsuta, E. salsugineum, S. parvula, B. rapa and A. arabicum constructed using 10,111 orthologous genes within these eight species. Bold branches have maximum confidence35. The number of gene families expanded (red) or contracted (blue) compared with the most recent ancestor common ancestor (MRCA) are indicated along each branch. b, The upper panel shows the position of genetic markers mapped to the C. hirsuta genome assembly; the y-axis shows the genetic distance; shaded regions are inferred centromeric or pericentromeric heterochromatic regions that show very rare recombinations. The lower panel shows a rainforest plot of long terminal repeat (LTR) genes in the C. hirsuta genome; the y-axis shows the minimum distance of each LTR gene to its neighbours in a logarithmic scale. Chromosomes are indicated on the x-axis. c,d, Circos plots showing synteny between the genomes of A. thaliana (ath) and C. hirsuta (chi) (c), and A. lyrata (aly) and C. hirsuta (chi) (d); the outer circle shows the gene density distribution with a window size of 100 kbp.
To sequence C. hirsuta, we used a shotgun sequencing strategy, combining paired end reads (197× assembled sequence coverage) and mate pair reads (66× assembled) from Illumina HiSeq (a total of 52 Gbp raw reads; see Supplementary Table 1). The short reads were first assembled with SOAPdenovo15 to generate contigs, which were further linked into superscaffolds using a custom Bayesian framework-based algorithm, which is a unified platform for genome assembly utilizing the mapping quality of the paired reads (BAMLINK; see Supplementary Methods). The superscaffolds were anchored and oriented with a further 8,249 bacterial artificial chromosome paired end sequences (6 Mbp physical coverage) and a genetic map with 328 markers, to obtain eight pseudomolecules of a total length of 183 Mbp (92.2% of the assembly, corresponding to the C. hirsuta chromosomes) and 614 unanchored fragments. The final assembly encompasses 198 Mbp, which is comparable to a previous flow cytometry estimate of 225 Mbp16. We demonstrated the high quality of the assembly using three independent datasets: by perfectly aligning 358 randomly selected, Sanger-sequenced regions (size 500–600 bp) to our assembly (Supplementary Table 2); by mapping a total of 1 Mbp of sequence derived from eight 454 shotgun sequence-assembled fragments and two Sanger-sequenced fragments with 99.98% identity to our assembly (Supplementary Fig. 1); and finally, we confirmed the co-linearity between the physical and genetic position of 328 genetic markers and 36 additional markers which were not used in the assembly procedure (Supplementary Methods; Fig. 1b, top, and Supplementary Fig. 2). Our results demonstrate that BAMLINK provides an efficient and accurate method to merge local information from different sequencing platforms with broad scale information from a genetic map.
We annotated the genome by a combination of ab initio gene prediction using Illumina transcriptome data collected from a range of tissues and heterologous homology evidence. A total of 29,458 protein-coding genes with 37,997 transcripts and 579 nuclear encoded tRNA were predicted in the C. hirsuta genome (Supplementary Table 3). We built a phylogeny based on the complete set of protein-coding genes for C. hirsuta, C. rubella, A. thaliana, A. lyrata, A. arabicum, Brassica rapa12, Schrenkiella parvula13 and Eutrema salsugineum14. This dates the divergence of C. hirsuta and A. thaliana to around 32 Myr ago, which is within the range of previous estimates17 (Fig. 1a). The C. hirsuta genome is largely syntenic to the genomes of A. thaliana and A. lyrata (Fig. 1c,d and Supplementary Fig. 3). Gene-rich regions are mostly confined to the chromosome arms and all chromosomes have long centromeric and pericentromeric regions, which collectively account for approximately 40% of the genome (78.9 Mbp). We assembled these typically challenging chromosome regions using BAMLINK and found that they are highly enriched in long terminal repeat (LTR) retrotransposons and exhibit very low recombination frequencies (Fig. 1b). In contrast, the centromeric regions account for only 14 Mbp of the A. thaliana genome, thereby explaining the inflated genome size of C. hirsuta compared with A. thaliana. A recent expansion of LTR retrotransposons also contributed to the increased genome size of A. lyrata compared to A. thaliana9 (Supplementary Fig. 4). However, whereas A. lyrata LTR retrotransposons are relatively young (∼0.8 Myr) and broadly distributed throughout the genome, C. hirsuta LTR retrotransposons are older (median age ∼4.8 Myr) (Supplementary Fig. 4). The centromeres of other sequenced Brassicaceae are similarly large9,10, indicating that the C. hirsuta genome retains more ancestral features, including karyotype18 and genome size, than A. thaliana. Predominant selfing in C. hirsuta18 is associated with loss of gene function at the self-incompatibility S locus (Supplementary Figs 5 and 6). The S locus in C. hirsuta is syntenic with other Brassicaceae genomes, but the distinct S locus that evolved secondarily in the closely related genus Leavenworthia19 did not exist in C. hirsuta (Supplementary Fig. 7). Self-compatibility probably evolved recently in C. hirsuta as the S locus maintains hallmarks of functional S haplotypes despite disruptive mutations in the SRK and SCR genes20 (Supplementary Fig. 5).
To identify species-specific gene families that might contribute to trait diversification, we clustered the annotated protein-coding genes of C. hirsuta, C. rubella, A. thaliana, A. lyrata, A. arabicum, B. rapa, S. parvula and E. salsugineum. We identified 10,871 core gene families comprising at least one gene from each species, and determined expansion and contraction of gene families in different evolutionary lineages (Fig. 1a and Supplementary Methods). A five-way comparison of four lineage I species, C. hirsuta, C. rubella, A. thaliana and A. lyrata, with additional species distributed across the Brassicaceae, E. salsugineum, S. parvula, B. rapa and A. arabicum, shows that C. hirsuta has 694 unique gene families (Fig. 2a). We identified 5,560 genes in 2,067 families in C. hirsuta as tandem duplicates, and 16 of these families were specific to C. hirsuta (Supplementary Fig. 8). Among the total number of gene families in common to all eight species, 53 were identified as significantly expanded in C. hirsuta based on a phylogenetically informed test21 (P ≤ 0.05) (Fig. 2b and Supplementary Fig. 9). Analysis of these expanded or unique gene families in C. hirsuta revealed an overrepresentation of transcription factor function (adjusted P = 2 × 10−5 for GO:0010468) (see Supplementary Tables 4–6 for enriched InterPro terms). Previous genetic studies have shown that transcription factors and tandem gene duplication contribute to morphological differences between C. hirsuta and A. thaliana leaves1–3. To test the significance of this observation genome wide, in an unbiased way, we identified differentially expressed genes (DEGs) between C. hirsuta and A. thaliana during early leaf development. We found a significant overrepresentation of both transcription factors (P = 1.9 × 10−4) and tandemly duplicated genes (P = 2.07 × 10−46) among these DEGs, indicating that these gene types are prevalent in the species-specific leaf transcriptomes.

a, Venn diagram comparing the number of gene families shared between four lineage I species, A. lyrata, C. hirsuta, A. thaliana and C. rubella, and four additional species distributed across the Brassicaceae, E. salsugineum, S. parvula, B. rapa and A. arabicum. b, Logarithmically scaled smooth scatterplot of gene families showing the number of species-specific members in A. thaliana (x-axis) and C. hirsuta (y-axis). Dots above the grey line represent gene families that are significantly expanded in C. hirsuta (pink) or contracted in A. thaliana (green), and dots below the grey line represent gene families that are expanded in A. thaliana (green), based on Hahn's test with eight species; pale pink dots represent families that are unique to C. hirsuta and pale green dots represent families that are unique to A. thaliana. The arrows indicate two families containing pectin methylesterase (PME) and PME inhibitor genes; one gene family has no members in A. thaliana. The arrowhead indicates the gene family that contains PLETHORA5/7 transcription factors (note this family does not correspond to the green dot and is not significantly expanded or contracted).
We used these transcriptome data to investigate the molecular causes of leaf shape diversity between C. hirsuta and A. thaliana in more depth. Following the premise that co-option of gene networks active in the shoot apical meristem contributes to leaf shape diversity1, we found 278 meristem genes22 upregulated in C. hirsuta relative to A. thaliana during early leaf development (fold change ≥2.0 times greater in C. hirsuta than in A. thaliana). Transcription factors were significantly enriched (P ≤ 0.05) among these upregulated meristem genes and comprised 44 genes including SHOOT MERISTEMLESS, BREVIPEDICELLUS and CUP-SHAPED COTYLEDON1, which were previously implicated in dissected leaf development2,3 (Fig. 3a, Supplementary Tables 7 and 8). These enriched transcription factors included the C. hirsuta orthologues of PLETHORA5 (PLT5) and PLT7, which are involved in meristem stem cell specification but have not been previously implicated in leaf diversity23. ChPLT5 and ChPLT7 are upregulated in C. hirsuta leaves relative to A. thaliana and their transcripts accumulate at the sites of emerging leaflets (Fig. 3b,c and Supplementary Fig. 10). We reduced ChPLT5/7 expression in C. hirsuta leaves by means of an artificial miRNA that targeted both genes and found a pronounced reduction in the number of leaflets formed per leaf (Fig. 3d–h and Supplementary Fig. 10). Moreover, expressing ChPLT7 in the simple leaf margin of A. thaliana under the CUC2 promoter was sufficient to cause ectopic leaflet formation (Fig. 3i,j). Therefore, ChPLT5/7 are necessary and ChPLT7 is sufficient for leaflet formation. Since PLT7 coding sequences from both C. hirsuta and A. thaliana were sufficient to cause leaflet production in A. thaliana (Fig. 3i,j and Supplementary Fig. 10), it is likely that regulatory sequence differences in PLT7 contributed to leaf shape divergence between these species.

a, Venn diagram showing the intersection of shoot meristem-expressed genes and transcription factor-encoding genes that are expressed more than twofold higher in C. hirsuta than in A. thaliana leaves. b,c, In situ hybridization of ChPLT5 (b) and ChPLT7 (c) expression in transverse and longitudinal (shown in inset) sections through C. hirsuta shoot apices. Arrows indicate signal in the shoot apical meristem and leaflets. Scale bars, 100 µm. d–g, Rosette leaf 5 (d,f) and cauline leaves (e,g) of C. hirsuta wild type (WT) (d,e) and 35S::amirChPLT5,7 (f,g). h, Quantification of leaflet number in rosette leaf 5 of wild type and 35S::amirChPLT5,7. Error bars show standard error of leaflet number. ***, regression of leaflet number on genotype (P < 0.001). i,j, Representative rosette leaves of A. thaliana vector-only control (i) and CUC2::ChPLT7 (j). N = 20/20 (i) and 4/27 (j) independent transgenic lines with similar phenotypes. Scale bars, 1 cm.
To exploit comparisons between C. hirsuta and A. thaliana more broadly, we determined DEGs during seed pod development in each species. Seeds are dispersed by explosive pod shatter in C. hirsuta; a trait that readily distinguishes it from A. thaliana and other species in the Brassicaceae family24. We found a significant overrepresentation of transcription factors (P = 2.6 × 10−8 for C. hirsuta and P = 1.2 × 10−4 for A. thaliana) and tandemly duplicated genes (P = 8.6 × 10−4 for C. hirsuta and P = 1.0 × 10−15 for A. thaliana) in both species. Among 319 orthologous genes that were differentially expressed only in C. hirsuta (adjusted P < 0.05 in C. hirsuta, adjusted P > 0.3 in A. thaliana), we found six highly enriched gene ontology (GO) terms related to cell wall and pectinesterase activity (Fig. 4a). The six GO term enrichments were largely attributed to ten genes encoding pectin methylesterases (PMEs) and PME inhibitors (PMEIs) (Supplementary Table 9). To investigate whether species-specific PME/I genes were differentially expressed in C. hirsuta seed pods, we identified two expanded PME/I families, which together contained eight upregulated genes (Fig. 2b, Supplementary Fig. 11 and Supplementary Table 9). A total of five DEGs within these expanded PME/I families were tandem duplicates present in C. hirsuta but not in A. thaliana or other sequenced genomes that we analysed in the Brassicaceae (Fig. 4b–d, Supplementary Figs 11 and 12). Three upregulated PMEI genes were highly expressed in C. hirsuta seeds, which had lower PME enzymatic activity per unit protein than A. thaliana seeds, and accumulated pectin with a high degree of methyl-esterification in asymmetrically thickened cell walls in the seed coat (Fig. 4e–h and Supplementary Fig. 13). Thus, our results provide an avenue to explore cell wall properties that distinguish the seeds and pods of C. hirsuta from A. thaliana.

a, GO terms that are enriched (dark blue) in the set of 319 DEGs specific to C. hirsuta and not A. thaliana seed pod development, and their parental terms (light blue). P values were obtained from exact Fisher tests after correcting for multiple hypothesis testing. Black arrows indicate an ‘is a’ and orange arrows indicate a ‘part of’ relationship between respective terms in the GO graph. b–d, Syntenic blocks in A. thaliana and C. hirsuta indicate that the C. hirsuta genes CARHR045830, CARHR045840 and CARHR045850 (b), CARHR213450 and CARHR213460 (c), and CARHR265360 and CARHR265370 (d) were derived by species-specific tandem duplication. e–h, Fluorescent signal of LM20 antibody labelling in seeds of C. hirsuta (e,f) and A. thaliana (g,h). Intense signal shows the asymmetrically thickened cell walls of the seed coat sub-epidermal layer in C. hirsuta (e,f), and the outer cell wall of the seed coat epidermis in A. thaliana (g,h); arrowheads indicate the difference in cell wall thickening of the sub-epidermal layer between A. thaliana and C. hirsuta. Scale bars, 100 µm (e), 20 µm (f,h), 50 µm (g).
Individual case studies have previously identified changes in transcription factors and tandemly duplicated genes as causes of morphological diversity in multicellular organisms1,25–29. Our results indicate that these are not isolated examples but rather that evolutionary changes in the expression of transcription factors and tandem gene duplicates may provide privileged molecular paths for the generation of diversity. For example, we identified previously unsuspected roles for PLT5/7 transcription factors and tandemly duplicated PMEI genes in divergent leaf and seed dispersal traits between C. hirsuta and A. thaliana. Notably, tandemly duplicated genes (including PMEIs) and transcription factors (including PLTs), as well as genes differentially expressed in a species-specific manner during fruit development and in young developing leaves, were enriched within gene families that showed evidence for positive selection (Supplementary Fig. 14), suggesting that some of these genes may have evolved non-neutrally to contribute to trait diversity. We found that these gene families under positive selection, together with expanded gene families and tandemly duplicated genes, have increased functional diversity as measured by the Shannon entropy of domain architecture (Supplementary Figs 15 and 16). However, tandemly duplicated genes show more domain conservation than expanded gene families (Kolmogorov–Smirnov test, P < 2.2 × 10−16). Taken together, these findings suggest that tandem gene duplication contributes to trait diversity while retaining stronger domain conservation than expanded gene families. This difference might reflect stronger evolutionary constraints30, gene conversion31 or simply younger age. Our study underscores how the comparison of high quality, annotated genomes and developmentally targeted transcriptomes between closely related species with high genetic tractability can establish causal links between genotypic and phenotypic variation above the species level29. This approach provides a valuable complement to linkage-based methods that rely on genetic crosses or association mapping32,33.
Methods
Plant material
DNA
C. hirsuta of the reference accession Oxford (Ox) (specimen voucher Hay 1 (OXF)4 was self-pollinated in the greenhouse for seven generations before being used for next generation sequence library preparation.
RNA
Leaf and fruit tissue was harvested from A. thaliana Col-0 and C. hirsuta Ox grown on soil in either a growth chamber under short day conditions (8 h light (20 °C) and 16 h dark (18 °C)) for leaves, or a greenhouse under long day conditions (16 h light (20 °C) and 8 h dark (16 °C)) for fruit. Total RNA from three biological replicates of microdissected young leaves (L5 and L6), or two biological replicates of whole fruits at two developmental stages (9 and 16), was isolated from each species using the RNeasy Plus Micro Kit (Qiagen) and reverse transcription carried out using the Superscript VILO cDNA Synthesis Kit (Life technologies).
Genome assembly and annotation
The Illumina short reads were first assembled with SOAPdenovo15 to generate contigs. These contigs were further linked into superscaffolds using BAMLINK, which is a unified platform for genome assembly utilizing the paired reads and genetic map information (see Supplementary Methods). Initial gene models were derived as statistically combined consensus models from both ab initio gene predictions and homologous evidence (see Supplementary Methods). These predictions were adjusted by aligning C. hirsuta RNA-Seq data from seedling, leaf, floral and fruit tissues, using the cufflinks suite34, to retrieve alternative splicing models. Gene models were annotated for Interpro domains, GO terms and a description line using the AHRD pipeline (https://github.com/groupschoof/AHRD/), and gene models with transposon signatures were removed.
Phylogenetic analysis
An ultrametric species tree of eight crucifers, A. thaliana, A. lyrata, C. hirsuta, C. rubella, A. arabicum, B. rapa, E. salsugineum and S. parvula, was generated from 10,111 concatenated multiple sequence alignments (MSA) of orthologous genes. This MSA was submitted to maximum likelihood phylogenetic reconstruction with FastTree v2.1.735. The maximum likelihood tree was then rescaled into an ultrametric tree using a penalized likelihood approach.
Quantification of gene expression
Paired-end reads were aligned to the reference genome (tair10 for A. thaliana and CHIV1 for C. hirsuta) using tophat with default parameters. Raw read counts per gene were quantified with HTSeq v0.5.4p1 (http://www-huber.embl.de/users/anders/HTSeq/) using the ‘–stranded = no –type = CDS’ option. To facilitate cross-species comparisons, reads within UTR regions were ignored since UTR regions are generally more divergent than CDS regions. Differential expression between samples from the same species was determined using DESeq. We found the most sensitive parameter settings for the function estimateDispersions were method = ‘blind’, and sharingMode = ‘fit-only’.
Data availability
The assembled genome sequence and annotation, the raw Illumina genomic DNA reads and the Illumina RNA-seq reads are available from GenBank (Biosample: SAMN02183597; Bioproject: PRJNA293154) and from our website http://chi.mpipz.mpg.de/assembly. Source code of BAMLINK is available at http://chi.mpipz.mpg.de/software. The data that support the findings of this study are also available from the corresponding author on request.
References
Vlad, D. et al. Leaf shape evolution through duplication, regulatory diversification, and loss of a homeobox gene. Science 343, 780–783 (2014).
Hay, A. & Tsiantis, M. The genetic basis for differences in leaf form between Arabidopsis thaliana and its wild relative Cardamine hirsuta. Nat. Genet. 38, 942–947 (2006).
Blein, T. et al. A conserved molecular framework for compound leaf development. Science 322, 1835–1839 (2008).
Hay, A. & Tsiantis, M. Cardamine hirsuta: a comparative view. Curr. Opin. Genet. Dev. 39, 1–7 (2016).
Bar, M. & Ori, N. Leaf development and morphogenesis. Development 141, 4219–4230 (2014).
The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815 (2000).
Beilstein, M. A., Al-Shehbaz, I. A. & Kellogg, E. A. Brassicaceae phylogeny and trichome evolution. Am. J. Bot. 93, 607–619 (2006).
Huang, C. H. et al. Resolution of Brassicaceae phylogeny using nuclear genes uncovers nested radiations and supports convergent morphological evolution. Mol. Biol. Evol. 33, 394–412 (2016).
Hu, T. T. et al. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat. Genet. 43, 476–481 (2011).
Slotte, T. et al. The Capsella rubella genome and the genomic consequences of rapid mating system evolution. Nat. Genet. 45, 831–835 (2013).
Haudry, A. et al. An atlas of over 90,000 conserved noncoding sequences provides insight into crucifer regulatory regions. Nat. Genet. 45, 891–898 (2013).
Wang, X. et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–1039 (2011).
Dassanayake, M. et al. The genome of the extremophile crucifer Thellungiella parvula. Nat. Genet. 43, 913–918 (2011).
Yang, R. et al. The reference genome of the halophytic plant Eutrema salsugineum. Front. Plant. Sci. 4, 46 (2013).
Li, R. et al. The sequence and de novo assembly of the giant panda genome. Nature 463, 311–317 (2010).
Johnston, J. S. et al. Evolution of genome size in Brassicaceae. Ann. Bot. 95, 229–235 (2005).
Beilstein, M. A., Nagalingum, N. S., Clements, M. D., Manchester, S. R. & Mathews, S. Dated molecular phylogenies indicate a Miocene origin for Arabidopsis thaliana. Proc. Natl Acad. Sci. USA 107, 18724–18728 (2010).
Hay, A. S. et al. Cardamine hirsuta: a versatile genetic system for comparative studies. Plant J. 78, 1–15 (2014).
Chantha, S. C., Herman, A. C., Platts, A. E., Vekemans, X. & Schoen, D. J. Secondary evolution of a self-incompatibility locus in the Brassicaceae genus Leavenworthia. PLoS Biol. 11, e1001560 (2013).
Shimizu, K. K. & Tsuchimatsu, T. Evolution of selfing: recurrent patterns in molecular adaptation. Ann. Rev. Ecol. Evol. Syst. 46, 593–622 (2015).
Hahn, B. H., Shaw, G. M., De Cock, K. M. & Sharp, P. M. AIDS as a zoonosis: scientific and public health implications. Science 287, 607–614 (2000).
Yadav, R. K., Girke, T., Pasala, S., Xie, M. & Reddy, G. V. Gene expression map of the Arabidopsis shoot apical meristem stem cell niche. Proc. Natl Acad. Sci. USA. 106, 4941–4946 (2009).
Aida, M. et al. The PLETHORA genes mediate patterning of the Arabidopsis root stem cell niche. Cell 119, 109–120 (2004).
Hofhuis, H. et al. Morphomechanical innovation drives explosive seed dispersal. Cell 166, 222–233 (2016).
Chan, Y. F. et al. Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science 327, 302–305 (2010).
Van de Peer, Y., Maere, S. & Meyer, A. The evolutionary significance of ancient genome duplications. Nat. Rev. Genet. 10, 725–732 (2009).
Hanikenne, M. et al. Evolution of metal hyperaccumulation required cis-regulatory changes and triplication of HMA4. Nature 453, 391–395 (2008).
Freeling, M. Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Annu. Rev. Plant. Biol. 60, 433–453 (2009).
Koenig, D. & Weigel, D. Beyond the thale: comparative genomics and genetics of Arabidopsis relatives. Nat. Rev. Genet. 16, 285–298 (2015).
Hasselmann, M., Lechner, S., Schulte, C. & Beye, M. Origin of a function by tandem gene duplication limits the evolutionary capability of its sister copy. Proc. Natl Acad. Sci. USA 107, 13378–13383 (2010).
Chen, J. M., Cooper, D. N., Chuzhanova, N., Ferec, C. & Patrinos, G. P. Gene conversion: mechanisms, evolution and human disease. Nat. Rev. Genet. 8, 762–775 (2007).
Gan, X. et al. Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature 477, 419–423 (2011).
Atwell, S. et al. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 465, 627–631 (2010).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5, e9490 (2010).
Acknowledgements
We thank J. Martinez-Garcia for critical reading, the Wellcome Trust Centre for Human Genetics, University of Oxford, for sequencing, R. Berndtgen, W. Faigl, S. Tabata and the Centre for Genomic Research, University of Liverpool for assistance, H. North for seeds, S. Theologis for DNA isolation and helpful discussions, and B. Scheres and S. Grigg for discussions on PLT genes. This work was supported by Biotechnology and Biological Sciences Research Council grants BB/H011455/1 (M.T.) and BB/H01313X/1 (An.H.), a Gatsby Charitable foundation grant (M.T.), a Deutsche Forschungsgemeinschaft priority programme ‘Adaptomics’ grant TS 229/1-1 and HA 6316/1-3 (M.T. and An.H.), a University of Oxford pump-priming grant (M.T.), a Human Frontiers Science Programme Young Investigators grant RGY0087/2011 (An.H. and K.S.S.), EU Cofund Plant Fellow to R.B., URPP Evolution in Action to K.S.S. and R.S.-I., MEXT KAKENHI 16H06469 and Swiss National Science Foundation to K.S.S. An.H. was supported by the Max Planck Society W2 Minerva programme and M.T. by the Cluster of Excellence on Plant Sciences and a core grant of the Max Planck Society.
Author information
Authors and Affiliations
Contributions
M.T. designed and directed the study. M.T. and X.G. coordinated the project with help from R.M. X.G. assembled the genome. G.H., As.H. and K.F.X.M. annotated the genome and identified the tandemly duplicated genes. B.P. constructed the genetic map and performed quality checks with M.C. As.H. performed gene family analysis and positive selection scans. X.G. and As.H. analysed RNAseq data. An.H., H.H. and U.N. performed seed pod experiments. M.K., R.D.I., L.A.N., D.V. and P.H. performed leaf experiments. R.B., K.S.S. and R.S.-I. performed S-locus analysis. E.K., S.B., M.H., S.S., K.M., D.L., D.K. and R.W. provided materials, B.S., J.H., M.I., D.F. and R.M. analysed data. M.T., An.H. and X.G. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Methods, Supplementary Tables 1-13 and Figures 1-18. (PDF 4305 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Gan, X., Hay, A., Kwantes, M. et al. The Cardamine hirsuta genome offers insight into the evolution of morphological diversity. Nature Plants 2, 16167 (2016). https://doi.org/10.1038/nplants.2016.167
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/nplants.2016.167
This article is cited by
-
Common evolutionary trajectory of short life-cycle in Brassicaceae ruderal weeds
Nature Communications (2023)
-
Ancestral self-compatibility facilitates the establishment of allopolyploids in Brassicaceae
Plant Reproduction (2023)
-
Genome-wide survey and identification of AP2/ERF genes involved in shoot and leaf development in Liriodendron chinense
BMC Genomics (2021)
-
Divergence of three BRX homoeologs in Brassica rapa and its effect on leaf morphology
Horticulture Research (2021)
-
The Cardamine enshiensis genome reveals whole genome duplication and insight into selenium hyperaccumulation and tolerance
Cell Discovery (2021)
