
Abstract
Studying the relationship between virus infection and cellular response is paradigmatic for our understanding of how perturbation changes biological systems. Immune response, in this context is a complex yet evolutionarily adapted and robust cellular change, and is experimentally amenable to molecular analysis. To visualize the full cellular response to virus infection, we performed temporal transcriptomics, proteomics, and phosphoproteomics analysis of vesicular stomatitis virus (VSV)-infected mouse macrophages. This enabled the understanding of how infection-induced changes in host gene and protein expression are coordinated with post-translational modifications by cells in time to best measure and control the infection process. The vast and complex molecular changes measured could be decomposed in a limited number of clusters within each category (transcripts, proteins, and protein phosphorylation) each with own kinetic parameter and characteristic pathways/processes, suggesting multiple regulatory options in the overall sensing and homeostatic program. Altogether, the data underscored a prevalent executive function to phosphorylation. Resolution of the molecular events affecting the RIG-I pathway, central to viral recognition, reveals that phosphorylation of the key innate immunity adaptor mitochondrial antiviral-signaling protein (MAVS) on S328/S330 is necessary for activation of type-I interferon and nuclear factor κ B (NFκB) pathways. To further understand the hierarchical relationships, we analyzed kinase–substrate relationships and found RAF1 and, to a lesser extent, ARAF to be inhibiting VSV replication and necessary for NFκB activation, and AKT2, but not AKT1, to be supporting VSV replication. Integrated analysis using the omics data revealed co-regulation of transmembrane transporters including SLC7A11, which was subsequently validated as a host factor in the VSV replication. The data sets are predicted to greatly empower future studies on the functional organization of the response of macrophages to viral challenges.
Similar content being viewed by others
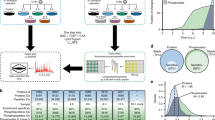


Introduction
To maintain host homeostasis and provide protection against invading pathogens, cells are constantly sensing the microenvironment through signaling machinery that, when activated, drives downstream inflammatory responses. Innate immune responses are initiated by germline encoded pattern recognition receptors such as Toll-like receptors and retinoic acid inducible gene (RIG)-like receptors expressed on the cell surface and throughout the cell cytoplasm, as well as integral membrane compartments.1 These receptors sense pathogen-associated molecular patterns to induce strong pathogen-specific innate immune responses. Antiviral responses activated downstream from virus-specific pathogen-associated molecular patterns include the activation of type-I interferons, transcriptional activation of subsets of inflammatory genes, and a cytokine/chemokine milieu that in turn contributes to the activation of the adaptive immune response.2 In order to activate a potent inflammatory immune response, cells employ different layers of regulation that range from chromatin remodeling and transcriptional or translational modulation to protein folding, post-translational modifications (PTMs), and localization in the appropriate subcellular compartment.3 It is plausible that these different regulatory levels enable self-governing and failsafe mechanisms, ensuring clearance of the pathogen while minimizing damage to the host.
Several systematic studies using transcriptomics and functional RNA interference (RNAi) screens have begun to identify (i) the genome-scale regulatory networks that underlie a pathogen response;4 (ii) a specific FOXO3-IRF7 feed-forward loop in antiviral response;5 (iii) development of a virus-specific regulatory map;6 (iv) identification of effector molecules of type-I interferon response7 and several key regulators of innate immunity such as cyclic GMP-AMP synthase (cGAS)8 and interferon induced protein with tetratricopeptide repeats 1 (IFIT1).9 Further, mass spectrometry (MS)-based quantitative proteomics has proven to be a powerful complementary method to study innate immune responses.10–12 These systems-level approaches have begun to reveal how viruses have evolved ways to tame the host immune system by modulating the key effector proteins and signaling pathways as well disrupting homeostatic sensing mechanisms.13–15 Together, these studies shows us that systems-level methodologies can assist in understanding the full spectrum of activation and regulation behind this complex machinery, and aid in defining the global and unbiased dynamics of the host response.16
In this study, we aimed to map the complex cellular immune response of murine macrophages to vesicular stomatitis virus (VSV) infection in an unbiased approach, profiling the transcriptome, proteome, and phosphoproteome changes in a quantitative and time-resolved manner (Figure 1a). This allowed the global evaluation of the dynamics of host genes that have a role in infection, commonly referred to as the ‘Infectome’, and to assess the extent and the logic of the cellular response. We chose VSV, a negative-sense single-stranded RNA virus, for its wide model usage, safety, convenience, and parsimonious molecular functions, encoding five proteins. An integrated bioinformatics analysis of these data sets allowed insights into infection-induced gene expression dynamics, identification of many known and novel differentially regulated genes and proteins during the infection process, as well as numerous altered known and novel phosphosites enabling to explore potential kinase–substrate associations and perturbed signaling pathways. Overall, our data revealed that (i) molecular responses can be clustered by their kinetics and nature in modules with different properties and distinctive features—interestingly, several genes associated with these clusters had no previous innate immune annotation; (ii) phosphorylation appears to occupy a hierarchically dominant challenge-specific function; and (iii) phosphorylation of the innate immunity adapter MAVS has a key role in controlling innate immunity response pathways.

Overview of the experimental outline, analyses, and clustering patterns. (a) Workflow outlining the experimental design, profiling, and bioinformatics analyses. (b) Transcriptome changes of selected genes upon vesicular stomatitis virus (VSV) infection. (c) Skewness coefficient for the distribution of fold changes based on Z score for transcriptome, proteome, and phosphoproteome for each time point after infection. (d) Venn diagram depicting the overlap of differentially expressed genes at transcriptome, proteome, and phosphoproteome levels. (e) Significantly enriched biological processes (P value⩽0.005) at transcriptome, proteome, and phosphoproteome. (f) The normalized mean expression changes for each time point after VSV infection with error bars depicting the extent of expression changes for the subset of genes associated with three representative biological processes that are significantly regulated at transcriptome/proteome/phosphoproteome. Heatmap depicts the expression changes of selected genes associated with the respective biological process.
Results
OMICS profiling reveals global insights into gene expression dynamics upon VSV infection
The cellular response of a murine macrophage cell line (RAW264.7) infected with a mutant strain of VSV (VSV-M2) was profiled over the period of time required for infection. Three time points were selected including 20 min, 3, and 6 h. VSV-M2 is not competent for suppression of cytoplasmic recognition17 and consequently triggers a robust interferon (IFN) response (Supplementary Figure S1A,B). The upregulation of type-I IFNs, IFITs, interleukins (ILs), and chemokines at the transcript level measured using RNA sequencing (RNA-Seq) confirmed that the cells responded to the viral infection (Figure 1b). In total, we were able to quantify 19,889 transcripts, 5,828 proteins, and 1,799 phosphoproteins (available as a resource at www.infectome-map.org). The phosphoproteins included 6,526 phosphoserine, 2,188 phosphothreonine, and 817 phosphotyrosine sites. Using an absolute robust Z score threshold of 2.5, we identified 2,002 differentially regulated genes (Supplementary Figure S1C). These included 1,432 transcripts, 652 proteins, and 401 phosphoproteins (Supplementary Table S1). The differentially regulated phosphoresidues were comprised 506 phosphoserine; 102 phosphothreonine; and 46 phosphotyrosine residues. We further used a rank-based normalization method to calculate a normalized fold change that allowed the comparison of changes in the transcriptome, proteome, and phosphoproteome (Supplementary Figure S1D).
To understand the expression dynamics of transcripts, proteins, and phosphoproteins, we estimated the skewness in the distribution of expression changes upon VSV infection. We found that there is a statistically significant level of skewness in the individual distributions revealing distinct dynamics in innate immune response upon VSV infection (Figure 1c). Transcriptome distribution revealed a large downregulation at 20 min and 3 h, whereas at 6 h a profound upregulation was observed (Figure 1c). Proteome distribution showed an upregulation at 3 and 6 h, and a subtle downregulation at 20 min. Strikingly, phosphoproteome showed a strong upregulation at all three time points (Figure 1c). The overlap between the genes that are differentially expressed at transcript/protein/phosphoprotein levels, taking into account all time points, was assessed. Interestingly, we observed that only a small number of genes changed at two or three molecular levels, thus revealing a discrete molecular-level footprint (Figure 1d). We also observed that differentially regulated proteins and phosphoproteins are significantly enriched for housekeeping genes (Supplementary Figure S1E).18 This is in line with the published observation that lipopolysaccharide (LPS)-induced changes in dendritic cells cause remodeling of housekeeping genes, primarily by protein translation or degradation.19
We performed enrichment analysis for biological processes using genes that were regulated at the transcriptome, proteome, and phosphoproteome levels, and identified 43 significantly enriched processes (P value⩽0.005; Figure 1e). We identified discrete cellular processes that were regulated at one or more molecular levels. For instance, we found genes associated with defense response regulated at the transcriptome level; lipid biosynthetic process at the proteome level; and mRNA processing at the phosphoproteome level (Figure 1f). We also performed k-means clustering of the transcript, protein, and phosphosite measurements, and were able to segregate the regulated genes into early, late, and mid-late responders to viral infection (Supplementary Figure S1F). The mid-to-late responders at the transcript level-cluster 1 (T-C1) included expected early antiviral genes such as type-I IFN and various ILs. In addition, T-C1 also contains unique genes such as Dusp8, a member of dual specificity phosphatases that regulate MAPK signaling,20 and MIR702, an anti-apoptotic mirtron that binds to 3’-untranslated repeat of ATF6, which is an endoplasmic reticulum stress-associated transcription factor.21,22 The late-responder cluster at the protein level, Pr-C2, further included known IFN-induced genes such as IFITs ISG15, MDA5 and RSAD2.2 In addition, GBP5, which is reported to promote NLRP3 inflammasome assembly upon bacterial infection,23 and CMPK2, a mitochondrial gene involved in the nucleotide salvage pathway,24 were also part of Pr-C2 cluster, hinting at possible involvement in antiviral responses. Phosphorylation sites in the early response cluster Ph-C2 included several innate immune-regulated genes with known sites, e.g., T7/S9 on GSK3B and several novel sites, e.g., S208 on OAS1A, S506 on IRAK2, and S3857 on UBR4. Further, we examined the overlap between phosphoproteome clusters to identify proteins with multiple differentially regulated phosphosites with varying kinetics. For instance, we found overlap between the phosphoproteome clusters Ph-C1 and Ph-C8, indicating a strong phosphorylation regulation (Supplementary Figure S1G). These included RB1, a master cell cycle regulator whose phosphorylation state determines its E2F-binding state;25 SPAG9, a scaffold protein that is required for JNK signaling;26 and ZC3HC1, a component of the SCF-type E3 ligase system involved in ubiquitination of cell cycle proteins.27
To address the specificity of VSV-induced transcriptome changes with other antiviral response, we compared our 6 h transcriptome data with the transcriptome of RAW264.7 cells infected with Murine norovirus (MNV1) for 9 h.28 There was a good correlation (Pearson correlation=0.53) between the differentially expressed VSV-induced transcriptome at 6 h with MNV1-induced transcriptome at 9 h. The transcript levels of type-I IFNs, IFN-stimulated genes (ISGs), and proinflammatory cytokines were upregulated in both VSV and MNV1 transcriptome (Supplementary Figure S1H). To elucidate the specificity of VSV-induced phosphoproteome change, we compared our data set with LPS-induced phosphoproteome changes in RAW264.7 cells.11 As these two data sets were generated in different technical settings, we used a robust Z score cutoff of ±2.5 for the comparison in order to create as little bias as possible. On the basis of this threshold, there were 425 and 469 differentially regulated phosphoproteins that are LPS- and VSV-induced, respectively (Supplementary Figure S1I). There was an overlap of 137 phosphoproteins common to LPS and VSV infection. Interestingly, only 82 phosphosites from these 137 proteins were common to both LPS and VSV (Supplementary Figure S1I). Overall, whereas there is a subset of phosphoproteome that is common to LPS and VSV, and transcriptome that is common to VSV and MNV1, a large portion of the changes are distinct depending on the stimulation, indicating the presence of yet unknown differential regulatory factors and events.
A top-down strategy to identify co-regulated gene modules reveals co-regulation of transmembrane transporters and SLC7A11 as a host factor in the VSV life cycle
Gene regulation during cellular perturbation events such as infection is critical for mounting an antimicrobial state while minimizing damage to the host. Systems-level approaches have aided the identification of some of these regulatory circuits associated with innate immune response.4,5,29 Using normalized RNA and protein fold changes of the differentially regulated genes (Supplementary Figures S1D and S2A), we constructed a gene–gene correlation matrix that allowed us to identify 12 co-regulated gene modules (Figure 2a and Supplementary Figure S2B). Gene ontology analysis showed several significantly enriched biological processes that were associated with each specific module, and some that were associated with multiple gene modules (Figure 2b). We performed enrichment analysis for transcription factor gene set based on target-binding sites using MSigDB30 and summarized the results as a network, where nodes comprises co-regulated modules and transcription factors; moreover, edges represent a regulatory relationship (Figure 2c). We found both common and unique transcription factors associated with the co-regulated modules. Several members of the E26 transcription factor family including ETS1, ZEB1, SPI1, TCF3, ETS2, and NFE2L1 were associated with one or more modules.

Gene co-regulation analysis. (a) Heatmap of gene–gene correlation matrix based on normalized RNA and protein fold changes of differentially regulated genes. The 12 identified co-regulated gene modules are highlighted on the diagonal of the matrix. (b) Gene ontology biological process enrichment of co-regulated gene modules. Significantly enriched processes are shown as colored circles and small black dots represent processes that were enriched, but not significant. (c) Network representation of transcription factor target enrichment data of co-regulated modules. Squares represent co-regulated modules and circles represent transcription factors. Size of the circles is proportional to the number of genes associated with a given module. Color of the edge denotes the level of significance for the enrichment. Transcription factors that are present more than once indicate different binding motifs. (d) Barplot showing the proteome fold change of the subset of genes significantly enriched for ‘transmembrane transport’ process, which is part of the co-regulated module M1. (e) Percent of green fluorescent protein (GFP)-positive HAP1 cells. CRISPR-Cas9-based knockout of SLC7A11 as compared with wild-type cells.
In addition, such a co-regulation network could also be used for tracking the temporal kinetics of specific family or group of genes triggered by specific perturbations such as ISGs. We investigated the kinetics of ISGs7 in the co-regulated gene modules and found four distinct groups based on changes in the transcript and protein levels after VSV infection (Supplementary Figure S2C), which could be the potential subsets of co-regulated antiviral effector genes. This network could serve as a point of departure to model host gene regulatory networks after viral infection. For instance, transcription factors ETS2 and FOXO4 that have a subset of common target genes in the co-regulated modules M1 and M7 are likely to share functional roles.
Gene ontology analysis for significantly enriched biological processes showed a group of transmembrane transporters as part of the co-regulated module M1 (Figure 2d). These included a number of genes from the understudied solute carrier family.31 One of the highly upregulated genes in this group was an amino-acid transporter SLC7A11 that transports cysteine/glutamate.32 SLC7A11 has also been described as a fusion-entry receptor of Kaposis’s sarcoma-associated herpes virus.33 More recently, SLC7A11 was reported to have a decisive role in reactive oxygen species-induced ferroptosis and p53-mediated tumor suppression.34 We generated a CRISPR-Cas9-based knockout of SLC7A11 in HAP1 cells35 that were subsequently infected with green fluorescent protein (GFP)-expressing VSV. Measurement of the GFP-positive cells after 6 h and multiplicity of infection (MOI) 10 showed a significant decrease in percent of GFP-positive cells in SLC7A11 knockout compared with the wild-type cells (Figure 2e), indicating that SLC7A11 does have a key role in the VSV infection life cycle.
Regulated novel kinases and substrates revealed by integrating publicly available kinase–substrate associations
To understand the regulated kinome landscape upon VSV infection, we integrated our OMICS data with publicly available kinase–substrate interactions36 and assembled a 638-interaction-containing kinase–substrate network (Figure 3a) that included 68 kinases, which were regulated at the transcript/protein/phosphoprotein level and 181 substrates, which were differentially phosphorylated (Supplementary Table S2). The regulated kinases were members of nine different kinase family subgroups (Supplementary Figure S3A and B). Comparing this list with a transcription factor database,37 we uncovered 13 transcription factors among the differentially phosphorylated substrates including Protein C-ets-2 (ETS2) at S294 and S297. To estimate the number of genes that are linked to innate immunity, we intersected this network with InnateDB.38 We found 29 kinases and 41 substrates annotated as innate immunity genes, thus highlighting the identified differentially regulated novel kinases and substrates having a role in phosphorylation signaling upon viral infection. Clustering the phosphorylated peptides, including the kinases and substrates in this network, into seven different groups (Figures 3a and b and Supplementary Figure S3C) allowed us to identify the potential key kinase–substrate associations that showed coherent phosphorylation kinetics (Figure 3c). This kinase–substrate network also included several differentially phosphorylated kinases (Supplementary Figure S3D). On the basis of the temporal phosphorylation pattern, JAK2 and WNK1 appear to be late-responding kinases after infection, whereas AKT2, SYK, PAK1, PAK2, ABL1, ABL2, and ARAF respond early. Within this kinase–substrate network, we observed a potential link between the early responding kinases PAK2/ABL1 and the transcription factor JUN (AP-1), which showed coherent differential phosphorylation (Figure 3d). We further investigated whether the phosphorylation kinetics of JUN correlated with the transcriptome changes of JUN target genes. To this end, we identified three clusters in the transcriptome (T-C1, T-C5, and T-C7; Supplementary Figure S1F) that showed similar temporal kinetics as compared with JUN phosphorylation. We performed motif analysis on these three clusters for enrichment of transcription factor-binding motifs using MSigDB and found statistically significant enrichment for JUN (P value<5.43E−15; Figure 3e). These temporal data along with the motif analysis provide a potential functional link between the kinases PAK2/ABL1 and activation of transcription factor JUN in antiviral response; moreover, the data set provides the feasibility to identify other potential regulatory mechanisms by integrating phosphorylation events of transcription factors with temporal transcriptome data.

Kinase–substrate interactions and validation of differentially regulated kinases ARAF and RAF1. (a) Kinase–substrate interaction network of differentially regulated kinases and substrates. Inner circle represents kinases with a differentially regulated phosphosites, middle circle represents kinases that are regulated at the RNA/protein level, and outer circle represents the substrates. Nodes are color-coded based on (b) temporal profiles of the regulated phosphosites. (c) Selected kinase–substrate subnetworks that show coherent phosphorylation changes overlaid with OMICS data. Heatmap strip on top, bottom-right, and bottom-left represent changes at transcriptome, proteome, and phosphoproteome, respectively. Data points with no available OMICS data are marked with an X. Phosphorylation sites are also marked. (d) Bar plots showing the log2 fold change of phosphorylation sites in ABL1, PAK2, and Jun. (e) Heatmap showing the transcriptome changes of JUN target genes. (f) Percent of green fluorescent protein (GFP)-positive HAP1 cells. CRISPR-Cas9-based knockout of AKT2 as compared with wild-type cells infected with vesicular stomatitis virus (VSV)-GFP at an multiplicity of infection (MOI) of 10 for 6 h. (g) Percent of GFP-positive HAP1 cells. Perifosine- or Ipatasertib-treated cells as compared with dimethylsulfoxide (DMSO)-treated cells infected with VSV-GFP at an MOI of 10 for 6 h. (h) Percent of GFP-positive HAP1 cells. CRISPR-Cas9-based knockout of ARAF or RAF1 as compared with wild-type cells expressing Renilla targeting sgRNA infected with VSV-GFP at an MOI of 2 for 4 h. (i) Western blot analysis for probing phospho-p65 from wild-type, ARAF knockout, and RAF1 knockout HAP1 cells unstimulated or stimulated with VSV at an MOI of 2 for 30 and 60 min.
We sought to validate the early phosphorylation role revealed above for AKT2, a key factor in PI3K/mTOR signaling,39 by infecting CRISPR-Cas9-based AKT2 knockout HAP1 cells with VSV-GFP. The AKT2 knockout cells showed a significant reduction in GFP-positive cells after 6 h of infection with MOI10, thereby hinting that AKT2 kinase may have a role in efficient VSV infection or replication (Figure 3f). HAP1 cells treated with the Pan-AKT inhibitors Ipatasertib and MK-2206, but not the AKT1-specific inhibitor, Perifosine, had a reduced infection after 6 h at an MOI of 10 when compared with dimethylsulfoxide-treated cells (Figure 3g and Supplementary Figure S3E). The inhibitor phenotypes support what the omics data integration indicates: AKT2, but not AKT1, was revealed to be differentially regulated by phosphorylation during virus infection.
Among the various differentially phosphorylated kinases upon VSV infection were two members of the RAF family of kinases, ARAF (A-Raf) and RAF1 (c-Raf), which were phosphorylated at S157 and S29, respectively. To investigate the roles of these kinases in regulating virus replication and immune response, we again made CRISPR-Cas9-based ARAF or RAF1 knockout in HAP1. RAF1 and, to a lesser extent, ARAF, has been linked to innate immune activity downstream from virus recognition,40,41 and, in fact, after 4 h of infection with VSV-GFP at MOI2, the RAF1 KO cells, but not the ARAF KO, had significantly more infection compared with the WT (Figure 3h). A reduced viral MOI and infection time was used here compared with other experiments in order to limit virus turnover duration as the knockout cell lines were more susceptible VSV-mediated lysis. The increased infection phenotype was mirrored by a significant decrease in IL-6 transcription in the RAF1 KO line at the same time point (Supplementary Figure S3F). Further, there was a decrease of P65 phosphorylation in both the RAF1 and the ARAF knockout lines compared with WT at 30 min, whereas only the RAF1 KO had a pronounced and consistent loss of the phosphorylation at a 60 min post infection (Figure 3i), whereas p65 transcription levels are unchanged (Supplementary Figure S3G). Transcriptional activation of IL-6 after phosphorylation of P65 is a known and well-studied pathway post-NFκB activation.42–44 Together, this indicates that RAF1 and, to a less extent, ARAF are potentially activated during early VSV infection, and may mediate a controlling immune response activated by VSV—as elucidated by mining the kinome landscape (Figure 3a).
Integrated analysis to understand the dynamics of perturbed signaling pathways
To identify the signaling pathways that are perturbed upon VSV infection, we performed gene-set enrichment for signaling pathways at transcriptome, proteome, and phosphoproteome levels. This enrichment, for instance, revealed that the RIG-I signaling pathway was perturbed at all three molecular levels: p53 signaling and cytosolic DNA-sensing pathway at transcriptome and proteome levels, and the MAPK signaling pathway at transcriptome and phosphoproteome levels. In addition, we found that pathways such as JAK-STAT signaling and NOD-like receptor signaling were enriched at the transcriptome level, whereas focal adhesion and tight junction signaling was enriched at the phosphoproteome level (Supplementary Figure S4A). This substantiated the possibility that the cellular repertoire of possible responses to viral perturbation is varied and that, although some part of the program required transcriptional activation followed by protein changes, others are purely dependent on phosphorylation within this time frame. To obtain a more quantitative measure of the perturbation, we carried out Signaling Pathway Impact Analysis45 using normalized fold changes of transcriptome, proteome, and phosphoproteome data (Supplementary Figure S1D). Signaling Pathway Impact Analysis analysis on 148 KEGG signaling pathways, taking into account the directionality and type of the interactions, showed enrichment for several pathways as seen in the classical enrichment method. In addition, we were able to derive the perturbation state of these pathways at each molecular level over time (Figure 4a and Supplementary Table S3). For instance, tracking RIG-I signaling showed a time-dependent activation, whereas signaling during Influenza A virus infection has a sustained activation soon after infection.

Pathway perturbation analysis and dynamics of RIG-I signaling pathway. (a) Scatter plot summarizing the results of the signaling pathway impact analysis of differentially regulated transcriptome, proteome, and phosphoproteome at 20 min, 3 h, and 6 h after vesicular stomatitis virus (VSV) infection, and heatmap representing the total perturbation values for signaling pathways with false discovery rate (FDR)⩽0.05 at the transcriptome/proteome/phosphoproteome level. (b) The RIG-I signaling pathway (adapted from KEGG database) overlaid with the OMICS data. Genes are marked in yellow filled rectangles. Heatmap strip on top, bottom-right, and bottom-left represent changes at transcriptome, proteome, and phosphoproteome, respectively. Data points with no available OMICS data are marked with an X. Phosphorylation sites are also marked.
The RIG-I signaling pathway is important for sensing several viruses including VSV, Influenza virus, Sendai virus, and Hepatitis C virus,46 and initiating strong IFN response. The data resolution from the OMICS profiling was able to capture more than 95% of the genes at one or more molecular level. Thus, we created a temporal map describing the transcriptome, proteome, and phosphoproteome of RIG-I signaling upon VSV infection (Figure 4b), including novel phosphorylation sites on the key molecules involved in this signaling pathway. DHX58 (also called LGP2), a member of the RIG-like receptor family of proteins that has a virus-specific role in mediating antiviral response,47–50 was lacking upon infection phosphorylation at S477 located in the repressor domain. We also uncovered two phosphorylation sites S645 and S648, also not previously reported, that are upregulated on another member of the RIG-like receptor family, IFIH1/MDA5. MAVS, a key adaptor protein in RIG-I signaling, and downstream IFN activation, was found to be hyperphosphorylated upon infection at S328 in our study. In addition, TBK1, a key regulator of IRF3 signaling,51,52 was hyperphosphorylated at S716 and IRF3 at S388. Altogether, the signaling pathway impact analysis indicates that individual pathways have strong time- and molecule-level-dependent regulation; moreover, the resolution of the data sets allows temporal tracking of the key signaling pathways.
MAVS S328-S330 is required for IL-6 and IFN response
Our phosphoproteomics screen revealed that a number of novel phosphosites of the key innate immune signaling proteins and clustering of these phosphosites allowed us to interrogate the subsets with different kinetics. Among the various phosphosites that followed the same temporal kinetics as IRF3 was S328 on MAVS, which was hyperphosphorylated at 3 and 6 h after infection without considerable change at the RNA or protein level (Figure 5a,b). Multiple sequence alignment of the MAVS proteins from mouse, human, and rat revealed that the amino acid S328 is specific to mouse and is replaced by Isoleucine in human and rat, whereas adjacent S330 was conserved in all these species (Supplementary Figure S5A). The region that we identified is different from a recent study53 that described the function of a novel phosphorylation site on human MAVS. To test the role of this phosphorylation cluster S328-S330 in antiviral signaling, we reconstituted MAVS−/− immortalized primary mouse macrophages with MAVS WT, MAVS S328A, or MAVS S328A/S330A (Supplementary Figure S5B). First, we infected the reconstituted cells with VSV engineered to express GFP under its major promoter.54 Flow cytometry quantification of the number of GFP-positive cells revealed the double-mutant S328A/S330A to have a significant increase in viral gene production while S328A had only a marginal increase (Figure 5c).

Functional validation of MAVS phosphorylation S328/S330. (a) A representative mass spectrometry (MS)/MS spectrum supporting the phosphorylation of Mavs at S328 with annotated fragment ions and intensity of iTRAQ labels. (b) Barplot showing the RNA, protein, and S328 phosphorylation changes of MAVS upon vesicular stomatitis virus (VSV) infection. (c–e) MAVS−/− immortalized mouse macrophages stably expressing wild-type or MAVS phosphosite mutants tested for (c) viral replication based on percent of VSV-green fluorescent protein (GFP)-positive cells after 16 h of infection. (d) Poly(I:C)-induced RNA levels of IFNβ, IFIT1, and interleukin (IL)-6. (e) interferon beta (IFNβ) protein expression by ELISA after 6 h Poly(I:C) stimulation. (f) Bar plots showing log2 fold changes of interferon-stimulated genes from RNA sequencing (RNA-Seq) analysis of MAVS−/− immortalized mouse macrophages stably expressing wild-type or MAVS phosphosite mutants stimulated with Poly(I:C) for 3 or 9 h as compared with unstimulated condition.
To further define the roles of the phosphosites in NFκB and IFNβ activation downstream of MAVS activation, we analyzed the mRNA expression levels of IFNβ, IL-6, and IFIT1 upon Poly(I:C) stimulation. mRNA expression of these genes was significantly reduced in both S328A and S328A/S330A mutants compared with cells reconstituted with MAVS WT (Figure 5d). Poly(I:C)-induced IFNβ protein secretion was also significantly reduced (Figure 5e). Upon VSV infection, the double mutant showed a significant decrease in IL-6 and IFNβ transcript levels, whereas the single mutant showed a significant decrease in IL-6 but not IFNβ transcript levels (Supplementary Figure S5C,D). In addition, both mutants showed a decrease in IRF3 phosphorylation, with the double mutant having a profound effect (Supplementary Figure S5E). We further investigated whether the MAVS phosphorylation had any impact on other activation of other antiviral genes. To this end, we performed RNA-Seq-based global transcriptome analysis upon stimulation with Poly(I:C) for 3 and 9 h. We found that ISGs were part of the differentially regulated transcripts upon Poly(I:C) stimulation. Detailed analysis of this subset showed that the mRNA levels of several ISGs such as IFIT1, IFIT2, IFIT3, MX1, ISG15, CXCL10, and ISG20 were significantly reduced in both the S328A and S328A/S330A mutants (Figure 5f). This shows that the MAVS phosphorylation is critical for triggering the transcriptional rewiring during innate immune response. The phospho-mutant MAVS-expressing cells had no significant difference in induction of IL-6 mRNA expression upon LPS stimulation (Supplementary Figure S5F). Confocal microscopy revealed that the MAVS constructs were properly localized to mitochondria in our reconstituted cells (Supplementary Figure S5G). The identification of this specific MAVS phosphorylation and subsequent validation to link its functional effect emphasizes the importance of using orthogonal technologies to profile the perturbation state of a cell to understand cellular response, in addition to validating the quality of the data made available through this study.
Discussion
How much of the response of mammalian cells to viral challenge is dedicated to recognition of the intrusion, measurement of the severity of the challenge, and mounting an adequate response? Are there processes that need to occur before others? Are there ‘housekeeping’ processes that would be triggered also by non-microbial perturbations or are most mechanisms evolved for safe-guarding homeostasis to withstand microbes? We joined the effort of several laboratories focused on gaining fundamental insights to answer questions such as these by attempting a global, unbiased survey of the events in macrophages triggered by VSV infection.55–57 The analysis and validation that ensued aimed at deconvoluting the response to discern distinct functional relationships and a possible unifying logic. This study describes discrete patterns of events and reports on observations that allow to formulate distinct, testable, hypotheses. Using an integrated transcriptome, proteome, and phosphoproteome profiling, we have created a quantitative infectome map describing the gene expression dynamics of VSV-infected macrophages. This has led to identification of several known and novel differentially regulated genes that have a potential role in antiviral innate immune response. These data indicate that the temporal dynamics of the host response is distinct, depending on the molecular level, likely reflecting protein translation/degradation rates but also suggestive of specialized regulatory options, which may each be best achieved at the level of RNA, protein, or PTM.19,58–61 Enrichment for so-called housekeeping genes at proteome and phosphoproteome levels may be indicative of the careful orchestration of host’s cellular response to perturbation scenarios such as viral infection, and a recent study19 validated this notion that housekeeping genes are regulated at the proteome level in a LPS-induced dendritic cell response. This study extends this observation to a viral response and also shows a similar response for PTMs such as phosphorylation. The observation that a large number of genes are exclusively regulated by phosphorylation of their products underscores the substantial role of PTM in innate immune response62 likely evolved because of the requirement of a fast response to pathogens. Analysis of other PTMs amenable to high-throughput identifications, such as acetylation and glycosylation,63 should shed light on their role in innate immune response. The phenotypic effect of MAVS phosphorylation, which we unraveled in this study, emphasizes on the advantages on using orthogonal measurements to study perturbations.
A data set as large as the one created here requires efficient bioinformatics methods for integration and to allow to detect general principles or formulate precise hypotheses. There is a great variety of possibilities to analyze the data, and in future it should be possible to unearth may other properties that are not touched upon here. The analysis methods that we used in this study, such as clustering of genes based on temporal kinetics, quantitative pathway impact analysis, kinase–substrate interaction networks, and co-regulation networks, provide different perspective depending on the biological question and plane of intellectual focus that is being sought. Whereas the analysis presented identifies biological objects that have expression behaviors that are similar to or undistinguishable from elements known to have important roles in innate immune signaling; the data offer the basis for analyses at the systems-level perspective of regulation of antiviral signaling and response.
A general question that arises during genome-wide expression profiling of host responses at the cellular level is how specific are these changes with respect to the type of perturbation. It is known that there is a transcriptional signature for various innate immune stimuli,4 but so far no recognizable patterns have been proposed for changes at the PTM level such as phosphorylation. We tried to address this by comparing VSV-induced phosphoproteome changes with LPS-induced changes. What emerges from this comparison is that, whereas there is small part that is common to both, a larger portion of the changes is very stimulus-specific, allowing the speculation that the various biochemical effects offered by phosphorylation (modulation of protein interfaces of the molecular machinery, intrasteric and allosteric regulation of enzymes) may provide the level of fine-tuning necessary to focus on specific challenges and responses.58,64 In comparison, at the protein level, one can detect a large overlap of commonly regulated proteins but a plethora of phosphosites that are ligand-specific. This shows that phosphoproteome changes will have common and ligand-specific signatures. It will certainly be interesting, in the future, to systematically assess the PTM dynamics of various innate immune stimuli.
Regulation of gene expression occurs at multiple levels such as chromatin remodeling,65 transcription,66 post-transcriptional modifications,67 translation,68 and PTMs.69 This study validates this notion that cells have multiple regulatory options under perturbation conditions and is important to profile multiple molecular layers to appreciate the cellular response in full. Without doubt, adding measurements of changes in the metabolism of infected cells should further allow to deconvolute the response.70,71 There are a number of genes with a potential role in antiviral innate immune response identified in this study, and they could serve as a reference point of future studies, as VSV is a commonly used model virus. Two independent studies have showed that phosphorylation status of ASC at Y146 (ref. 72) and NLRC4 at S533 (ref. 73) is essential for its inflammasome activation.74 Along these lines, the validation of the phosphosites on MAVS in this study is a clear demonstration of the significance of phosphorylation in regulating innate immune responses. In general, we envisage that the data set provided by this study together with its analytical strategy will prove to be useful to the community of scientists dedicated to the elucidation of the cellular processes involved in recognizing and responding to viral infections. The analysis strategies used in our study could be applied to characterize any immunological perturbation scenarios to get a systems-level overview. Overall, these unique data sets from this study could serve as a useful resource for generating interesting hypotheses.
Materials and methods
Cells and viruses
RAW264.7 cells (from American Type Cell Culture), infected with VSV-M2 at an MOI of 5 were used for profiling transcriptome, proteome, and phosphoproteome. Cells were grown in DMEM (Sigma, St Louis, MO, USA) containing 10% fetal bovine serum (Gibco, Carlsbad, CA, USA), 100 U/ml Penicillin (Sigma), and 100 μg/ml Streptomycin (Sigma) at 37 °C and 5% CO2. HAP1 cells were cultured in IMDM (GIBCO) containing serum and antibiotics as described above. Primary immortalized Mavs−/− mouse macrophages were a gift from the Professor Katherine A Fitzgerald lab (UMass Medical School, Worcester, USA). Validation experiments were also carried out in HAP1 cells, a near-haploid human cell line derived from chronic myeloid leukemia cell line KBM-7, containing single copy of all chromosomes except a 30MB chromosome 15 fragment. The cells were checked for their haploid status before the experiments.
Phosphopeptide enrichment and offline fractionation
Uninfected and VSV-infected RAW264.7 cells at ~70% confluency were washed in cold phosphate-buffered saline (Sigma), cells were scraped, centrifuged, and the cell pellets were snap-frozen in liquid nitrogen. Frozen cell pellets were lysed in 8 M urea lysis buffer containing phosphatase and kinase inhibitors (Roche Diagnostics GmbH, Mannheim, Germany). Total protein concentration was normalized across the samples and a total of 7.7 mg protein per condition was used for tryptic digestion and phosphotyrosine enrichment. Peptide digest (100 μg) was used for whole-cell proteome and global phosphopeptide enrichment. iTRAQ 4-plex labeling was performed according to the instructions of the manufacturer. The whole-cell proteome was separated into 50 fractions and the global phosphopeptide-enriched samples were separated into 10 fractions.
Online LC-MS and data processing
MS was performed on a hybrid LTQ-Orbitrap Velos mass spectrometer (ThermoFisher Scientific, Waltham, MA, USA) using the Xcalibur version 2.1.0.1140 coupled to an Agilent 1200 HPLC nanoflow system via a nanoelectrospray ion source using liquid junction (Proxeon, Odense, Denmark). The analyses of the phosphopeptides were performed in a data-dependent acquisition mode using a top 10 hybrid method with a multistage-activation collision-induced dissociation MS2 event, followed by a higher-energy dissociation MS2 event for each selected precursor ion. The analysis of the normal proteome was performed with a top 10 higher-energy dissociation method.
Peak list data were extracted using the Proteowizard software (http://proteowizard.sourceforge.net/-release 3.0.3201) and searched against the SwissProt mouse database (release 2012.05) appended with common contaminants, using Mascot (version 2.3.02; MatrixScience, London, UK) and Phenyx (version 2.5.14; GeneBio, Geneva, Switzerland). Fixed modifications included cysteine carbamidomethylation and iTRAQ labels on lysine and N terminus. Variable modifications included methionine oxidation and phosphorylation of serine, threonine, and tyrosine. One tryptic missed cleavage was allowed. A two-pass search strategy was used to apply a final false discovery rate of 1% at the protein level and 0.1% at the peptide level as detailed in Bennett et al.75 Protein-level quantitation for total cellular proteome and peptide-level quantification for phosphoproteome data were performed using isobar R package,76 and robust Z score were calculated. Protein (for total cellular proteome) or peptide (for phosphoproteome) with an absolute Z score⩾2.5 and P value⩽0.05 were considered differentially expressed.
RNA-Seq
Total RNA was extracted using TRIzol reagent (Life Technologies) following the manufacturer's protocol from 1×107 cells. Quality of RNA was measured using BioRad’s Experion RNA Analysis chip, and samples with a RNA integrity number greater than 7 were used for whole-transcriptome analysis. RNA (2 μg) was used for complementary DNA library synthesis using the TruSeq RNA Sample Prep kit v2 (Illumina, San Diego, CA, USA) according to the manufacturer's recommendations. RNA-Seq libraries were sequenced on an Illumina HiSeq 2000 machine with a 50 bp single-end protocol. The sequence reads were aligned to mouse reference genome build mm9 using TopHat2 (ref. 77) and Bowtie2.78 RPKM (reads per kilobase million) and FPKM (fragments per kilobase million) values were computed using RSeQC package79 and Cufflinks,80 respectively. Fold changes were calculated using DEGseq R package81 applying the MARS (MA-plot-based method with Random Sampling model) algorithm. Robust Z scores were also computed based on the fold change. Transcripts with an absolute Z score⩾2.5 and false discovery rate⩽0.001 were considered differentially expressed.
Bioinformatics analyses
Gene ontology biological process and KEGG pathway enrichment analysis were carried out using DAVID web service.82 Signaling pathway impact analysis was analyzed using Signaling Pathway Impact Analysis package implemented in R statistical environment (http://www.r-project.org/). The list of kinases was downloaded from UniProt database (http://www.uniprot.org/docs/pkinfam). Multiple sequence alignment was carried out using ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/). Networks were visualized using Cytoscape (http://www.cytoscape.org/). Pearson’s correlation method was used for constructing the gene–gene correlation matrix. Optimal numbers of clusters were calculated using the statistical method clusGap, available under the cluster package in R. Transcription factor target site enrichment was performed using the GSEA software from MSigDB (version 5.0; http://www.broadinstitute.org/gsea/msigdb) and top 10 gene sets with false discovery rate q-value⩽0.05 were considered significant. All statistical analyses, data processing, and visualization were performed in the R statistical environment.
Rank-based normalization
Rank-based normalization of ratio distribution was used to map proteomics and phosphoproteomics ratios to the range of observed mRNA ratios and make them comparable. mRNA ratios mr1, …, mrN were sorted from largest negative to largest positive. The same sorting operation was carried out with protein abundance ratios pr1, …, prM. Then, the proteins were assigned ratios from the mRNA ratio distribution according to the formula pri → mrtrunc(i/M×N), trunc(x) being the function returning the largest integer smaller than or equal to x. The same transformation was imposed to phosphoproteomics ratios ppr1, …, pprK: ppri → mrtrunc(i/K×N).
CRISPR-Cas9-mediated generation of HAP1 knockout cells
SLC7A11: pX459 plasmid83 (gift from Feng Zhang lab—Addgene, Cambridge, MA, USA, #48139) was ligated with guide sequence inserts 5ʹ- CACCGCTCCAAAGGAGGTTACCTGC-3ʹ and 5ʹ-AAACGCAGGTAACCTCCTTTGGAGC-3ʹ. HAP1 cells were transiently transfected with for 24 h, followed by selection with Puromycin (5 μg/ml). Single-cell clone was generated by limiting dilution. ARAF and RAF1: lentiCRISPRv2 plasmid84 (gift from Feng Zhang lab—Addgene #52961) was ligated with 5ʹ- CACCGCCGTGCGTTGCTTGTTGGGC-3ʹ and 5ʹ-AAACGCCCAACAAGCAACGCACGGC-3ʹ in case of ARAF; 5ʹ-CACCGAGCAGTTTGGCTATCAGCGC-3ʹ and 5ʹ-AAACGCGCTGATAGCCAAACTGCTC-3ʹ in case of RAF1. Lentiviruses were produced using second-generation packaging plasmids pMD2.G and psPAX2. HEK293T cells were co-transfected with packaging and lentiCRISPRv2 plasmids. Cells were washed 16 h after transfection. Virus-containing supernatants were collected 24 h after washing, filtered, and used for infection of HAP1 cells. Twenty-four hours later, a second round of infection was performed. Forty-eight hours after the last infection, HAP1 cells were selected with Puromycin (5 μg/ml). HAP1 cells deficient in ARAF, RAF1, and AKT2 were also obtained from Haplogen GmbH (Vienna, Austria) produced with CRISPR technology.
Cell stimulations and infections
Wild-type or mutant mouse Mavs were constitutively expressed under a retrovirus promoter in immortalized bone marrow-derived mouse macrophages lacking the Mavs gene (immortal cells were a gift from K Fitzgerald, UMass Medical School). Macrophages or HAP1 cells were stimulated or infected with Poly(I:C) (Sigma) transfected with Lipofectamine RNAiMAX (Life Technologies) or with VSV with or without GFP expression, or left naive, at indicated time points. MK-2206, Ipatasertib, and Perifosine (Selleckchem, Houston, TX, USA) were incubated for 1 h prior to virus infection. Total RNA was isolated from cell lysates with RNeasy Mini Kit (Qiagen, Hilden, Germany) and complementary DNA produced with oligo (DT) primers using RevertAid (Fermentas, Boston, MA, USA) following the manufacturer's directions. The following primers were used to determine the transcript level of genes in the mouse macrophages: mIFNβ (5′- TCAGAATGAGTGGTGGTTGC, 3′- GACCTTTCAAATGCAGTAGATTCA), mIFIT1 (5′- CATGGGAGAGAATGCTGATG, 3′- GTCAAGGAACTGGACCTGCT), mIL-6 (5ʹ- GTAGCTATGGTACTCCAGAAGAC, 3′- ACGATGATGCACTTGCAGAA), mMAVS (5′- CGCCCACGCAGCAAAT, 3′- GTTGGAGACACAGGTCCATAGGT), and CycB (5′- CAGCAAGTTCCATCGTGTCATCAAGG, 3′- GGAAGCGCTCACCATAGATGCTC), and in the HAP1: hIL6 (5′- TGGACTGCAGGAACTCCTTA, 3′- ACCTAGAGTACCTCCAGAAC), hRELA (5′ -AGCACCATCAACTATGATGAGTTTC, 3′ -GAGTTATAGCCTCAGGGTACTCCAT), and hGAPDH (5′ -GAAGGTGAAGGTCGGAGT, 3′- GAAGATGGTGATGGGATTTC) was determined with SensiMix SYBR Hi-ROX (Bioline, Taunton, MA, USA) on a RotorGene RG-600 (Qiagen). Mouse MAVS (M300, Santa Cruz, Dallas, TX, USA) or IκBα (R&D, Minneapolis, MN, USA) protein levels were determined by western blot analysis with rabbit anti with donkey anti-rabbit IgG (H+L) 680 (Licore, Lincoln, NE, USA) secondary, and imaged with a LI-COR imager. ELISA for mIL-6 (Ready-SET-Go concentration levels GFP expression from virus infection was determined with flow cytometry (BD LSRFortessa with the Diva software (San Jose, CA, USA) and analyzed with FlowJo (Ashland, OR, USA)) after fixation.
Confocal microscopy
Immortalized bone marrow-derived mouse MAVS−/− macrophages reconstituted with hemagglutinin (HA)-tagged MAVS WT or mutants were seeded on glass coverslips overnight. Before fixing, cells were stained with Mitotracker Orange (invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocols. Cells were fixed in 2% formaldehyde with 0.1% triton-X114. Proteins of interest were visualized with the following: anti-HA (Santa Cruz Biotechnology, Y-11)/anti-rabbit AlexaFlour 633 (invitrogen). Slides were visualized using an LSM780 (Carl Zeiss, Oberkochen, Germany) with a GaAsP multidetector unit and two PMTs. Pinhole was set to 1 AU on each channel. Images were taken sequentially. Images were taken at ×100 and analyzed with ImageJ (NIH, open source).
References
Kumar, H., Kawai, T. & Akira, S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 30, 16–34 (2011).
Brubaker, S. W., Bonham, K. S., Zanoni, I. & Kagan, J. C. Innate immune pattern recognition: a cell biological perspective. Annu. Rev. Immunol. 33, 257–290 (2015).
Rebsamen, M., Kandasamy, R. K. & Superti-Furga, G. Protein interaction networks in innate immunity. Trends Immunol. 34, 610–619 (2013).
Amit, I. et al. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science 326, 257–263 (2009).
Litvak, V. et al. A FOXO3-IRF7 gene regulatory circuit limits inflammatory sequelae of antiviral responses. Nature 490, 421–425 (2012).
Shapira, S. D. et al. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell 139, 1255–1267 (2009).
Schoggins, J. W. et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485 (2011).
Sun, L., Wu, J., Du, F., Chen, X. & Chen, Z. J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013).
Pichlmair, A. et al. IFIT1 is an antiviral protein that recognizes 5'-triphosphate RNA. Nat. Immunol. 12, 624–630 (2011).
Meissner, F., Scheltema, R. A., Mollenkopf, H. J. & Mann, M. Direct proteomic quantification of the secretome of activated immune cells. Science 340, 475–478 (2013).
Weintz, G. et al. The phosphoproteome of toll-like receptor-activated macrophages. Mol. Syst. Biol. 6, 371 (2010).
Weekes, M. P. et al. Quantitative temporal viromics: an approach to investigate host-pathogen interaction. Cell 157, 1460–1472 (2014).
Pichlmair, A. et al. Viral immune modulators perturb the human molecular network by common and unique strategies. Nature 487, 486–490 (2012).
Rozenblatt-Rosen, O. et al. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 487, 491–495 (2012).
Jager, S. et al. Global landscape of HIV-human protein complexes. Nature 481, 365–370 (2012).
Zak, D. E., Tam, V. C. & Aderem, A. Systems-level analysis of innate immunity. Annu. Rev. Immunol. 32, 547–577 (2014).
Stojdl, D. F. et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 4, 263–275 (2003).
Eisenberg, E. & Levanon, E. Y. Human housekeeping genes, revisited. Trends Genet. 29, 569–574 (2013).
Jovanovic, M. et al. Immunogenetics. Dynamic profiling of the protein life cycle in response to pathogens. Science 347, 1259038 (2015).
Martell, K. J., Seasholtz, A. F., Kwak, S. P., Clemens, K. K. & Dixon, J. E. hVH-5: a protein tyrosine phosphatase abundant in brain that inactivates mitogen-activated protein kinase. J. Neurochem. 65, 1823–1833 (1995).
Li, M. et al. ATF6 as a transcription activator of the endoplasmic reticulum stress element: thapsigargin stress-induced changes and synergistic interactions with NF-Y and YY1. Mol. Cell Biol. 20, 5096–5106 (2000).
Zhang, W. G. et al. Mmu-miR-702 functions as an anti-apoptotic mirtron by mediating ATF6 inhibition in mice. Gene 531, 235–242 (2013).
Shenoy, A. R. et al. GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science 336, 481–485 (2012).
Xu, Y., Johansson, M. & Karlsson, A. Human UMP-CMP kinase 2, a novel nucleoside monophosphate kinase localized in mitochondria. J. Biol. Chem. 283, 1563–1571 (2008).
Knudsen, E. S. & Knudsen, K. E. Tailoring to RB: tumour suppressor status and therapeutic response. Nat. Rev. Cancer 8, 714–724 (2008).
Kelkar, N., Standen, C. L. & Davis, R. J. Role of the JIP4 scaffold protein in the regulation of mitogen-activated protein kinase signaling pathways. Mol. Cell Biol. 25, 2733–2743 (2005).
Bassermann, F. et al. NIPA defines an SCF-type mammalian E3 ligase that regulates mitotic entry. Cell 122, 45–57 (2005).
Waugh, E., Chen, A., Baird, M. A., Brown, C. M. & Ward, V. K. Characterization of the chemokine response of RAW264.7 cells to infection by murine norovirus. Virus Res. 181, 27–34 (2014).
Gilchrist, M. et al. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature 441, 173–178 (2006).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 (2005).
Cesar-Razquin, A. et al. A call for systematic research on solute carriers. Cell 162, 478–487 (2015).
Gasol, E., Jimenez-Vidal, M., Chillaron, J., Zorzano, A. & Palacin, M. Membrane topology of system xc- light subunit reveals a re-entrant loop with substrate-restricted accessibility. J. Biol. Chem. 279, 31228–31236 (2004).
Kaleeba, J. A. & Berger, E. A. Kaposi's sarcoma-associated herpesvirus fusion-entry receptor: cystine transporter xCT. Science 311, 1921–1924 (2006).
Jiang, L. et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62 (2015).
Carette, J. E. et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477, 340–343 (2011).
Colinge, J. et al. Building and exploring an integrated human kinase network: global organization and medical entry points. J. Proteomics 107, 113–127 (2014).
Wilson, D., Charoensawan, V., Kummerfeld, S. K. & Teichmann, S. A. DBD--taxonomically broad transcription factor predictions: new content and functionality. Nucleic Acids Res. 36, D88–D92 (2008).
Breuer, K. et al. InnateDB: systems biology of innate immunity and beyond--recent updates and continuing curation. Nucleic Acids Res. 41, D1228–D1233 (2013).
Laplante, M. & Sabatini, D. M. mTOR signaling at a glance. J. Cell Sci. 122, 3589–3594 (2009).
Gringhuis, S. I. et al. Dectin-1 directs T helper cell differentiation by controlling noncanonical NF-kappaB activation through Raf-1 and Syk. Nat. Immunol. 10, 203–213 (2009).
Popik, W. & Pitha, P. M. Binding of human immunodeficiency virus type 1 to CD4 induces association of Lck and Raf-1 and activates Raf-1 by a Ras-independent pathway. Mol. Cell Biol. 16, 6532–6541 (1996).
Chen, L. F. & Greene, W. C. Shaping the nuclear action of NF-kappaB. Nat. Rev. Mol. Cell Biol. 5, 392–401 (2004).
Ghosh, S. & Hayden, M. S. New regulators of NF-kappaB in inflammation. Nat. Rev. Immunol. 8, 837–848 (2008).
Medzhitov, R. & Horng, T. Transcriptional control of the inflammatory response. Nat. Rev. Immunol. 9, 692–703 (2009).
Tarca, A. L. et al. A novel signaling pathway impact analysis. Bioinformatics 25, 75–82 (2009).
Bowie, A. G. & Unterholzner, L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 8, 911–922 (2008).
Bruns, A. M., Leser, G. P., Lamb, R. A. & Horvath, C. M. The innate immune sensor LGP2 activates antiviral signaling by regulating MDA5-RNA interaction and filament assembly. Mol. Cell 55, 771–781 (2014).
Saito, T. et al. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc. Natl. Acad. Sci. USA 104, 582–587 (2007).
Satoh, T. et al. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc. Natl. Acad. Sci. USA 107, 1512–1517 (2010).
Venkataraman, T. et al. Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J. Immunol. 178, 6444–6455 (2007).
Fitzgerald, K. A. et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4, 491–496 (2003).
Goncalves, A. et al. Functional dissection of the TBK1 molecular network. PLoS One 6, e23971 (2011).
Liu, S. et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630 (2015).
Boritz, E., Gerlach, J., Johnson, J. E. & Rose, J. K. Replication-competent rhabdoviruses with human immunodeficiency virus type 1 coats and green fluorescent protein: entry by a pH-independent pathway. J. Virol. 73, 6937–6945 (1999).
Lee, M. N. et al. Identification of regulators of the innate immune response to cytosolic DNA and retroviral infection by an integrative approach. Nat. Immunol. 14, 179–185 (2013).
Schoggins, J. W. et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505, 691–695 (2014).
Yoh, S. M. et al. PQBP1 Is a proximal sensor of the cGAS-dependent innate response to HIV-1. Cell 161, 1293–1305 (2015).
Cohen, P. The regulation of protein function by multisite phosphorylation--a 25 year update. Trends Biochem. Sci. 25, 596–601 (2000).
Mann, M. et al. Analysis of protein phosphorylation using mass spectrometry: deciphering the phosphoproteome. Trends Biotechnol. 20, 261–268 (2002).
Papin, J. A., Hunter, T., Palsson, B. O. & Subramaniam, S. Reconstruction of cellular signalling networks and analysis of their properties. Nat. Rev. Mol. Cell Biol. 6, 99–111 (2005).
Rabani, M. et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat. Biotechnol. 29, 436–442 (2011).
Seth, R. B., Sun, L. & Chen, Z. J. Antiviral innate immunity pathways. Cell Res. 16, 141–147 (2006).
Olsen, J. V. & Mann, M. Status of large-scale analysis of post-translational modifications by mass spectrometry. Mol. Cell Proteomics 12, 3444–3452 (2013).
Creixell, P. & Linding, R. Cells, shared memory and breaking the PTM code. Mol. Syst. Biol. 8, 598 (2012).
Wyrick, J. J. et al. Chromosomal landscape of nucleosome-dependent gene expression and silencing in yeast. Nature 402, 418–421 (1999).
Wang, Z. et al. Evolution of gene regulation during transcription and translation. Genome Biol. Evol. 7, 1155–1167 (2015).
Filipowicz, W., Bhattacharyya, S. N. & Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat. Rev. Genet. 9, 102–114 (2008).
Ingolia, N. T., Lareau, L. F. & Weissman, J. S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147, 789–802 (2011).
Mann, M. & Jensen, O. N. Proteomic analysis of post-translational modifications. Nat. Biotechnol. 21, 255–261 (2003).
Koberlin, M. S. et al. A conserved circular network of coregulated lipids modulates innate immune responses. Cell 162, 170–183 (2015).
Pearce, E. L. & Pearce, E. J. Metabolic pathways in immune cell activation and quiescence. Immunity 38, 633–643 (2013).
Hara, H. et al. Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat. Immunol. 14, 1247–1255 (2013).
Qu, Y. et al. Phosphorylation of NLRC4 is critical for inflammasome activation. Nature 490, 539–542 (2012).
Martinon, F., Burns, K. & Tschopp, J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426 (2002).
Bennett, K. L. et al. Proteomic analysis of human cataract aqueous humour: comparison of one-dimensional gel LCMS with two-dimensional LCMS of unlabelled and iTRAQ(R)-labelled specimens. J. Proteomics 74, 151–166 (2011).
Breitwieser, F. P. et al. General statistical modeling of data from protein relative expression isobaric tags. J. Proteome Res. 10, 2758–2766 (2011).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Wang, L., Wang, S. & Li, W. RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185 (2012).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Wang, L., Feng, Z., Wang, X. & Zhang, X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138 (2010).
Jiao, X. et al. DAVID-WS: a stateful web service to facilitate gene/protein list analysis. Bioinformatics 28, 1805–1806 (2012).
Ran, F. A. et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389 (2013).
Sanjana, N. E., Shalem, O. & Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014).
Acknowledgements
We thank Katherine A Fitzgerald for providing MAVS−/− immortalized macrophages; members of the GSF laboratory, Florian Pauler and Florian Breitwieser, for discussion. The work was funded by the Austrian Academy of Sciences; i-FIVE European Research Council grant and the FP7 ERC ASSET to GS-F; European Molecular Biology Organization long-term fellowship to RKK (ALTF 314–2012), GIV (ALTF 1543–2012) and MR (ALTF-1346–2011); and Swiss-National Science Foundation advanced postdoc mobility fellowship to BS (P300P3-147897).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Systems Biology and Applications website (http://www.nature.com/npjsba)
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Kandasamy, R., Vladimer, G., Snijder, B. et al. A time-resolved molecular map of the macrophage response to VSV infection. npj Syst Biol Appl 2, 16027 (2016). https://doi.org/10.1038/npjsba.2016.27
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/npjsba.2016.27
