
Abstract
Proximal spinal muscular atrophy (SMA) is the predominant form of motor neuron disease in children and young adults. In contrast to other neurodegenerative disorders, SMA is a genetically homozygous autosomal recessive disease that is caused by deficiency of the survival motor neuron (SMN) protein. This homogeneity should in principle facilitate therapy development. Previous therapy approaches have focused on upregulation of SMN expression from a second SMN (SMN2) gene that gives rise to low amounts of functional SMN protein. Drug development to target disease-specific mechanisms at cellular and physiological levels is in its early stages, as the pathophysiological processes that underlie the main disease symptoms are still not fully understood. Mouse models have helped to make conceptual progress in the disease mechanism, but their suitability in the search for therapeutic agents remains to be validated—an issue that is ubiquitous to the translational therapeutic research of other neurodegenerative diseases. Human induced pluripotent stem cell technology for generation of large numbers of human motor neurons could help to fill this gap and advance the power of drug screening. In parallel, advances in oligonucleotide technologies for engineering SMN2 pre-mRNA splicing are approaching their first clinical trials, whose success depends on improved technologies for drug delivery to motor neurons. If this obstacle can be overcome, this could boost therapy development, not only for SMA but also for other neurodegenerative disorders.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others

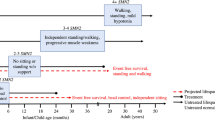
References
Valdmanis, P.N., Daoud, H., Dion, P.A. & Rouleau, G.A. Recent advances in the genetics of amyotrophic lateral sclerosis. Curr. Neurol. Neurosci. Rep. 9, 198–205 (2009).
Simpson, C.L. & Al-Chalabi, A. Amyotrophic lateral sclerosis as a complex genetic disease. Biochim. Biophys. Acta 1762, 973–985 (2006).
Crawford, T.O. & Pardo, C.A. The neurobiology of childhood spinal muscular atrophy. Neurobiol. Dis. 3, 97–110 (1996).
Burghes, A.H. & Beattie, C.E. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 10, 597–609 (2009).
Rossoll, W. et al. Specific interaction of Smn, the spinal muscular atrophy determining gene product, with hnRNP-R and gry-rbp/hnRNP-Q: a role for Smn in RNA processing in motor axons? Hum. Mol. Genet. 11, 93–105 (2002).
Wirth, B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum. Mutat. 15, 228–237 (2000).
Cartegni, L. & Krainer, A.R. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet. 30, 377–384 (2002).
Kashima, T. & Manley, J.L. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat. Genet. 34, 460–463 (2003).
Mattis, V.B. et al. Novel aminoglycosides increase SMN levels in spinal muscular atrophy fibroblasts. Hum. Genet. 120, 589–601 (2006).
Cho, S. & Dreyfuss, G. A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity. Genes Dev. 24, 438–442 (2010).
Schrank, B. et al. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc. Natl. Acad. Sci. USA 94, 9920–9925 (1997).
Jablonka, S., Schrank, B., Kralewski, M., Rossoll, W. & Sendtner, M. Reduced survival motor neuron (Smn) gene dose in mice leads to motor neuron degeneration: an animal model for spinal muscular atrophy type III. Hum. Mol. Genet. 9, 341–346 (2000).
Monani, U.R. et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn−/− mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet. 9, 333–339 (2000).
Rossoll, W. et al. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J. Cell Biol. 163, 801–812 (2003).
McWhorter, M.L., Monani, U.R., Burghes, A.H. & Beattie, C.E. Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J. Cell Biol. 162, 919–931 (2003).
Kong, L. et al. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J. Neurosci. 29, 842–851 (2009).
Jablonka, S., Beck, M., Lechner, B.D., Mayer, C. & Sendtner, M. Defective Ca2+ channel clustering in axon terminals disturbs excitability in motoneurons in spinal muscular atrophy. J. Cell Biol. 179, 139–149 (2007).
Le, T.T. et al. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 14, 845–857 (2005).
Jedrzejowska, M. et al. Unaffected patients with a homozygous absence of the SMN1 gene. Eur. J. Hum. Genet. 16, 930–934 (2008).
Jarecki, J. et al. Diverse small-molecule modulators of SMN expression found by high-throughput compound screening: early leads towards a therapeutic for spinal muscular atrophy. Hum. Mol. Genet. 14, 2003–2018 (2005).
Marks, P.A., Richon, V.M., Miller, T. & Kelly, W.K. Histone deacetylase inhibitors. Adv. Cancer Res. 91, 137–168 (2004).
Chang, J.G. et al. Treatment of spinal muscular atrophy by sodium butyrate. Proc. Natl. Acad. Sci. USA 98, 9808–9813 (2001).
Sumner, C.J. et al. Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann. Neurol. 54, 647–654 (2003).
Avila, A.M. et al. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J. Clin. Invest. 117, 659–671 (2007).
Tsai, L.K., Tsai, M.S., Lin, T.B., Hwu, W.L. & Li, H. Establishing a standardized therapeutic testing protocol for spinal muscular atrophy. Neurobiol. Dis. 24, 286–295 (2006).
Swoboda, K.J. et al. Phase II open label study of valproic acid in spinal muscular atrophy. PLoS ONE 4, e5268 (2009).
Garbes, L. et al. LBH589 induces up to 10-fold SMN protein levels by several independent mechanisms and is effective even in cells from SMA patients non-responsive to valproate. Hum. Mol. Genet. 18, 3645–3658 (2009).
Butchbach, M.E. et al. Effects of 2,4-diaminoquinazoline derivatives on SMN expression and phenotype in a mouse model for spinal muscular atrophy. Hum. Mol. Genet. 19, 454–467 (2010).
Rak, K. et al. Valproic acid blocks excitability in SMA type I mouse motor neurons. Neurobiol. Dis. 36, 477–487 (2009).
Artsma-Rus, A. & van Ommen, G.J. Progress in therapeutic antisense applications for neuromuscular disorders. Eur. J. Hum. Genet. 18, 146–153 (2010).
Pan, W.H. & Clawson, G.A. Antisense applications for biological control. J. Cell. Biochem. 98, 14–35 (2006).
van Deutekom, J.C. et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N. Engl. J. Med. 357, 2677–2686 (2007).
Goyenvalle, A. et al. Rescue of dystrophic muscle through U7 small nuclear RNA-mediated exon skipping. Science 306, 1796–1799 (2004).
Khoo, B. & Krainer, A.R. Splicing therapeutics in SMN2 and APOB. Curr. Opin. Mol. Ther. 11, 108–115 (2009).
Zaiss, A.K. & Muruve, D.A. Immunity to adeno-associated virus vectors in animals and humans: a continued challenge. Gene Ther. 15, 808–816 (2008).
Meyer, K. et al. Rescue of a severe mouse model for spinal muscular atrophy by U7 small nuclear RNA-mediated splicing modulation. Hum. Mol. Genet. 18, 546–555 (2009).
Hua, Y., Vickers, T.A., Okunola, H.L., Bennett, C.F. & Krainer, A.R. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet. 82, 834–848 (2008).
Singh, N.N., Singh, R.N. & Androphy, E.J. Modulating role of RNA structure in alternative splicing of a critical exon in the spinal muscular atrophy genes. Nucleic Acids Res. 35, 371–389 (2007).
Hua, Y., Vickers, T.A., Baker, B.F., Bennett, C.F. & Krainer, A.R. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 5, e73 (2007).
Ralph, G.S. et al. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat. Med. 11, 429–433 (2005).
Azzouz, M. et al. Lentivector-mediated SMN replacement in a mouse model of spinal muscular atrophy. J. Clin. Invest. 114, 1726–1731 (2004).
Towne, C., Raoul, C., Schneider, B.L. & Aebischer, P. Systemic AAV6 delivery mediating RNA interference against SOD1: neuromuscular transduction does not alter disease progression in fALS mice. Mol. Ther. 16, 1018–1025 (2008).
Foust, K.D. et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 28, 271–274 (2010).
Passini, M.A. et al. CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J. Clin. Invest. 120, 1253–1264 (2010).
Towne, C., Schneider, B.L., Kieran, D., Redmond, D.E. Jr. & Aebischer, P. Efficient transduction of non-human primate motor neurons after intramuscular delivery of recombinant AAV serotype 6. Gene Ther. 17, 141–146 (2010).
Foust, K.D. et al. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 27, 59–65 (2009).
Thoenen, H. & Sendtner, M. Neurotrophins: from enthusiastic expectations through sobering experiences to rational therapeutic approaches. Nat. Neurosci. 5 (suppl.), 1046–1050 (2002).
Storkebaum, E., Lambrechts, D. & Carmeliet, P. VEGF: once regarded as a specific angiogenic factor, now implicated in neuroprotection. Bioessays 26, 943–954 (2004).
Dimos, J.T. et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 321, 1218–1221 (2008).
Ebert, A.D. et al. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 457, 277–280 (2009).
Acknowledgements
I thank R. Blum and R. Götz for critical reading and many helpful comments. Work in my laboratory on spinal muscular atrophy was supported by the SMA Foundation, the Hermann und Lilly Schilling Stiftung im Stifterverband der Deutschen Industrie and the Deutsche Forschungsgemeinschaft, grant SFB 581, B1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declares no competing financial interests.
Rights and permissions
About this article
Cite this article
Sendtner, M. Therapy development in spinal muscular atrophy. Nat Neurosci 13, 795–799 (2010). https://doi.org/10.1038/nn.2565
Published:
Issue Date:
DOI: https://doi.org/10.1038/nn.2565
This article is cited by
-
Induced Pluripotent Stem Cells for Disease Modeling and Drug Discovery in Neurodegenerative Diseases
Molecular Neurobiology (2015)
-
The changing scene of amyotrophic lateral sclerosis
Nature Reviews Neuroscience (2013)
-
A new postal code for dendritic mRNA transport in neurons
EMBO reports (2011)
