
Abstract
Studies on the relationships between humans and microbes in space habitation environments are critical for success in long-duration space missions, to reduce potential hazards to the crew and the spacecraft infrastructure. We performed microbial monitoring in the Japanese Experiment Module “Kibo”, a part of the International Space Station, for 4 years after its completion, and analyzed samples with modern molecular microbiological techniques. Sampling was performed in September 2009, February 2011, and October 2012. The surface of the incubator, inside the door of the incubator, an air intake, air diffuser, and handrail were selected as sampling sites. Sampling was performed using the optimized swabbing method. Abundance and phylogenetic affiliation of bacteria on the interior surfaces of Kibo were determined by quantitative PCR and pyrosequencing, respectively. Bacteria in the phyla Proteobacteria (γ-subclass) and Firmicutes were frequently detected on the interior surfaces in Kibo. Families Staphylococcaceae and Enterobacteriaceae were dominant. Most bacteria detected belonged to the human microbiota; thus, we suggest that bacterial cells are transferred to the surfaces in Kibo from the astronauts. Environmental bacteria such as Legionella spp. were also detected. From the data on bacterial abundance and phylogenetic affiliation, Kibo has been microbiologically well maintained; however, the microbial community structure in Kibo may change with prolonged stay of astronauts. Continuous monitoring is required to obtain information on changes in the microbial community structure in Kibo.
Similar content being viewed by others

Introduction
Research on microbial dynamics in crewed habitats in space is indispensable to achieve safe and healthy long-duration space habitation. According to the previous research, microgravity and spaceflight affect bacterial growth, virulence, biofilm formation, and so on (reviewed in Yamaguchi et al.1). For instance, Wilson et al.2 reported that spaceflight-grown Salmonella enterica serovar Typhimurium increased their virulence when compared with control bacteria on Earth. Kim et al.3 reported that Pseudomonas aeruginosa showed different structure and biomass of biofilms between normal and spaceflight conditions. Furthermore, immune dysfunction occurs associated with spaceflight.4 Therefore, the relationship between humans and microbes may change in space habitation. For successful manned space missions, it is necessary to investigate the relationship between humans and microbes, and how microbes influence the systems and materials in such space environments. That is, we have to understand how and where microbes proliferate in a confined environment in space.
Since 2009, we have been continuously performing bacterial monitoring in Kibo, the Japanese Experiment Module of the International Space Station (ISS), in cooperation with the Japan Aerospace Exploration Agency (research title: “Microbe”: http://iss.jaxa.jp/en/kiboexp/news/101101_microbe-2_start.html). The objective of this research is to monitor microbes and analyze their dynamics in Kibo from environmental microbiological viewpoints. Previously, many studies on microbial contamination in space habitats have been based on culture-based approaches, despite the fact that the majority of microbes in natural environments are hard to culture under conventional culture methods.5,6 In environmental microbiology, some culture-independent techniques are available, and these techniques have been introduced in recent research on microbial contamination in space habitats. Ichijo et al.7 used total direct counting, quantitative PCR and PCR denaturant gradient gel electrophoresis for determination of the abundance and phylogenetic affiliation of bacteria on the interior surfaces in Kibo. Venkateswaran et al.8 used quantitative PCR and pyrosequencing followed by propidium monoazide treatment to understand the distribution and diversity of viable microbes in debris collected from the ISS. These reports provided the useful information that the ISS environmental microbiota comprised human-related microbes.
For long space missions, understanding changes in microbial dynamics in a confined environment is essential, as described above. In this research, we collected samples from interior surfaces in Kibo every ca. 500 days after Kibo began operation. Bacterial abundance and phylogenetic affiliation were determined by fluorescent staining, 16S ribosomal RNA (rRNA) gene-targeted quantitative PCR and pyrosequencing. This is the first report on continuous monitoring of bacterial abundance and phylogenetic affiliation in a space habitat determined by culture-independent molecular microbiological methods.
Results
Bacterial abundance
Bacterial abundance on the interior surfaces in Kibo is shown in Table 1. We determined bacterial abundance with different approaches, fluorescent microscopy and quantitative PCR targeting the bacterial 16S rRNA gene, to confirm the reliability of the results. Quantitative PCR outputs the copy number of the 16S rRNA gene in a sample. To convert the copy number to a cell number, we need to know the copy number of the target gene in a single cell. However, bacterial cells carry 1–15 copies of the 16S rRNA gene in their genomes.9 We therefore calculated the average copy number of 16S rRNA genes in the bacterial community in Kibo as 5 copies/cell, based on the community structure determined by pyrosequencing as described below and the ribosomal RNA operon copy number database rrnDB.9 As shown in Table 1, ca. 103 cells/cm2 of bacteria were detected on the interior surfaces in Kibo in Microbe-I (Table 2). In Microbe-II and Microbe-III (Table 2), bacterial abundance was ca. 102 cells/cm2, or less than the quantification limit. Overall, bacterial abundance did not exceed 104 cells/cm2.
Bacterial community structure
Bacterial community structure on the interior surfaces in Kibo was analyzed by 16S rRNA gene-targeted pyrosequencing. Among 99,967 raw reads, 71,190 were high-quality reads after processing with the Quantitative Insights into Microbial Ecology (QIIME) pipeline. These reads were distributed in each sample (13 samples) with an average of 5,472 reads. At first, we estimated sufficient read number for bacterial community analysis using chao1 index (Supplementary Table S1) and a rarefaction curve (Supplementary Figure S1). As shown in Supplementary Table S1, we found that observed operational taxonomic units (OTUs) revealed 73–100% of bacterial family-level OTUs (equivalent to OTU at 90% similarity) that were present in our sample. We therefore confirmed the number of high-quality reads obtained was sufficient to reveal the bacterial community structure in the Kibo sample at the family level.
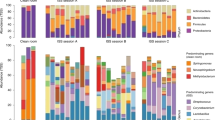
Figure 1a shows the bacterial community structure at the phylum level on the interior surfaces in Kibo. Bacteria of the phyla Proteobacteria (beta- and gamma-subclasses), Firmicutes, and Actinobacteria were frequently detected. The families Enterobacteriaceae and Staphylococcaceae were dominant in each sample (Figure 1b).

Bacterial community structure on the interior surfaces in Kibo. (a) At the phylum level. (b) Expanding beta- and gamma-Proteobacteria and Firmicutes to the family level.
Discussion
Studies on the relationship between humans and microbes in space habitation environments are critical for success in long-duration space missions. To reduce potential hazards to the crew and the spacecraft infrastructure, indoor quality control is important, as defined by the roadmap or scenario of each agency. In this study, we performed microbial monitoring in the ISS—“Kibo” for 4 years after its completion, and analyzed samples with culture-independent techniques.
The majority of bacteria in environments are hard to culture under conventional culture condition. Therefore, bacterial number is usually underestimated with culture methods. To obtain bacterial numbers more accurately, bacterial abundance was determined with culture-independent techniques. We used both total direct counting and quantitative PCR to obtain more reliable results. Total direct counting potentially gives us the absolute cell numbers. In case the number of contaminants (e.g., non-biological fluorescent particles or detritus) is relatively large in the sample, the accuracy of total direct counting tends to decrease. In this study, we set up a criterion when we enumerate bacterial cells by total direct counting; tiny particles whose size are smaller than bacteria generally distributed in environments are omitted. Under this criterion, cell number might be underestimated. The results show that the bacterial number was below 104 cells/cm2. Therefore, we conclude that the bacterial abundance in Kibo was well controlled during the 1,596-day operation covered by our study. However, Kibo is a module for performing experiments and not for living activities. It is therefore important to compare results obtained in Kibo with those in modules for living activities to evaluate the whole microbial world in the ISS.
Bacterial community structure was determined by amplicon sequencing with a high-throughput sequencer. In this study, we used a two-step PCR protocol to generate amplicons. Amplicon sequencing with two-step PCR has several advantages.10 For example, this method increases reproducibility and recovers higher genetic diversity. Selection of the amplified region can influence the profile of the bacterial community generated by pyrosequencing.11 Various regions such as V1–V2,12,13 V3–V5,14,15 V4–V5,16,17 and V6–V8 (refs 18,19) have been analyzed for bacterial community structure by pyrosequencing. In this study, before the experiment, we evaluated several primer sets (targeting V1–V3, V3–V5, V7–V8, and V6–V8) by analyzing bacterial diversity on the incubator surface of Microbe-I by denaturing gradient gel electrophoresis. We selected a primer set of 968f and 1401r, which can amplify the V6–V8 region of the 16S rRNA gene based on greater diversity (data not shown).
The results of bacterial community analysis show that Actinobacteria, Firmicutes, and Proteobacteria were frequently detected on the surfaces of equipment in Kibo. In particular, Staphylococcaceae belonging to the phylum Firmicutes, Enterobacteriaceae belonging to the Proteobacteria (gamma-subclass), and Neisseriaceae belonging to the Proteobacteria (beta-subclass) were dominant taxonomic groups on the equipment surfaces (Figure 1b). Actinobacteria are commonly found in both terrestrial and aquatic ecosystems.20 This phylum is known as the dominant species of human gut microbiota.21,22 Enterobacteriaceae, Staphylococcaceae, and Neisseriaceae are also human related. Enterobacteriaceae, a large group of the gamma-Proteobacteria, are commonly found in the human gut microbiota.23 Fierer et al.12 reported that Actinobacteria, Firmicutes, and Proteobacteria contain the majority of the sequences retrieved from human palm surfaces. According to this report,12 Staphylococcaceae, one of the most abundant taxonomic groups collected from Kibo, are relatively highly abundant on human palm surfaces; Neisseriaceae made up ca. 3% of the sequences from human palm surfaces; Enterobacteriales were also commonly found there. As described above, most of the bacteria detected on the Kibo equipment surfaces belong to human microbiota; thus, we suggest that bacterial cells are transferred to the surfaces in Kibo from the astronauts. Kibo was disinfected with isopropyl alcohol before launch. We measured bacterial numbers on the surfaces in Kibo before launch by fluorescent microscopy, but the bacterial number was below the quantification limit (<2×102 cells/cm2). Our results show that immediately after beginning the operation of Kibo, few bacteria were present there. The bacterial community in Kibo formed from human-related bacteria through astronaut activities in the early stage of operation.
Microbial community structure on the equipments in Kibo varied during the first 4-year operation (Figure 1b). For example, on the surfaces of diffuser, handrail, and inside of incubator, Neiseriaceae occupied 3.4–48% of total bacterial communities on Microbe-I and Microbe-II; however, on Microbe-III, Neiseriaceae was rarely detected there (<1%). Relative abundance of Enterobacteriaceae seemed to increase in Microbe-III. Although human-related bacteria occupied majority of bacterial community in Kibo in the early stage of operation, their bacterial community was not stable during this period (Figure 1b). Because bacterial community in the space habitat was not established, it is considered that the community is easy to change affected by external factors such as astronauts’ activities and experiments in Kibo.
Legionella were dominant in the sample collected from the outer surface of the incubator on Microbe-I (Figure 1b). Legionella is normally found in aquatic and terrestrial environments,23 but not among human microbiota. Therefore, it seems unlikely that Legionella cells were transferred to the surface via the astronauts. In the monitoring of space habitat, Legionella species was only found in the Russian Space Station Mir.24 As environmental bacteria have been detected in Kibo in this study and they are unlikely to have derived from astronaut activities, humans may not be the only source of bacteria in Kibo, and resupply materials could be the vehicles of microbes to the space habitats. We have to monitor such microbes.
As mentioned above, Kibo was microbiologically well maintained during the first 4-year operation. Microbiological cleanliness was controlled by wiping interior surfaces with disinfectant (benzalkonium chloride) once a week. However, benzalkonium chloride may increase the antimicrobial resistance of bacteria.25,26 Prolonged stay of astronauts may increase microbial abundance and affect the microbial community in Kibo. In February 2015, “Microbe-IV”, the next phase of “Microbe”, was started. Four sampling are scheduled in fiscal year 2014–2016. Four new sampling points were added to the six points used in “Microbe I–III”: a foot hold (high frequency of human contact); the MELFI1 door (high frequency of human contact; located on the floor); the Intermodule Ventilator Fan (air of Kibo; located between Kibo and the module next to Kibo); and the wall of Kibo (low frequency of human contact). Continuous monitoring provides further information on changes in the microbial community structure in Kibo and the stability of the microbial ecosystem during prolonged stay in confined environments in space, and leads to assure crew safety and the success of long-duration missions.
Materials and methods
Sampling in Kibo
Sampling in Kibo was performed three times by an astronaut as part of the “Microbe” research program (Table 2). The sampling locations inside the Kibo module were the internal and outer surface of the incubator named CBEF (Cell Biology Experiment Facilities; low frequency of astronaut contact), an air return grill and an air diffuser (reflection of the bacterial community of the air in Kibo), and handrail (high frequency of astronaut contact; Figure 2). The air return grill and the inside of the incubator were selected as sampling points from Microbe-II onwards. An optimized swabbing protocol27 was used for sampling the interior surfaces. Samples were stored in the freezer (−95 °C) installed in Kibo and transferred to our laboratory via the NASA Kennedy Space Center and Japan Aerospace Exploration Agency Tsukuba Space Center at −80 °C. We previously confirmed that long-term storage with freezing does not affect the number of bacteria collected with swab method.27

Photographs of sampling points in Kibo ((c) NASA/JAXA).
Total direct counting of microbes
Bacteria collected with swabs were suspended in 10 ml of particle-free sterilized water and trapped on an autoclaved polycarbonate membrane filter (pore size 0.2 μm; Advantec, Tokyo, Japan) in a funnel (filtration area 13 mm2; KGS-04, Advantec). Bacterial cells on the filter were stained with filter-sterilized 1× SYBR Green II nucleic acid staining dye (Invitrogen, Carlsbad, CA, USA). We enumerated fluorescently stained bacterial cells under an epifluorescent microscope (E-400; Nikon, Tokyo, Japan) with the Nikon filter sets B2-A (EX450–490, DM505, BA520). One-hundred microscopic fields (1 mm2) per sample were observed.
DNA extraction
Following microscopic observation, DNA was extracted from bacterial cells on the polycarbonate filter as described previously.6 Extracted DNA was suspended in TE buffer (10 mmol/l Tris-HC1, 1 mmol/l EDTA, pH 8.0) and stored at −20 °C before use. To estimate the DNA recovery rate during extraction, known amounts of PCR products of the luciferase gene (luc) fragment were inoculated into the samples as an internal standard and quantified after DNA extraction according to Nishimura et al.28 The DNA recovery rate was calculated by comparing the copy number of the inoculated luc gene before and after DNA extraction.
Quantitative PCR for quantification of bacteria
For quantification of bacteria, the bacterial 16S rRNA gene was quantified with the primer set EUB f933 and EUB r1387 (Table 3) using a LightCycler (Roche Diagnostics, Mannheim, Germany) according to the protocol reported by Yamaguchi et al.6 The copy number of the 16S rRNA gene in a sample was calibrated based on the DNA recovery rate.
16S rRNA gene-targeted amplicon sequencing
16S rRNA gene fragments, including V6–8 regions, were amplified and analyzed by amplicon sequencing with the GS FLX System (Roche Diagnostics). In this study, we used a two-step PCR protocol, as previously described by Sutton et al.29 The first PCR amplification was performed using primers 968f and 1401r (Table 3), which are specific for conserved bacterial 16S rRNA gene sequences. The PCR mixture, containing 1.25 U of AmpliTaq Gold (Applied Biosystems, Foster City, CA, USA), 25 pmol of each primer, 75 nmol of MgCl2, 10 nmol of each deoxyribonucleoside triphosphate, 5 μl of 10× PCR buffer, and 0.25 μl of 2.5 mg/ml 8-methoxypsoralen (Sigma-Aldrich, St Louis, MO) in dimethyl sulfoxide, was made up to 48 μl with DNA-free water. A DNA suspension was added last in a 2-μl volume after irradiation of the PCR mixture with ultraviolet light (λ=365 nm).30,31 A hot-start PCR was performed at 95 °C for 9 min, and touchdown PCR was performed as follows: the annealing temperature was initially set at 63 °C and was then decreased by 1 °C every cycle until it was 53 °C. Twenty additional cycles were carried out at 53 °C. Denaturing was carried out at 94 °C for 1 min. Annealing was performed using the scheme described above for 1 min, and extension was performed at 72 °C for 3 min. The final extension step was 7 min at 72 °C. The first PCR product was purified with a Wizard SV Gel and PCR Clean-Up System (Promega, Madison, WI, USA) and eluted with nuclease-free water. The second PCR used the purified products of the first PCR as templates. Amplification was performed as described above, except that primers with an adapter and 10-nt-length barcode were used, and the total number of amplification cycles was reduced to 18. The second PCR product was also purified with a Wizard SV Gel and PCR Clean-Up System and eluted with nuclease-free water. PCR product was quantified with a Qubit dsDNA HS Assay Kit (Invitrogen) and mixed in approximately equal concentrations (5×1010 copies/μl). The sample was sequenced with a GS FLX System at Hokkaido System Science (Hokkaido, Japan).
Data analysis
Sequencing data were processed using the QIIME pipeline.32 Sequences were quality filtered by removing reads that (a) did not contain the primer sequence, (b) contained an uncorrectable barcode, (c) were <250 nt in length, (d) had homopolymers >8 nt, or (e) had a quality score of <25, and then demultiplexed using the respective sample nucleotide barcodes.8 These sequences were clustered into OTUs based on their similarity (97%). The taxonomic affiliation of each OTU was determined using the RDP Classifier at a confidence threshold of 80%.33 Chao1 index and rarefaction curves based on family-level OTUs were estimated with the RDPipeline34 (Ribosomal Database Project). The sequences obtained from amplicon sequencing were deposited in the DNA Data Bank of Japan Sequence Read Archive under the accession number DRA004228.
References
Yamaguchi N. et al. Microbial monitoring of crewed habitats in space -Current status and future perspectives. Microbes Environ. 29, 250–260 (2014).
Wilson J. W. et al. Space flight alters bacterial gene expression and virulence and reveals a role for global regulator Hfq. Proc. Natl Acad. Sci. USA; 104, 16299–16304 (2007).
Kim W. et al. Spaceflight promotes biofilm formation by Pseudomonas aeruginosa. PLoS One 8, e62437 (2013).
Borchers, A. T., Keen, C. L. & Gershwin, M. E. Microgravity and immune responsiveness. Nutrition 18, 889–898 (2002).
Kawai, M., Yamaguchi, N. & Nasu, M. Rapid enumeration of physiologically active bacteria in purified water used in the pharmaceutical manufacturing process. J. Appl. Microbiol. 86, 496–504 (1999).
Yamaguchi, N., Ichijo, T., Sakotani, A., Baba, T. & Nasu, M. Global dispersion of bacterial cells on Asian dust. Sci. Rep. 2, 525 (2012).
Ichijo, T., Hieda, H., Ishihara, R., Yamaguchi, N. & Nasu, M. Bacterial monitoring with adhesive sheet in the International Space Station-‘Kibo’, the Japanese Experiment Module. Microbes Environ. 28, 264–268 (2013).
Venkateswaran K. et al. International Space Station environmental microbiome—microbial inventories of ISS filter debris. Appl. Microbiol. Biotechnol. 98, 6453–6466 (2014).
Stoddard, S. F., Smith, B. J., Hein, R., Roller, B. R. K. & Schmidt, T. M. rrnDB: improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Res. 43, D593–D598 (2014).
Berry, D., Ben Mahfoudh, K., Wagner, M. & Loy, A. Barcoded primers used in multiplex amplicon pyrosequencing bias amplification. Appl. Environ. Microbiol. 77, 7846–7849 (2011).
Kumar, P. S., Brooker, M. R., Dowd, S. E. & Camerlengo, T. Target region selection is a critical determinant of community fingerprints generated by 16S pyrosequencing. PLoS One 6, e20956 (2011).
Fierer, N., Hamady, M., Lauber, C. L. & Knight, R. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc. Natl Acad. Sci. USA 105, 17994–17999 (2008).
Li J. et al. Bacterial dynamics within the mucus, tissue and skeleton of the coral Porites lutea during different seasons. Sci. Rep. 4, 7320 (2014).
Hoppe B. et al. A pyrosequencing insight into sprawling bacterial diversity and community dynamics in decaying deadwood logs of Fagus sylvatica and Picea abies. Sci. Rep. 5, 9456 (2015).
Yu, Z., Yang, J., Amalfitano, S., Yu, X. & Liu, L. Effects of water stratification and mixing on microbial community structure in a subtropical deep reservoir. Sci. Rep. 4, 5821 (2014).
Chen, W. Y., Wu, J. H. & Chang, J. E. Pyrosequencing analysis reveals high population dynamics of the soil microcosm degrading octachlorodibenzofuran. Microbes Environ. 29, 393–400 (2014).
Ding, L. J., Su, J. Q., Xu, H. J., Jia, Z. J. & Zhu, Y. G. Long-term nitrogen fertilization of paddy soil shifts iron-reducing microbial community revealed by RNA-(13)C-acetate probing coupled with pyrosequencing. ISME J. 9, 721–734 (2015).
Mohit, V., Archambault, P., Toupoint, N. & Lovejoy, C. Phylogenetic differences in attached and free-living bacterial communities in a temperate coastal lagoon during summer, revealed via high-throughput 16S rRNA gene sequencing. Appl. Environ. Microbiol. 80, 2071–2083 (2014).
Nakayama J. et al. Diversity in gut bacterial community of school-age children in Asia. Sci. Rep. 5 8397 (2015).
Ventura M. et al. Genomics of Actinobacteria: tracing the evolutionary history of an ancient phylum. Microbiol. Mol. Biol. Rev. 71, 495–548 (2007).
Arumugam M. et al. Enterotypes of the human gut microbiome. Nature 473, 174–180 (2011).
Andersson A. F. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One 3, e2836 (2008).
Brenner D. J., Krieg N. R. & Staley J. T. (eds) Bergey’s Manual of Systematic Bacteriology. Vol. 2 (Springer, 2005).
Ott, C. M., Bruce, R. J. & Pierson, D. L. Microbial characterization of free floating condensate aboard the Mir space station. Microb. Ecol. 47, 133–136 (2004).
Mc Cay, P. H., Ocampo-Sosa, A. A. & Fleming, G. T. Effect of subinhibitory concentrations of benzalkonium chloride on the competitiveness of Pseudomonas aeruginosa grown in continuous culture. Microbiology 156, 30–38 (2010).
Tandukar, M., Oh, S., Tezel, U., Konstantinidis, K. T. & Pavlostathis, S. G. Long-term exposure to benzalkonium chloride disinfectants results in change of microbial community structure and increased antimicrobial resistance. Environ. Sci. Technol. 47, 9730–9738 (2013).
Yamaguchi, N., Hieda, H. & Nasu, M. Simple and reliable swabbing procedure for monitoring microbes in the International Space Station. Eco-Engineering 22, 27–30 (2010).
Nishimura Y. et al. Similarity of bacterial community structure between Asian dust and its sources determined by rRNA gene-targeted approaches. Microbes Environ. 25, 22–27 (2010).
Sutton N. B. et al. Impact of long-term diesel contamination on soil microbial community structure. Appl. Environ. Microbiol. 79, 619–630 (2013).
Meier, A., Persing, D. H., Finken, M. & Bottger, E. C. Elimination of contaminating DNA within polymerase chain reaction reagents: implications for a general approach to detection of uncultured pathogens. J. Clin. Microbiol. 31, 646–652 (1993).
Kawai M. et al. 16S Ribosomal DNA-based analysis of bacterial diversity in purified water used in pharmaceutical manufacturing processes by PCR and denaturing gradient gel electrophoresis. Appl. Environ. Microbiol.; 68: 699–704 (2002).
Caporaso J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336 (2010).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267 (2007).
Cole J. R. et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, D633–D642 (2014).
Iwamoto T. et al. Monitoring impact of in situ biostimulation treatment on groundwater bacterial community by DGGE. FEMS Microbiol. Ecol. 32, 129–141 (2000).
Felske, A., Engelen, B., Nubel, U. & Backhaus, H. Direct ribosome isolation from soil to extract bacterial rRNA for community analysis. Appl. Environ. Microbiol. 62, 4162–4167 (1996).
Acknowledgements
We thank the following for technical assistance: Dr Hatsuki Hieda, Ms Rie Ishihara and Ms Nozomi Inoue at Osaka University. We also thank Mr Yuuichi Takahashi and Mr Masamitsu Hida (Japan Aerospace Exploration Agency, JAXA), and Dr Toru Shimazu and Dr Keiji Fukui (Japan Space Forum, JSF) for their cooperation with this research program. This study was supported by the JAXA and the JSF as a theme for Kibo Pressurized Module use in the second period.
Author information
Authors and Affiliations
Contributions
TI and NY contributed equally to this work. MN conceived the study; TI performed quantitative PCR and phylogenetic analysis; NY performed fluorescent microscopy; FT and MS managed sampling programs; TI, NY, and MN wrote the paper; all authors discussed the results in the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Ichijo, T., Yamaguchi, N., Tanigaki, F. et al. Four-year bacterial monitoring in the International Space Station—Japanese Experiment Module “Kibo” with culture-independent approach. npj Microgravity 2, 16007 (2016). https://doi.org/10.1038/npjmgrav.2016.7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/npjmgrav.2016.7
This article is cited by
-
Exposure of Legionella pneumophila to low-shear modeled microgravity: impact on stress response, membrane lipid composition, pathogenicity to macrophages and interrelated genes expression
Archives of Microbiology (2024)
-
Solar ultraviolet light collector for germicidal irradiation on the moon
Scientific Reports (2023)
-
Microbial Tracking-2, a metagenomics analysis of bacteria and fungi onboard the International Space Station
Microbiome (2022)
-
Bacterial bioburden and community structure of potable water used in the International Space Station
Scientific Reports (2022)
-
Passive limitation of surface contamination by perFluoroDecylTrichloroSilane coatings in the ISS during the MATISS experiments
npj Microgravity (2022)
