
Abstract
The usefulness of graphene for electronics has been limited because it does not have an energy bandgap. Although graphene nanoribbons have non-zero bandgaps, lithographic fabrication methods introduce defects that decouple the bandgap from electronic properties, compromising performance. Here we report direct measurements of a large intrinsic energy bandgap of ∼50 meV in nanoribbons (width, ∼100 nm) fabricated by high-temperature hydrogen-annealing of unzipped carbon nanotubes. The thermal energy required to promote a charge to the conduction band (the activation energy) is measured to be seven times greater than in lithographically defined nanoribbons, and is close to the width of the voltage range over which differential conductance is zero (the transport gap). This similarity suggests that the activation energy is in fact the intrinsic energy bandgap. High-resolution transmission electron and Raman microscopy, in combination with an absence of hopping conductance and stochastic charging effects, suggest a low defect density.
Similar content being viewed by others

Main
A key goal of research into graphene, a two-dimensional carbon sheet1,2, is to introduce an energy bandgap to enable its use in conventional semiconductor device operations, because pristine graphene is a zero-bandgap material. At least two approaches have been used for the introduction of such a bandgap: the formation of few-layer graphenes3,4,5 and the formation of graphene-nanoribbon structures6,7,8,9,10,11,12.
A non-zero bandgap can be induced in graphene by breaking the inversion symmetry of the AB-stack in bi- or trilayer graphenes3,4,5, but it is very difficult to control the exact number of layers during fabrication. Alternatively, quasi-one-dimensional confinement of the carriers in graphene nanoribbons induces an energy gap in the single-particle spectrum13,14,15,16,17. However, as it is difficult to fabricate graphene nanoribbons using lithographic methods without producing damage (disorder, defects), most of the experimental results reported to date are for disordered graphene nanoribbons6,7,8, except for one group18,19,20. Disorder-induced phenomena (such as hopping conductance, stochastic single electron charging effects in series-coupled quantum dots introduced by defects) obstruct observation of the intrinsic physical properties of graphene nanoribbons6,7,8. Theoretical models to explain the observed disorder-induced gaps have been reported9,10,11,12, such as the stochastic single electron charging effect, Anderson localization due to edge disorder and the contribution of edge irregularity. These energy gaps are not, however, the same as intrinsic energy bandgaps in defect-free graphene nanoribbons.
Reference 6 reports transport gaps that are inversely proportional to the width of lithographically formed disordered graphene nanoribbons at the charge neutrality point. However, these transport gaps were not attributed to the intrinsic energy bandgap or to simple quasi-one-dimensional confinement of the carriers but rather to edge disorder that introduced localized states, causing one-dimensional nearest-neighbour hopping at high temperatures and variable-range hopping at low temperatures. Therefore, the formation of damage-free graphene nanoribbons is key to revealing the intrinsic physical properties (energy bandgaps) of graphene nanoribbons and applying them to bandgap-tuned electronic devices.
Sample fabrication and characterization
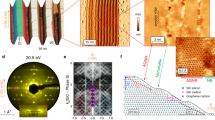
Graphene nanoribbons were prepared through the oxidization and longitudinal unzipping of multiwalled carbon nanotubes. This was achieved by suspending them in concentrated sulphuric acid, followed by treatment with KMnO421,22. The acid and KMnO4 were removed as described elsewhere21,22. Figure 1a (main panel) shows a field-emission scanning electron microscope (SEM) image of an ensemble of the as-grown graphene nanoribbons, which are entangled with one another and not sufficiently exfoliated. The present air-blowing method realized individual graphene nanoribbons forming single-layered rectangular structures (Fig. 1a, insets) (see Methods).

a, Field-emission SEM image of an ensemble of the as-grown nanoribbons, which were dispersed on a silicon oxide substrate in a water droplet containing a suspension of nanoribbons. The nanoribbons are entangled and not sufficiently exfoliated. Insets (right): atomic force microscopy images of individual nanoribbons spread by applying a strong air flow to the droplet (see Methods). Inset (left): height of the nanoribbon measured along the longitudinal direction. The thickness of ∼0.8 nm indicates a single-layer nanoribbon. b, HRTEM image of the as-grown nanoribbon (no annealing; see Methods), showing a clear hexagonal lattice of graphene with few defects and some oxidized parts. Annealing (see Methods) strongly enhances the quality of the nanoribbons and also results in carrier doping. c, Typical Raman spectrum of an annealed bilayer nanoribbon taken with a laser excitation of 532 nm and 0.14 mW incident power at room temperature. The width of the nanoribbon is ∼100 nm and the G/D band ratio is as high as ∼2.2. d, FET fabricated using a nanoribbon end-bonded by two metal electrodes, which eliminates single-electron charging effects (see Supplementary Section 1). The thickness of the SiO2 layer on the gold/titanium backgate electrode was 300 nm, and the spacing of source and drain electrodes was 500 nm in all samples.
A high-resolution transmission electron microscope (HRTEM) image (Fig. 1b) of the as-grown graphene nanoribbon shows the presence of a clear hexagonal graphene lattice with few defects (because of its non-lithographic formation)18. Some areas of the graphene nanoribbon were oxidized because the sample had not yet undergone an annealing process (see Methods). Moreover, both zigzag-edged18,19,20 and armchair-edged structures, both with little disorder, were observed in some areas of the as-grown nanoribbons.
The biggest problem with the oxidative chemical unzipping method is that the graphene nanoribbons are initially oxidized. To entirely remove the oxygen from the bulk and edges of the nanoribbons and also effectively dope charge carriers, we found that three annealing steps were necessary (see Methods). The first step comprised annealing at 800 °C immediately after dispersion of the nanoribbons on the substrate, to remove the bulk of the oxygenation. The second step was carried out at 750 °C in a H2 atmosphere to produce edge termination and charge carrier doping (before creating the marking patterns for electron-beam lithography as were necessary for fabricating the field-effect transistor (FET) electrodes). In the third step, annealing was conducted at 300 °C to clean the surface immediately before the formation of the electrodes.
The Raman spectrum for the bilayer graphene nanoribbon annealed using these processes shows a high-intensity G band at ∼1,580 cm−1, a low-intensity D band at ∼1,350 cm−1, and a low-intensity 2D band at ∼2,700 cm−1 (Fig. 1c). The peak positions are in good agreement with those from previous reports, and the peak ratio of the G/D bands is as high as ∼2.2. This strongly suggests that the present graphene nanoribbon is of high quality. The D-band peak, which is less intense or absent in high-quality graphene, is present only because of residual edge disorder (for example, residual oxygen atoms, missing carbon atoms, and so on), as mentioned in the discussion. In contrast, the peak widths are broader than those in conventional graphenes, in particular for the 2D band. Because the diameter of the laser beam used for measurements is 1 µm, and the width of each graphene nanoribbon is ∼100 nm, the spectra are affected by the nanoribbon edge structures, so the observed small and broad 2D band could be due to the large number of these edge structures in the graphene nanoribbon.
Both ends of a nanoribbon were bonded to the electrodes of a FET structure (Fig. 1d) to form ohmic contacts at the interface of the metal electrodes and the nanoribbon, in a similar process to that carried out in carbon nanoubes27 to suppress the single-electron charging effect (see Supplementary Section 1). This is supported by the tunnel barrier layers being very thin or even absent.
Electronic transport of wide graphene nanoribbon FETs
To explore their electrical properties, graphene nanoribbons with different widths (W), both monolayer and bilayer (N = 1, 2), were selected for FET formation (see Supplementary Section 11). The abovementioned annealing process significantly improved the conductivity of the FETs, resulting in ohmic behaviour at room temperature (Fig. 2a, inset). However, at T = 1.5 K it results in nonlinear behaviour, with a strong zero-bias (G0) anomaly (Fig. 2, main panel) that is at least 10 times greater than that found in lithographically formed graphene nanoribbons of the same length6. The G0 anomaly is not attributable to a single-electron charging effect, as mentioned in relation to Fig. 5 (see Supplementary Section 2).

a, Typical drain current (ISD) versus drain voltage (VSD) relationship for a sample (W = 75 nm and N = 1) at room temperature (inset) and T = 1.5 K (main panel), revealing a strong zero-bias (G0) anomaly with a voltage width of ΔVSD ≈ ± V, even at a backgate voltage (VBG) of ±20 V. ΔVSD was determined from the VSD range for differential conductance (dISD/dVSD) ≈ 0. The temperature dependence of the G0 anomaly does not follow the formula for single-electron charging effect26,27. This is consistent with end-bonding of the nanoribbon. b, Differential conductance (dISD/dVSD) as a function of VBG at T = 1.5 K for the sample in a. A transport gap of the backgate voltage of ΔVBG ≈ 1 V is observed. At higher VSD, the gap becomes ambiguous (Fig. 3). In contrast, at lower VSD, the same gap was observed in most cases, although it also becomes ambiguous due to insulating behaviour at values of VBG outside the gap. The energy estimated from this in the single-particle energy spectrum, Δm, is ∼70 meV, which is smaller than that for lithographically formed nanoribbons (∼100 meV) with the same W.
The transport gap of ΔVBG = 1 V, which is the width of the backgate voltage region for which differential conductivity (dI/dV) is zero, is observed around the graphene charge neutrality (Dirac) point (Fig. 2b). Applying a backgate voltage (VBG) can shift the Fermi level in graphene nanoribbons. When the Fermi level is located between the bottom of the conduction band and the top of the valence band (that is, around the charge neutrality point), the conductance disappears, resulting in a transport gap. Conventionally, this gap can correspond to an energy in the single-particle energy spectrum17 given by

where vF = 106 m s−1 is the Fermi velocity of graphene6 and CBG is the capacitive coupling of the graphene nanoribbon to the backgate electrode. This is because the applied VBG modulates electronic states of the graphene nanoribbons via the silicon substrate, so electronic charging in the substrate (QBG = CBGΔVBG) gives rise to the true energy values for transport gaps. When CBG is taken to be 690 aF µm−2, which is the same as that in lithographically formed graphene nanoribbons6, Δm in the present nanoribbons is estimated to be ∼70 meV. This value is smaller than that of lithographically formed nanoribbons (∼100 meV)6, and is believed to be appropriate through comparison with theoretical predictions averaged over many configurations of edge disorder13. Thus, the small Δm value for the present graphene nanoribbon strongly suggests that the degree of edge disorder is less than that in lithographically formed nanoribbons. This supports the conclusion that the present non-lithographic formation of graphene nanoribbons, with annealing, results in a very small amount of disorder22.
When the Δm of 70 meV is normalized by the number of carbon atoms per W (W/a0 = 75 nm/0.142 nm, where a0 is the carbon–carbon bond length), one obtains Δm0 = 11.3 eV. This is three times smaller than the value of Δm0 = 36.3 eV found in lithographically formed graphene nanoribbons, and is close to that of disorder-free nanoribbons. This result suggests a strong intrinsic contribution of quasi-one-dimensional confinement of the carriers to the observed transport gap13,14,15,17 in the present nanoribbons.
A typical drain current (ISD) versus VBG relationship for four different samples (Fig. 3) reveals a drain voltage (VSD) dependence, with (i) p-type semiconducting behaviour at high VSD (>> 1 V; the transport gap of Fig. 2b disappears), (ii) slight ambipolar behaviour in the middle of the VSD range (∼1 V; this corresponds to Fig. 2b) and (ii) predominantly insulating behaviour at very low VSD (<<1 V; approximately maintaining the same transport gap as in Fig. 2b). The ISD on–off ratio24 also exhibits a strong dependence on VSD. Such a dependence is highly sensitive to annealing conditions (see Methods and Supplementary Section 3), and can be interpreted as being due to edge reconstruction25,28 and carrier doping to the edge (edge effect) in the high VSD region (Supplementary Section 7).

a–d, IDS versus VBG in four different nanoribbon FETs. VSD was changed in steps of 1 V. A significant increase in ISD in the −VBG region suggests hole-dominant transport. The ratio of ISD at VBG = –20 V to the minimum ISD strongly depends on VSD, and becomes large for large absolute values of VSD. This suggests the appearance of different states driven by high VSD (such as edge reconstruction; Supplementary Section 7). The transport gap and slight ambipolar feature shown in Fig. 2b remain at low VSD (∼1 V) in d. (W, width (nm); N, number of layers.).
In typical Arrhenius plots for minimum differential conductance (Gmin) versus temperature in the three samples (Fig. 4a; with the exception of the W = 75 nm sample in Fig. 3d), an activation energy (Ea) value of ∼50 meV is obtained from a linear fit for high temperatures (Fig. 4a, dotted lines). The proportional relationship of Ea versus 1/W is very weak in the region of large W (>100 nm), and Ea is independent of N (Supplementary Section 12). This is more obvious in Fig. 4b.

a, Arrhenius plot for minimum conductance (Gmin) versus temperature of the samples shown in Fig. 3. Gmin represents the values at the charge neutrality point (VBG ≈ 0–2 V). An Ea of ∼50 meV was obtained from the dotted lines, and Gmin ∝ exp(−Ea/2kBT) at high temperatures is mostly independent of W and N, except for the W = 75 nm sample, where Ea increases slightly to ∼55 meV. This value is seven times greater than those in lithographically formed nanoribbons and is close to the Δm value (∼80%). b, Ea and Δm values as a function of W for two different values of N. The trend in a is more evident.
In a graphene nanoribbon with W = 75 nm, the value of Ea increases slightly to ∼55 meV (Fig. 4a,b). Remarkably, this value of ∼55 meV is approximately seven times greater than those in lithographically formed graphene nanoribbons with the same width of 75 nm (ref. 6), and is quite close to the abovementioned Δm value of ∼70 meV. This trend was common for other samples (Fig. 4b). Moreover, Gmin becomes largely independent of temperature, and no signature of variable-range hopping is detected below T ≈ 100–60 K in all samples (that is, Gmin = exp(A), where A is a constant). Because the constant Gmin values are extremely small below T = 100–60 K, the present graphene nanoribbons can be insulating. Such characteristics are significantly different from those of lithographically formed nanoribbons6.

The colours represent differential conductance. ΔVBG is 5 mV. The transport gap is represented by the region of 20 × 10−12 S. No stochastic Coulomb diamonds can be observed inside the transport gap (Supplementary Section 1). A transport gap only exists around the charge neutrality point. The measured sample, with W = 80 nm and N = 1, is different from that in Figs 2 and 3.
As mentioned in relation to the transport gap, the fact that the Δm values are smaller than those in lithographically formed nanoribbons suggests a smaller level of edge disorder in the present graphene nanoribbons. The Ea values observed in lithographically formed nanoribbons were well below Δm (near 10%) and were therefore attributed to one-dimensional nearest-neighbour hopping through edge disorder6. In this regard, the value of Ea for the present nanoribbons, being seven times greater than those in lithographically formed nanoribbons and close to the value of Δm (∼80%), suggests that activation does not originate from one-dimensional nearest-neighbour hopping, but instead from an intrinsic energy bandgap within the nanoribbons. As the temperature decreases, this activation via bandgap rapidly decreases, and electron transport becomes insulating because of the absence of both edge disorder and variable-range hopping at low temperatures (<∼100–60 K). All of these interpretations are qualitatively consistent with a low defect density in the studied nanoribbons.
Moreover, the signature of the single-electron charging effect26,27 cannot be observed in any measurements in the present graphene nanoribbons. Single-electron spectroscopy measurements (for example, the so-called Coulomb diamond) comprise a very effective method with which to detect any small defects in nanomaterials (Supplementary Section 1)26,27. When very small defects exist in a sample, they act as quantum dots with the charging energies of single electrons. Where there are many defects, they resemble quantum dots coupled in series. These quantum dots lead to the so-called stochastic Coulomb diamond, in which the Coulomb diamonds are very large and non-uniform, and a series of such Coulomb diamonds can overlap. Previous reports of electronic transport in lithographically formed graphene nanoribbons have shown this stochastic Coulomb diamond effect6,7,8. In contrast, in examinations of the present nanoribbons, no indication of such stochastic diamonds inside the transport gaps is observed (Fig. 5). Only a transport gap (as in Figs 2b and 3) is seen around the charge neutrality point. This result strongly supports the conclusion that there is only a small number of defects between the two electrodes in these graphene nanoribbons.
The results also suggest that the observed Ea values are intrinsic bandgaps in the present graphene nanoribbons, in contrast to the case for lithographically formed nanoribbons, where the single-electron charging energy accounts for 60% of the Ea value6. This means that the large ΔVSD (Fig. 2a) can also be attributed to the intrinsic energy bandgap (Supplementary Section 2).
Away from the charge neutrality point (out of the transport gap), ISD oscillation is seen to occur with VBG period (Δosc), as large as ∼5–6 V (ISD oscillation: Supplementary Section 13). This suggests the presence of discrete energy levels in the present graphene nanoribbons and supports the absence of defect-induced levels.
Origin of large bandgaps in wide nanoribbons
We next discuss the origins of the large bandgap values observed. In previous theoretical reports about graphene nanoribbons, widths of less than ∼10 nm were used13,14,15,16,17, but we have used larger widths of W ≈ 100 nm. In spite of this, large Ea values were determined. We consider two factors influencing this observation: (i) there is quantum confinement of electrons into quasi-one-dimensional space, in which enhanced electron–electron interactions remain due to a large mean free path; (ii) the effective width of the present graphene nanoribbons is much smaller (of the order 10 nm). The second factor does not appear to be dominant based on our analysis (Supplementary Section 10).
The first factor is taken to be the dominant reason for the observation of large bandgap values. It is known that the origins of bandgaps vary in low-disorder graphene nanoribbons with different types of homogeneous edges (Supplementary Section 4)13,14,15,16. The bandgaps of graphene nanoribbons with armchair edges originate mainly from quantum confinement of electrons into quasi-one-dimensional space. In contrast, in graphene nanoribbons with zigzag-shaped edges, the bandgaps arise from a staggered sublattice potential due to spin-ordered states at the edges (that is, spin gaps), as a peculiar case. Because the zigzag edge states localize within a few nanometres of the edge, showing an exponential decay towards the centre of the nanoribbons, and the present nanoribbons are as wide as ∼100 nm, the possibility that zigzag graphene nanoribbons make a major contribution to the observed large bandgaps is minimized (Supplementary Section 6). It is probable, therefore, that the observation of such large bandgaps corresponds to major effects arising from the presence of armchair-edged graphene nanoribbons. Quantum confinement of electrons into quasi-one-dimensional space and enhanced electron–electron interactions thus become very important.
Reference 13 has reported that the electron-electron interaction enlarges the self-energy correction in the first-principles calculation based on many-body perturbation theory (the GW approximation), leading to large bandgap values. Following this approach, reference 13 also calculated bandgap values of 3 eV for a W = 1 nm armchair-edged graphene nanoribbon with hydrogen passivation. Based on this value and the conventional inverse proportional relationship between the bandgap and W, the bandgap can be estimated to be ∼30 meV for W ≈ 100 nm in an armchair-edged nanoribbon. This value is in approximate agreement with the observed bandgap of ∼50 meV for W ≈ 75 nm if it is considered that the relationship of bandgap versus 1/W becomes weaker in large W regions. Moreover, because our graphene nanoribbons are also hydrogen-passivated, our data tend to fit the theory of reference 13.
As mentioned above, graphene nanoribbons must be one-dimensional if the theory of reference 13 is to be applied. It might therefore seem that the present nanoribbons, with W ≈ 100 nm, are too large and no longer one-dimensional. However, the strength of quantum confinement in the one-dimensional regime and the electron–electron interaction are determined by comparison of the mean free path le with the size of the one-dimensional space (that is, the W value of the graphene nanoribbons). When le is on the order of 100 nm, which is comparable to the W values of the present nanoribbons (W ≈ 75–350 nm), the nanoribbons are within one-dimensional charge transport regimes and provide strong one-dimensional electron–electron interaction, resulting in large bandgaps. In fact, we have confirmed the le value over 100 nm due to low defects (Supplementary Section 5). As the nanoribbon length decreased, the resistivity decreased linearly. The resistivity saturated at lengths shorter than ∼100–300 nm and became only slightly dependent on a change in length, showing a near constant value of quantum resistance (h/4e2). This suggests le ≈ 100–300 nm. Therefore, the present graphene nanoribbons are one-dimensional and, therefore, the large bandgaps are reasonable. When the W values become much larger than le, this correction will disappear and the bandgaps will also disappear.
The present graphene nanoribbons still have a small number of residual defects. As discussed earlier, edge disorder enhances bandgaps by means of many mechanisms (such as localization)9. Although the contribution from the localization effect becomes weaker in large-W nanoribbons, and is sensitive to the degree of disorder, it will nonetheless make a contribution to the present large bandgaps. In fact, the observed le value and the small influence of the localization effect are in good agreement with theoretical calculations9,10. Reference 9 reports the correlation of le values with the number of defects on each edge (P × L/a, where P is defect probability, L is edge length and a = 2.46 Å is the lattice parameter) of graphene nanoribbons with four different types of disorder. The observed le values of ∼100–300 nm in our nanoribbons correspond to the low-defect edge for Klein defects or single missing hexagon defects, with P as low as ∼2.5%.
Reference 10 reports the correlation of P and W with bandgaps and local density of states in armchair-edged graphene nanoribbons under the localization effect, with a calculated presence of high local density of states in the bulk of the armchair-edged nanoribbons (P = 5% and W = 74 nm). This supports a small contribution of the localization effect in the present nanoribbons with P ≈ 2.5% and W > 75 nm. Furthermore, a bandgap of ∼25 meV was calculated for an armchair-edged nanoribbon with P = 5% and W = 74 nm. This suggests that the contribution of the localization effect to bandgaps in the present nanoribbons is ∼10 meV, at most.
Conclusions
Low-defect graphene nanoribbons, formed by unzipping of carbon nanotubes followed by annealing, are promising materials for bandgap engineering and applications in conventional semiconductor device operations. Further study is anticipated regarding correlation with edge states (Supplementary Section 8) and also the formation of narrow graphene nanoribbons (W < ∼10 nm) by applying the present unzipping method to single-walled carbon nanotubes21.
Methods
Spreading the as-grown nanoribbons (Supplementary Video)
To diminish the entanglement of the as-grown nanoribbons (Fig. 1a, main panel) and form sufficient rectangular structures of individual nanoribbons on the SiO2 substrate (Fig. 1a, right insets), an air gun or conventional heat dryer was used. Air flow was thus applied to the water droplet containing the as-grown nanoribbons, changing directions every 30 s, 50 times. The strength of the air flow was tuned by controlling the distance between the droplet and the air gun. In this way, the droplet was moved around on the substrate and the entanglement of as-grown nanoribbons caused to diminish. Subsequently, the surface was dried at 300 °C in high vacuum (10−6 torr).
TEM observations
Only the HRTEM image of the as-grown graphene nanoribbon before annealing is shown in Fig. 1b, because it is extremely difficult to carry out HRTEM observations on FET structures fabricated on silicon substrates. Moreover, because the available TEM grid is not stable at high temperatures (for example, 800 °C), changes in the edge structure cannot be observed by HRTEM after high-temperature annealing. (See Supplementary Section 9 for more details.)
Annealing
The first step was to anneal the sample at 800 °C for 15 h under high vacuum (10−6 torr) to reduce the bulk of the oxygen immediately after dispersion of nanoribbons on the substrate. In the second step, before forming the marking patterns required for electron-beam lithography in the fabrication of the FET electrodes, annealing was carried out at 750 °C for 30 min in a hydrogen atmosphere with pre-annealing at 750 °C for 20 min (in high vacuum, 10−6 torr) to terminate edges and any residual dope charge carriers. A final annealing at 300 °C for 30 min (in high vacuum, 10−6 torr) was performed for surface cleaning immediately before patterning of the FET electrodes (gold/titanium) by electron-beam lithography on a highly doped silicon substrate. (See Supplementary Sections 3 and 7 for more details.)
References
Novoselov, K. S. et al. Electric field effect in atomically thin carbon films. Science 306, 666–669 (2004).
Geim, A. K. & Novoselov, K. S. The rise of graphene, Nature Mater. 6, 183–191 (2007).
Zhao, Y. et al. Symmetry breaking of the zero energy Landau level in bilayer graphene. Phys. Rev. Lett. 104, 066801 (2010).
Zhang, Y. et al. Direct observation of a widely tunable bandgap in bilayer graphene. Nature 459, 820–823 (2009).
Craciun, M. F. et al. Trilayer graphene is a semimetal with a gate-tunable band overlap. Nature Nanotech. 4, 383–388 (2009).
Han, M. Y. et al. Electron transport in disordered graphene nanoribbons. Phys. Rev. Lett. 104, 056801 (2010).
Han, M. Y. et al. Energy band-gap engineering of graphene nanoribbons. Phys. Rev. Lett. 98, 206805 (2007).
Stampfe, C. et al. Energy gaps in etched graphene nanoribbons. Phys. Rev. Lett. 102, 056403 (2009).
Cresti, A. & Roche, S. Edge-disorder-dependent transport length scales in graphene nanoribbons: from Klein defects to the superlattice limit. Phys. Rev. B 79, 233404 (2009).
Evaldsson, M. et al. Edge-disorder-induced Anderson localization and conduction gap in graphene nanoribbons. Phys. Rev. B 78, 161407(R) (2008).
Mucciolo, E. R. et al. Conductance quantization and transport gaps in disordered graphene nanoribbons. Phys. Rev. B 79, 075407 (2009).
Lherbier, A. et al. Transport length scales in disordered graphene-based materials: strong localization regimes and dimensionality effects. Phys. Rev. Lett. 100, 036803 (2008).
Yang, L. et al. Quasiparticle energies and band gaps in graphene nanoribbons. Phys. Rev. Lett. 99, 186801 (2007).
Son, Y-W., Cohen, M. L. & Louie, S. G. Energy gaps in graphene nanoribbons. Phys. Rev. Lett. 97, 216803 (2006).
Son, Y-W., Cohen, M. L. & Louie, S. G. Half-metallic graphene nanoribbons. Nature 444, 347–349 (2006).
Nakada, K., Fujita, M., Dresselhaus, G. & Dresselhaus, M. S. Edge state in graphene ribbons: nanometer size effect and edge shape dependence. Phys. Rev. B 54, 17954–17961 (1996).
CastroNeto, H. et al. The electronic properties of graphene. Rev. Mod. Phys. 81, 109–163 (2009).
Jiao, L. et al. Facile synthesis of high-quality graphene nanoribbons. Nature Nanotech. 5, 321–325 (2010).
Li, X. et al. Chemically derived, ultrasmooth graphene nanoribbon semiconductors. Science 319, 1229–1232 (2008).
Wang, X. et al. Room-temperature all-semiconducting sub-10-nm graphene nanoribbon field-effect transistors. Phys. Rev. Lett. 100, 206803 (2008).
Kosynkin, D. V. et al. Longitudinal unzipping of carbon nanotubes to form graphene nanoribbons. Nature 458, 872–877 (2009).
Higginbotham, A. L. et al. Low-defect graphene oxide nanoribbons from multiwalled carbon nanotubes. ACS Nano 4, 2059–2069 (2010).
Takesue, I. et al. Superconductivity in entirely end-bonded multiwalled carbon nanotubes. Phys. Rev. Lett. 96, 057001 (2006).
Bail, J. et al. Graphene nanomesh. Nature Nanotech. 5, 190–194 (2010).
Girit, C. O. et al. Graphene at the edge: stability and dynamics. Science 323, 1705–1708 (2009).
Grabert, H. & Devoret, M. H. (eds) Single Charge Tunneling, NATO ASI Series B, Vol. 294 (Plenum, 1991).
Haruyama, J. et al. Coulomb blockade related to a localization effect in a single tunnel-junction carbon nanotubes system. Phys. Rev. B 63, 073406 (2001).
Jia, X. M. et al. Controlled formation of sharp zigzag and armchair edges in graphitic nanoribbons. Science 323, 1701–1705 (2009).
Niimi, Y. et al. Scanning tunneling microscopy and spectroscopy of the electronic local density of states of graphite surface near monoatomic step edges. Phys. Rev. B 73, 085421 (2006).
Cervenka, J., Katsnelson, M. I. & Flipse, C. F. J. Room-temperature ferromagnetism in graphite driven by two-dimensional networks of point defects. Nature Phys. 5, 840–844 (2009).
Rangel, N. L., Sotelo, J. C. & Seminario, J. M. Mechanism of carbon nanotubes unzipping into graphene ribbons. J. Chem. Phys. 131, 031105 (2009).
Acknowledgements
The authors thank S. Tarucha, M. Yamamoto, T. Otsuka, Y. Iye, S. Katsumoto, T. Osada, H. Fukuyama, T. Ando, A. Endo, Y. Hashimoto, N. Miyazaki, Y. Yagi, H. Kodama, A. Sawabe, X.M. Jia, M.S. Dresselhaus, M.H. Han, J. Kohno and P. Kim for technical support, fruitful discussions and encouragement. The work at Aoyama Gakuin was partly supported by a Grant-in-aid for Scientific Research and a High-Technology Research Center Project for private universities in MEXT. The work at Rice University was supported by the AFOSR (FA9550-09-1-0581), the Alliance for Nanohealth, the AFRL through the University Technology Corporation (09-S568-064-01-C1) and the Office of Naval Research Multidisciplinary Research Program of the University Research Initiative (MURI) program.
Author information
Authors and Affiliations
Contributions
J.H., J.M.T. and K.S conceived and designed the experiments. T.S., D.C.M., D.V.K., and K.H. performed the experiments. J.H. analysed the data. J.H. and J.M.T. co-wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary information
Supplementary information (PDF 813 kb)
Rights and permissions
About this article
Cite this article
Shimizu, T., Haruyama, J., Marcano, D. et al. Large intrinsic energy bandgaps in annealed nanotube-derived graphene nanoribbons. Nature Nanotech 6, 45–50 (2011). https://doi.org/10.1038/nnano.2010.249
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nnano.2010.249
This article is cited by
-
Electron–phonon interaction toward engineering carrier mobility of periodic edge structured graphene nanoribbons
Scientific Reports (2023)
-
Synthesis of a magnetic π-extended carbon nanosolenoid with Riemann surfaces
Nature Communications (2022)
-
Room-temperature quantum spin Hall phase in laser-patterned few-layer 1T′- MoS2
Communications Materials (2020)
-
Phonon Thermal Properties of Heterobilayers with a Molecular Dynamics Study
International Journal of Thermophysics (2020)
-
Lithographic band structure engineering of graphene
Nature Nanotechnology (2019)
