
Abstract
The healthy human brain is a mosaic of varied genomes. Long interspersed element-1 (LINE-1 or L1) retrotransposition is known to create mosaicism by inserting L1 sequences into new locations of somatic cell genomes. Using a machine learning-based, single-cell sequencing approach, we discovered that somatic L1-associated variants (SLAVs) are composed of two classes: L1 retrotransposition insertions and retrotransposition-independent L1-associated variants. We demonstrate that a subset of SLAVs comprises somatic deletions generated by L1 endonuclease cutting activity. Retrotransposition-independent rearrangements in inherited L1s resulted in the deletion of proximal genomic regions. These rearrangements were resolved by microhomology-mediated repair, which suggests that L1-associated genomic regions are hotspots for somatic copy number variants in the brain and therefore a heritable genetic contributor to somatic mosaicism. We demonstrate that SLAVs are present in crucial neural genes, such as DLG2 (also called PSD93), and affect 44–63% of cells of the cells in the healthy brain.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others

Change history
13 July 2017
In the version of this article initially published, NIH grant T32 CA009370 to F.H.G. was missing from the Acknowledgments. The error has been corrected in the HTML and PDF versions of the article.
27 April 2018
In the version of this article initially published, NIH grant U01 MH106882 to F.H.G. was missing from the Acknowledgments. The error has been corrected in the HTML and PDF versions of the article.
References
Campbell, I.M., Shaw, C.A., Stankiewicz, P. & Lupski, J.R. Somatic mosaicism: implications for disease and transmission genetics. Trends Genet. 31, 382–392 (2015).
Shirley, M.D. et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N. Engl. J. Med. 368, 1971–1979 (2013).
Poduri, A. et al. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron 74, 41–48 (2012).
Muotri, A.R. et al. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 435, 903–910 (2005).
Erwin, J.A., Marchetto, M.C. & Gage, F.H. Mobile DNA elements in the generation of diversity and complexity in the brain. Nat. Rev. Neurosci. 15, 497–506 (2014).
Evrony, G.D. et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 151, 483–496 (2012).
Evrony, G.D. et al. Cell lineage analysis in human brain using endogenous retroelements. Neuron 85, 49–59 (2015).
Evrony, G.D., Lee, E., Park, P.J. & Walsh, C.A. Resolving rates of mutation in the brain using single-neuron genomics. eLife 5, e12966 (2016).
Upton, K.R. et al. Ubiquitous L1 mosaicism in hippocampal neurons. Cell 161, 228–239 (2015).
Coufal, N.G. et al. L1 retrotransposition in human neural progenitor cells. Nature 460, 1127–1131 (2009).
Muotri, A.R. et al. L1 retrotransposition in neurons is modulated by MeCP2. Nature 468, 443–446 (2010).
McConnell, M.J. et al. Mosaic copy number variation in human neurons. Science 342, 632–637 (2013).
Cai, X. et al. Single-cell, genome-wide sequencing identifies clonal somatic copy-number variation in the human brain. Cell Rep. 8, 1280–1289 (2014).
Dean, F.B. et al. Comprehensive human genome amplification using multiple displacement amplification. Proc. Natl. Acad. Sci. USA 99, 5261–5266 (2002).
Witherspoon, D.J. et al. Mobile element scanning (ME-Scan) by targeted high-throughput sequencing. BMC Genomics 11, 410 (2010).
Iskow, R.C. et al. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell 141, 1253–1261 (2010).
Brouha, B. et al. Hot L1s account for the bulk of retrotransposition in the human population. Proc. Natl. Acad. Sci. USA 100, 5280–5285 (2003).
Lasken, R.S. & Stockwell, T.B. Mechanism of chimera formation during the multiple displacement amplification reaction. BMC Biotechnol. 7, 19 (2007).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at arXiv 1303.3997v2 (2013).
Breiman, L. Random forests. Mach. Learn. 45, 5–32 (2001).
White, T.B., McCoy, A.M., Streva, V.A., Fenrich, J. & Deininger, P.L. A droplet digital PCR detection method for rare L1 insertions in tumors. Mob. DNA 5, 30 (2014).
Gilbert, N., Lutz-Prigge, S. & Moran, J.V. Genomic deletions created upon LINE-1 retrotransposition. Cell 110, 315–325 (2002).
Morrish, T.A. et al. DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nat. Genet. 31, 159–165 (2002).
Garvin, T. et al. Interactive analysis and assessment of single-cell copy-number variations. Nat. Methods 12, 1058–1060 (2015).
Jurka, J. Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc. Natl. Acad. Sci. USA 94, 1872–1877 (1997).
Feng, Q., Moran, J.V., Kazazian, H.H. Jr. & Boeke, J.D. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell 87, 905–916 (1996).
Yu, D.X. et al. Modeling hippocampal neurogenesis using human pluripotent stem cells. Stem Cell Rep. 2, 295–310 (2014).
Moran, J.V. et al. High frequency retrotransposition in cultured mammalian cells. Cell 87, 917–927 (1996).
d'Adda di Fagagna, F. et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature 426, 194–198 (2003).
Gasior, S.L., Wakeman, T.P., Xu, B. & Deininger, P.L. The human LINE-1 retrotransposon creates DNA double-strand breaks. J. Mol. Biol. 357, 1383–1393 (2006).
Nithianantharajah, J. et al. Synaptic scaffold evolution generated components of vertebrate cognitive complexity. Nat. Neurosci. 16, 16–24 (2013).
Kirov, G. et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry 17, 142–153 (2012).
Fromer, M. et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184 (2014).
Lim, J.S. et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat. Med. 21, 395–400 (2015).
Kempermann, G., Kuhn, H.G. & Gage, F.H. Genetic influence on neurogenesis in the dentate gyrus of adult mice. Proc. Natl. Acad. Sci. USA 94, 10409–10414 (1997).
Aimone, J.B., Deng, W. & Gage, F.H. Adult neurogenesis: integrating theories and separating functions. Trends Cogn. Sci. 14, 325–337 (2010).
Hosono, S. et al. Unbiased whole-genome amplification directly from clinical samples. Genome Res. 13, 954–964 (2003).
Needleman, S.B. & Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 48, 443–453 (1970).
Langdon, W.B. Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. BioData Min. 8, 1 (2015).
Quinlan, A.R. & Hall, I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Untergasser, A. et al. Primer3--new capabilities and interfaces. Nucleic Acids Res. 40, e115 (2012).
Kent, W.J. et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Acknowledgements
We thank J. Moran, M. Gage, M. McConnell, C. Benner, R. Herai and D. O'Keefe for critical reading and discussions of the manuscript. We thank C. Fitzpatrick and C. O'Connor for FACS. J.A.E. was supported by the George E. Hewitt Foundation for Medical Research, and A.C.M.P. was supported by a training grant from the California Institute for Regenerative Medicine. The Gage Laboratory, and this project, was partially funded by NIH MH095741 (F.H.G.), NIH MH088485 (F.H.G.), NIH T32 CA009370 (F.H.G.), NIH U01 MH106882 (F.H.G.), The G. Harold & Leila Y. Mathers Foundation (F.H.G.), The Engman Foundation (F.H.G.), The Leona M. and Harry B. Helmsley Charitable Trust (F.H.G.), Paul G. Allen Family Foundation (F.H.G.), Glenn Center for Aging Research at the Salk Institute (F.H.G.) and JPB Foundation (F.H.G.). This work was supported by the Flow Cytometry Core Facility of the Salk Institute with funding from NIH-NCI CCSG: P30 014195.
Author information
Authors and Affiliations
Contributions
J.A.E. and A.C.M.P. contributed equally to this work. J.A.E., A.C.M.P. and F.H.G. planned, directed and designed the experiments. J.A.E. prepared sequencing libraries, and A.C.M.P. directed and designed computational analysis. T.S. designed and performed the initial L1 targeted sequencing protocol which was further developed into SLAV-seq. I.G., M.N., C.R.B., J.R.H., C.Q. and T.A.B. performed validation experiments. A.S. provided neural precursor cell lines, and F.I.A.A. and A.C.M. performed RNA-seq analysis. R.S.L., A.R.M. and F.H.G. directed the project. J.A.E., A.C.M.P. and F.H.G. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Efficient FACS isolation of neuronal and non-neuronal nuclei for single cell genomic analysis.
For each sample, 94 single nuclei were FACS sorted into individual wells of a 384-well qPCR plate with two negative control wells per plate. All amplified wells were surrounded by empty wells, and the two negative control wells per plate were also subjected to MDA amplification. We validated single cell sorting in replicate plates of unamplified single nuclei using a taqman qPCR copy number assay to measure the amount of genomic DNA within each well. (A) FACS analysis of hippocampal nuclei immunolabeled for NeuN shows two populations: NeuN+ and NeuN-. (B) Reverse transcription PCRs on pools of sorted NeuN+ and NeuN- nuclei confirm neuronal (NeuN) and glial (GFAP) identities from sorted nuclei. (C) Multiplexed LINE-1 and SATA taqman qPCR copy number assays (Hosono, S. et al. (2003) Genome research 13, 954-964) on single nuclei sorted into 384-well plates confirm that more than 95% of wells contain single nuclei. (D) Quantification of the number of single cell MDA products passing qPCR quality control assay.
Supplementary Figure 2 SLAV-seq obtains high enrichment for L1 3′ sequences.
(A) SLAV-seq protocol: Biotinylated (represented by *) L1HS-specific oligo was hybridized separately to sheared genomic DNA. A single extension, followed by capture and on-bead ligation of an amino-modified asymmetric adapter, is performed. Paired-end products (100 bp) from ligation-associated PCR are then sequenced on an Illumina HiSeq2000 with read 2 beginning with the 3' end of L1 sequence (magenta arrow indicates hemispecific nested PCR oligo). (B) Density plots of reads from all libraries displaying the proportion of reference germline insertion reads, non-reference L1 insertions reads, and non-L1 non-reference reads. The x axis represents the alignment score for the first 30 bp of read 2 when aligned to hg19. The y axis represents the alignment score for the first 30 bp of read 2 when aligned to Alu/L1 consensus. Reference insertions have high alignment scores to L1 and hg19 (*). Non-reference insertions have high alignment scores to L1 and low alignment scores to hg19 (arrow). Alignment score parameters are +1 for match, -1 for mismatch, -5 for gap opening, -1 for gap extension, and 0 for terminal gaps.
Supplementary Figure 3 The random forest classifier achieves accuracy identifying L1 insertions.
(A,B) Performance of each classifier for the specified library, trained on 75% of the known non-reference germline insertion loci and tested on the remaining 25% of loci. (A) All classifiers have a high precision, high positive predictive value, and a high fraction of loci predicted to be insertions that are true KNRGL insertions, as indicated by boxplots for all libraries. (B) All classifiers have above a median of 50% recall, that is, sensitivity of the fraction of known insertions that are predicted to be insertions. (C) Average feature importance scores for the five highest-ranking features for the L1 classifiers.
Supplementary Figure 4 Sensitivity of the digital PCR assay for the Chr. 7: 45646250 variant.
(A) Single cell amplified DNA was mixed with a bulk human DNA lacking the chr7 variant in the specified amounts in a multiplexed digital PCR assay. An assay specific for the 3’ junction of L1 Chr7:45646250 variant (detected in VIC) was performed in multiplex with an RPP30 single copy human control region (detected in FAM). Amplitude of detected fluorescence for each singular drop are reported on the Y axis by a single dot. Individual wells are separated by a yellow line. (B) The amount of L1 detected in each reaction normalized to the concentration of RPP30 detected, with the single cell set to 100%.
Supplementary Figure 5 Sensitivity of the L1-flanking PCR validation on single-cell DNA.
The flanking PCR validation assay for L1 insertions has approximately 80% sensitivity in single cells. Flanking PCR assays were performed on genomic DNA and amplified single cell DNA for specified polymorphic germline non-reference L1 loci identified in individual 5125 and absent from individual 1079. Single cells with ≥5 L1 junction reads at the specified loci were randomly selected. Red arrows indicate the insertion alleles; stars indicate the empty alleles. Ten nanograms of DNA were used for each PCR reaction.
Supplementary Figure 6 Comparison of SLAV-seq with two recent L1-targeted sequencing methods.
(A) Performing L1 capture coupled to library preparation with biotinylated primers leads to more purity in and less waste of L1 DNA. (B) Paired-end libraries allow the confirmation of presence of L1 sequence in one end, making sure the reads come from a DNA fragment actually containing L1 and not a miscapture. (C) The physical fragmentation of DNA generates molecules starting at virtually any position, whereas the use of a pool of primers limits them to the sites that match the primer sequening. Since PCR of the DNA is performed during library preparation, if two reads start at the same position, it is impossible to distinguish if they come from different fragments in the original material or if they result from PCR amplification. By generating many read starts we know for sure these are not PCR duplicates. This reflects on the number of unique read start positions detected by each method (D). In comparison with the other two studies, SLAV-Seq is capable of getting a much higher number of unique read starts per KNRGL locus, which allow us to identify somatic insertions with higher confidence. In addition, the combination of having a higher number of unique reads per locus and the fact that they are paired end enables us to extract a rich set of features and use a machine learning-based classifier to better distinguish true somatic L1 events from technical artifacts. (E) Denotes the type of single cell whole genome amplification that was performed. Multiple Displacement amplification (MDA) or Multiple Annealing and Looping Based Amplification Cycles (MALBAC). (F) Compares the main features used for somatic L1 prediction.
Supplementary Figure 7 Normalized read counts for 500-kb bins on whole-genomic sequencing obtained from MDA-amplified DNA from 3 single cells.
Normalized counts were calculated with the Ginkgo program (Garvin, T. et al. (2015) Nature methods 12, 1058-1060)
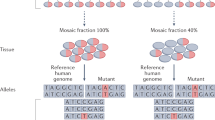
Supplementary Figure 8 SLAVs are composed of two classes of variants.
For somatic L1 insertions, a germline-inherited LINE-1 sequence is transcribed into RNA. The L1 endonuclease and reverse transcriptase protein nicks the genomic DNA and reverse transcribes the L1 RNA, resulting in the insertion of a new copy of Line-1 sequence. For retrotransposition-independent SLAVs, L1 endonuclease preferentially cuts a a germline-inherited LINE-1 sequence and recombination with a downstream A microsatellite results in a microhomology-mediated deletion. The A microsatellite regions may be nicked by the L1 endonuclease or a fragile site within the genome of neural progenitor cells.
Supplementary Figure 9 Display of full-length gel images of cropped gel images which appear in main text.
All ladders are 1Kb+ DNA ladder from Biorad. A. Full length gel associated to figure 2A left. B. Full length gel associated to figure 2A right. C. Full length gel associated to figure 2B left. D. Full length gel associated to figure 2B right. E. Full length gel associated to figure 4A. F. Full length gel associated to figure 4B.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–9 (PDF 1735 kb)
Supplementary Methods Checklist
Supplementary Methods Checklist (PDF 417 kb)
Supplementary Table 1
List of samples (CSV 7 kb)
Supplementary Table 2
List of features used for Random Forest classifiers (CSV 4 kb)
Supplementary Table 3
List of germline insertion loci and PCR validation information (XLSX 67 kb)
Supplementary Table 4
Counts of single cell SLAVs per cell with and without 10-kb filter (CSV 5 kb)
Supplementary Table 5
Table of singe cell SLAV events, PCR validation and Sanger sequencing information (XLSX 103 kb)
Supplementary Table 6
List of significantly differentially expressed genes in PWRN2 knockdown samples (XLSX 64 kb)
Rights and permissions
About this article

Cite this article
Erwin, J., Paquola, A., Singer, T. et al. L1-associated genomic regions are deleted in somatic cells of the healthy human brain. Nat Neurosci 19, 1583–1591 (2016). https://doi.org/10.1038/nn.4388
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nn.4388
This article is cited by
-
Proceedings of the inaugural Dark Genome Symposium: November 2022
Mobile DNA (2023)
-
Genomic data resources of the Brain Somatic Mosaicism Network for neuropsychiatric diseases
Scientific Data (2023)
-
Prevalence and mechanisms of somatic deletions in single human neurons during normal aging and in DNA repair disorders
Nature Communications (2022)
-
Genomic approaches to trace the history of human brain evolution with an emerging opportunity for transposon profiling of ancient humans
Mobile DNA (2021)
-
Cas9 targeted enrichment of mobile elements using nanopore sequencing
Nature Communications (2021)
