
Abstract
Eukaryotic microbial pathogens are major contributors to illness and death globally. Although much of their impact can be controlled by drug therapy as with prokaryotic microorganisms, the emergence of drug resistance has threatened these treatment efforts. Here, we discuss the challenges posed by eukaryotic microbial pathogens and how these are similar to, or differ from, the challenges of prokaryotic antibiotic resistance. The therapies used for several major eukaryotic microorganisms are then detailed, and the mechanisms that they have evolved to overcome these therapies are described. The rapid emergence of resistance and the restricted pipeline of new drug therapies pose considerable risks to global health and are particularly acute in the developing world. Nonetheless, we detail how the integration of new technology, biological understanding, epidemiology and evolutionary analysis can help sustain existing therapies, anticipate the emergence of resistance or optimize the deployment of new therapies.
Similar content being viewed by others


The identification and use of antibiotics are some of the great medical achievements of the twentieth century, saving countless lives by controlling the risk of infection from contagion, after injury, surgery or in immunosuppressed individuals. However, in only 80 years since the introduction of penicillin, resistance to a broad range of antibiotic drugs has become widespread, with the compounded risk from multidrug-resistant bacterial infections severely limiting treatment options. This has created justified concern and global attention, not only in the medical community but also at government level, in the media and the general public1.
While predominantly applied to control prokaryotic microbial infections, the threat of disease from eukaryotic microorganisms has also been contained by therapeutic drugs — preventing or controlling disease caused by eukaryotic parasites and fungi in both a human and animal health setting. These represent some of the most important disease-causing agents (Table 1), particularly in the tropics, where the distribution of a pathogen is frequently linked to the distribution of the arthropods that act as disease vectors. Such vector-borne parasites include malaria (Plasmodium spp.) and kinetoplastid parasites (Trypanosoma cruzi, which causes Chagas disease; Trypanosoma brucei gambiense and T. b. rhodesiense, which cause human African trypanosomiasis (HAT); and 17 Leishmania spp., which cause a variety of cutaneous and visceral diseases). Other clinically important protozoan parasite species not considered here are transmitted either orally (Toxoplasma, Giardia and Entamoeba) or venereally (Trichomonas). Distinct from the many obligate eukaryotic unicellular parasites, opportunistic fungal pathogens are global in distribution and include Candida, Aspergillus spp., Cryptococcus and Pneumocystis spp.
The control of these eukaryotic pathogens has often involved therapies whose development predates the use of penicillin and which, in some cases, have unacceptable toxicity profiles2. Nonetheless, as with the rise of antimicrobial resistance in bacteria, resistance has emerged or is emerging against therapies targeting these eukaryotic microorganisms, with potentially devastating consequences for exposed populations. This has received far less attention than antibacterial resistance, despite some commonality in its underlying causes. Here, we detail how the control of eukaryotic microorganisms poses both similar and distinct challenges to that of bacterial pathogens, the drugs used to combat these pathogens, and the resistance mechanisms they are evolving. Finally, we discuss how the latest methodological approaches can anticipate the emergence of drug resistance and support new therapeutic approaches, either through the development of new drugs, the maintenance of existing therapies or through the use of alternative approaches to limit the spread of drug resistance.
Common challenges for the control of microbial pathogens
The challenges of controlling eukaryotic microbial pathogens share many similarities with bacterial infections. Both replicate more rapidly than their hosts, such that resistance can be selected in a relatively short timescale in a treated host population. This is exacerbated by inappropriate treatment profiles, leading to sub-curative exposure in the context of infection3. Problems of suboptimal dosing are particularly acute when applied to tropical parasites. For example, in the case of antimalarial therapies, up to 35% of drugs may be of low quality, or have packaging and labelling that is poor or may be falsified4. The lower than optimal concentrations of the active agent found in these low-quality antimalarial drugs can rapidly select for resistance in exposed populations, as can under-dosing resulting from self-prescription. For zoonoses, parasite selection in livestock populations treated with antimicrobials in a context in which there is poor supply chain management, fraudulent provision or cost barriers to optimal dosing can also lead to the emergence of resistance. For African trypanosomes, this represents a considerable threat, since up to 50 million doses of trypanocides are used in sub-Saharan livestock annually, mainly as a prophylactic, and trypanocides represent 45% of animal health costs. Mirroring the situation with antibiotic use in veterinary contexts for bacterial infections, in which environmental contamination has generated substantial regulatory concern5, the agricultural use of fungicides might also contribute to the selection of azole-resistant Aspergillus fumigatus6.
A further similarity between bacterial and eukaryotic microbial pathogens is the phenomenon of persister populations7. Persisters are a subpopulation of pathogen that survives after exposure to a chemotherapeutic agent (or vaccine). They can then re-establish patent infection while remaining drug sensitive (reviewed in ref. 8). The state of persistence is not heritable and resistance is not due to genetic alterations directly linked to rendering a drug ineffective. Rather, persistence is a physiological state in which the pathogen actively responds to drug assault. Persistence ensures incidental survival but does not future-proof a pathogen as genetically heritable resistance would. However, the combination of a persister population and suboptimal drug dosage could provide a reservoir from which resistance can emerge and may even provide a population predisposed to evolve resistance more readily. An example of this relating to parasite dormancy is the resistance of Plasmodium falciparum to artemisinin (and other antimalarials such as mefloquine and atovaquone), which was first characterized by degrees of persistence followed by the emergence of genomic changes now causally associated with resistance (see later). Similarly, fungal infections (for instance with Candida albicans) that are associated with biofilms are a good example of persister populations analogous to those found in bacterial communities9–11. The duration of persistence can range from days (P. falciparum) to lifelong (for example, C. albicans). Mechanisms of persistence vary — persistence may emerge spontaneously, possibly through stochastic changes in gene expression that prepare a population of pathogens for survival in varying environmental conditions (‘bet hedging’). This is best described in bacteria12 but is a phenomenon recently characterized in P. falciparum13. Furthermore, environmental signals may induce persistence, such as the nutrient starvation typically encountered by C. albicans in biofilms9,14.
Distinct challenges for the control of microbial pathogens
Although bacterial and eukaryotic microorganisms share common features with respect to their responses to drug exposure, there are also differences, with some challenges specific to the control of eukaryotic pathogens. First, eukaryotic microorganisms are more similar to their hosts than prokaryotic pathogens in terms of their biochemistry and metabolism, genetic composition, cell architecture and biology. Consequently, drugs targeting eukaryotic microorganisms must focus on differences from the eukaryotic norm, or particular specialisms of each pathogen group. This can restrict the cross-specificity of drugs to a point where there are distinctions in sensitivity between different apicomplexans (malaria, toxoplasma) or between the evolutionarily divergent trypanosomes, T. brucei spp. and T. cruzi. Comprising a different evolutionary kingdom, fungi have many differences from other eukaryotic microbial pathogens, again necessitating drugs to be developed for, and targeted to, a particular pathogen. This increases the challenges for drug development and inevitably constricts the new drug pipeline.
Second, many eukaryotic microbial pathogens have evolved a parasitic lifestyle distinct from the opportunistic infections characteristic of most bacterial pathogens (but also fungi). The evolution of parasitism is often accompanied by the development of sophisticated immune evasion mechanisms, which increases the impact of the persister phenotypes described earlier. Specifically, bacteriostatic drugs can operate to clear infection in concert with the immune system15. However, drugs that generate cytostatic rather than cytocidal responses to infection with an immunosuppressive parasite can lead to recrudescence upon the removal of drug exposure. This, in turn, can predispose the population to selection for drug resistance. Similarly, adaptation to an intracellular lifestyle or particular body niche can protect parasites from drug exposure, a feature shared with some bacterial pathogens that have evolved to survive in cells rather than systemically (Legionella, Mycobacteria).
A third challenge relates to clinical diagnosis and screening for drug resistance in eukaryotic microbial pathogens16. In bacterial infections, screening for sensitivity to antibiotics is straightforward and routine. By contrast, eukaryotic parasites can require highly specialized growth media and considerable growth periods to determine their susceptibility or otherwise to potential drug therapies. Also, unlike bacterial susceptibility testing, in which a minimum inhibitory concentration (MIC) is determined, most parasitologists report half-maximum effective concentration (EC50) values without providing the Hill slope of the growth inhibition curve or calculating the 90% effective concentration (EC90) value. It is perfectly possible to obtain a resistant line with an identical EC50 to the susceptible isolate, yet that is still resistant owing to a shallower Hill slope. As a consequence, clinical diagnosis and the selection of appropriate clinical management can be slow or practically impossible in the context of all but the most specialized laboratories.
A fourth distinction from common bacterial infections is the economic challenge of treating diseases of the developing world. Diseases such as malaria, trypanosomiasis, leishmaniasis and cryptococcosis are common in the poorest parts of the world, where the economic capacity to develop or deliver treatments is very limited, often restricted to philanthropic and charitable donations or the concerted actions of intergovernmental agencies. This makes the threat of drug resistance even more acute since there is no financial incentive to develop new drugs to replace those to which resistance emerges. Nonetheless, some pharmaceutical companies are increasingly engaged in public–private partnerships, providing access to chemical compound collections and other resources helpful for discovering and developing new drugs for neglected tropical diseases. Excellent examples of this collaborative spirit include the Medicines for Malaria Venture (http://www.mmv.org/), the Drugs for Neglected Diseases initiative (http://www.dndi.org/) and the Tres Cantos Open Lab Foundation (http://www.openlabfoundation.org/).
One route to limit the impact of drug resistance has been the exploitation of combination therapies for parasitic infections. This approach has proved useful for cancer therapy as well as for the treatment of tuberculosis, leprosy and viral infections such as HIV. Results have also been encouraging for parasitic infections; for example, through artemisinin combination therapy (ACT)17,18 to limit the emergence and spread of artemisinin-resistant malaria, and for trypanosomes, for which nifurtimox–eflornithine combination therapy19 is proving more robust than eflornithine-based therapy alone. However, combination therapies for parasitic diseases require the availability of more than one effective drug or drug class, which is not always the case. Moreover, combination therapies have often been embraced only when resistance has already been detected to one of the frontline monotherapies, allowing multidrug-resistant parasites to be selected. In a situation like this, the use of drug combinations with different pharmacokinetics in plasma, as with artemisinin and piperaquine, can limit the emergence of resistance20. However, the cost of drugs for many parasites seen in the developing world can generate geographical discrepancies in the use of mono and combination therapies. In this scenario, the efficacy of combination therapies can be threatened by ingression of resistant parasites selected under monotherapy.
The final challenge for eukaryotic microorganisms that differs from many prokaryotic and viral pathogens has been the failure to formulate and use effective vaccines to prevent infection21. Malaria research has focused intensively on vaccine development without transformative success, whereas for African trypanosomes the immune evasion mechanism used by the parasite (antigenic variation) effectively renders vaccine approaches impossible. It has also proved challenging to produce safe, effective vaccines for other kinetoplastids, despite the widespread early use of ‘leishmanization’ for the cutaneous form of leishmaniasis, which has the risk of virulence in some individuals and immunosuppression22. Fungal pathogens have their greatest impact in immunocompromised individuals, rendering vaccines potentially less useful. At present there are no licenced fungal vaccines; nonetheless, there are promising developments for adhesion-like substance 3 (Als3)- and secreted aspartic protease 2 (Sap2)-based vaccines, although concerns have been raised over their univalency and the potential for C. albicans to circumvent their efficacy23.
Drugs and resistance mechanisms
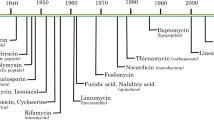
Microorganisms have evolved numerous strategies to counteract cellular toxicity induced by diverse chemical stresses (xenobiotics, metals, reactive oxygen and reactive nitrogen species, and so on). Many of these generic defences have been co-opted for drug resistance. Figure 1 summarizes the major therapeutic agents used to target malaria, kinetoplastid parasites and fungi, highlighting the dates of introduction and the appearance of resistance for each. The principal methods of resistance (Fig. 2) involve either reduction of the free drug level at the target site of action, alterations in the drug target, reducing its drug-binding affinity, or overexpression of the target, restoring its essential function. In the case of inhibition of a metabolic pathway, the essential end product can be produced either by induction of an alternative pathway or by upregulation of a salvage pathway to obtain an essential metabolite from the host. Downstream consequences of target inhibition include damage to DNA, proteins and lipids such that upregulation of repair pathways can also contribute to resistance. Unlike bacteria, acquisition of resistance genes by lateral gene transfer on plasmids has not been observed for protozoan parasites or fungal pathogens. In Table 2 we summarize the drugs used to treat eukaryotic microbial pathogens, their mode of action and mechanisms of resistance where known. Here, we highlight specific examples in which drug resistance or the threat of resistance challenges current control efforts.

The solid bars represents the time from first widespread clinical use to the first year drug resistance was suspected or confirmed. The shading gradient indicates that certain drugs are still in use for particular indications or in specific geographical locations. NECT, nifurtimox–eflornithine combination therapy; VL visceral leishmaniasis. For fungal pathogens, insensitive or resistant strains have been identified shortly after the introduction of all of the major classes of antifungal agents. In the case of amphotericin B, there remains very little resistance — and differences in sensitivity mainly reflect the relative inherent sensitivity of different species to this agent.

Eukaryotic microbial pathogens can exhibit drug resistance through reducing the overall intracellular concentration of the drug (less uptake, more efflux), by inactivating or failing to activate the drug, or by sequestering the drug away from its target. Resistance can also be mediated by reducing affinity of the drug for the target by mutation or by reducing the drug effect by overexpression of the target. Salvage and bypass pathways can also lower the overall impact of the drug action, as can the activation of pathways to repair any damage caused.
Malaria. The most successful antimalarial in history to date has been chloroquine, a 4-aminoquinoline derivative of quinine (itself the world's first mass-distributed antimalarial), first synthesized in 1934 (ref. 24). Chloroquine was cheap and remained effective for decades. However, due to massive overuse and suboptimal compliance, resistance to chloroquine emerged in Southeast Asia in 1957 and in South America in 1960, and — by the mid-1980s — it was barely possible to use even in Africa25. Although disputed by some26, the leading candidate for resistance to chloroquine is the P. falciparum chloroquine resistance transporter PfCRT (ref. 27).However, despite reports that PfCRT functions as a chloride channel, a proton pump, an activator of Na+/H+ exchangers or a cation channel, the physiological function of PfCRT remains unclear28. Nonetheless, PfCRT is central to much antimalarial resistance, the precise profile of which is modulated by associated mutations in other genes.
Artemisinin and its derivatives are fast-acting but short-lived antimalarials that have been globally successful. In particular, ACTs (for example, artemether–lumefantrine, artesunate–amodiaquine and dihydroartemisinin–piperaquine) were recommended by the World Health Organization in 2001 to ensure high cure rates of falciparum malaria and to reduce the spread of drug resistance to other frontline drugs. However, clinical resistance was confirmed in 2008 (ref. 29), characterized by a failure to clear parasites rapidly in patients around the Thai–Cambodian border30,31. Resistant parasites were characterized by transcriptomics32, large-scale whole-genome sequencing (WGS) of clinical isolates33,34 and classical generation of resistant mutants by in vitro culture followed by WGS (ref. 35). This pinpointed multiple independent mutations in a gene encoding a Kelch propeller protein (KELCH13), which was then causally linked to resistance by reverse genetics36,37. Large-scale genomic epidemiological evidence suggests that artemisinin resistance is not as straightforward as the simple acquisition of mutations in kelch13. Indeed, nonsynonymous mutations in ferredoxin, apicoplast ribosomal protein S10, multidrug resistance protein 2 and PfCRT also showed strong associations with artemisinin resistance29. These mutations appear to act as markers of a genetic landscape upon which artemisinin-resistance-conferring kelch13 mutations are more likely to occur. These landscape mutations also correlate with the current geographical limits of artemisinin resistance29. This concept is further supported by additional genomic epidemiological evidence that demonstrates that many of the 20 or so mutations in kelch13 that have been implicated in the Southeast Asian manifestation of artemisinin resistance are also found in African P. falciparum isolates. However, these mutations are present at no greater frequency in the African strains than other P. falciparum genes, indicating a lack of selective pressure in that continent and that these strains lack the enabling genetic background observed in Southeast Asia38.
Kelch propeller domain proteins are subcellular organizers of multiprotein complexes, and indeed artemisinin-resistance-associated mutation of KELCH13 results in its reduced association with phosphatidylinositol-3-OH kinase (PI(3)K)39. Experimental overexpression of PI(3)K results in enhanced artemisinin resistance, and phosphatidylinositol 3-phosphate (PI(3)P) levels are predictive of resistance to artemisinin39. In addition, upregulation of the chaperonin complexes PROSC and TRiC, involved in the unfolded protein stress response in other eukaryotes, may contribute to artemisinin resistance32. Worryingly, resistance to some of the various ACT regimens (involving lumefantrine, amodiaquine and PfCRT) is becoming evident40–43. However, the framework for the rapid evaluation of genome evolution in the face of drugs is in place and will hopefully swiftly indicate any further potential mechanisms.
Human African trypanosomiasis. The vast majority of reported cases of HAT are caused by T. b. gambiense, with less than 2% caused by T. b. rhodesiense44. Treatment involves either pentamidine or suramin for stage 1 infection (before central nervous system involvement), whereas melarsoprol, eflornithine or nifurtimox/eflornithine combination therapy are used once the parasite crosses the blood–brain barrier2, with the combination therapy reducing the duration of treatment regimens. Given the limited chemotherapeutic options for the treatment of HAT (Table 2), drug resistance could seriously compromise efforts to eliminate this epidemic disease as a public health problem44. Fortunately, resistance emergence for pentamidine has not been marked, despite continuous use of pentamidine since the 1940s, including a mass chemoprophylactic campaign in the 1950s in the then Belgian Congo. However, cross-resistance to pentamidine and melarsoprol, used for stage 2 of infection, is frequently observed. Melarsoprol is a trivalent melaminophenyl arsenical that has a propensity to react covalently with vicinal dithiols, including the parasite-specific dithiol, trypanothione45, to form a cyclic complex known as MelT (ref. 46). Melarsoprol has a high incidence of severe (lethal) toxicities and high rates of treatment failures have been reported in the Democratic Republic of Congo, Uganda, Angola and Sudan2. Although therapeutic failure does not necessarily equate with drug resistance, it appears that the high relapse rate in northwest Uganda is associated with reduced susceptibility to melarsoprol47,48. The recent report that the aquaglyceroporin AQP2 appears to function as a transporter for large drugs such as pentamidine and melarsoprol was surprising, given that aquaglyceroporins are channels facilitating the passive transport of water and small neutral molecules across cell membranes. Nonetheless, there is strong evidence that AQP2 is synonymous with the high-affinity pentamidine transporter (HAPT1)49, with a recent report indicating that pentamidine binds and inhibits the transporter and is then internalized via endocytosis50.
Chagas disease. For Trypanosoma cruzi, an intracellular parasite with a wide tissue tropism, infection has three phases: an acute phase associated with high parasitaemia; an asymptomatic (indeterminate) phase lasting anywhere between 10 to 30 years, during which parasitaemia is controlled by the immune response; and a chronic phase in about 30–40% of patients characterized by either cardiac disease or digestive disease (mega-oesophagus and mega-colon). For treatment, benznidazole and nifurtimox have considerable activity in the acute phase51 and benznidazole also eliminates parasitaemia in the indeterminate and chronic phases of the disease52,53. However, a large multi-centre, randomized trial of benznidazole for chronic Chagas cardiomyopathy failed to significantly reduce cardiac clinical deterioration at up to 5 years follow-up53. Whether this is due to differences in drug susceptibility, pharmacokinetic/pharmacodynamic issues or the pathophysiology of the disease is not known. The results of two recent clinical trials with azole ergosterol inhibitors, posaconazole and E1224 (a pro-drug of ravuconazole) have been equally disappointing52,54,55.
Visceral leishmaniasis. Treatment of visceral leishmaniasis, cutaneous and mucocutaneous leishmaniasis is limited to four main drugs: pentavalent antimonial complexes (sodium stibogluconate and meglumine antimonate); amphotericin B (as deoxycholate or liposomal formulations); the aminoglycoside paromomycin; and the alkylphosphocholine miltefosine56,57. Treatment varies according to geographical location, the immune status and other co-morbidities of the patient, and the disease classification58.
Of these treatments, widespread resistance to antimonial drugs is specific to South Asia and is not seen in sub-Saharan Africa or Brazil. Indeed, antimonial drugs are not recommended in India or Nepal owing to treatment failures starting in the 1990s and now reported to be as high as 60% in some regions59. This has been attributed to inappropriate treatment in an unregulated private health system or to the use of substandard antimonial drugs. However, South Asia is the only region where arsenic exposure and widespread antimonial resistance co-exist. Thus, environmental pollution and exposure of patients to arsenic in food and drinking water was proposed as an alternative hypothesis for the high rates of resistance60. Arsenic and antimony are both metalloids and selection of leishmania parasites for resistance to trivalent arsenic results in cross-resistance to trivalent antimony in vitro61, but its physiological relevance is uncertain. Chronic exposure of infected mice to arsenic in drinking water at environmentally relevant levels showed that it is possible to generate resistance to pentavalent antimony in vivo62. A retrospective clinico-epidemiological study identified a trend towards increased treatment failure in arsenic-exposed patients, but failed to reach statistical significance63.
Resistance to antimonials is multifactorial, and most of the mechanisms shown in Fig. 2 have been implicated in Leishmania. Studies on experimental and clinical resistant isolates strongly support the hypothesis that trypanothione has a pivotal role in antimonial resistance. However, none of the following mechanisms are universal in resistant isolates. Decreased biological reduction of SbV to SbIII has been reported in resistant leishmania amastigotes64 and two candidate ‘antimony reductases’ have been identified, although genetic65,66 and proteomic studies67,68 have not identified any changes in either TDR1 (ref. 69) or ArsC (ref. 70). The mechanism of uptake of SbV is not known, but modulation of expression of AQP1 affects SbIII susceptibility71–73. AQP1 copy number and expression levels correlate with susceptibility to SbIII in some, but not all, clinical isolates74,75. However, interpretation of this observation is complicated by the fact that AQP1 is located on chromosome 31, which is frequently trisomic or tetrasomic76 in these mosaic aneuploid parasites77. Upregulation of trypanothione and ancillary biosynthetic pathways has also been observed in genomic65,66 and metabolomic78,79 studies. MRPA is responsible for ATP-dependent efflux of SbIII as a thiol conjugate into membrane vesicles80 and a homodimeric ABC half-transporter (ABCI4) is one possible candidate for efflux across the plasma membrane81.
Miltefosine, the only oral treatment for visceral leishmaniasis, was first approved for use in India in 2002. However, a decade on there is an increasing rate of clinical relapse82,83, which threatens to undermine the Kala-Azar Elimination Program in the Indian subcontinent. Stable resistance is readily generated in the laboratory with no cross-resistance to other anti-leishmanial drugs84,85.
Fungi. Several classes of antifungals are used clinically (Tables 1 and 2) — each with very different drug resistance profiles. The oldest antifungals are the polyene macrolide antibiotics, exemplified by amphotericin B, which remains a frontline choice for a broad-spectrum agent for fungal infections of unknown aetiology. Amphotericin deoxycholate is toxic to the kidneys, a side effect that is substantially ameliorated in lipid carrier formulations such as AmBisome, which also has potent anti-Leishmania activity. As with other eukaryotic pathogens, resistance to antifungal drugs has become an increasingly important clinical problem86,87. A few recognized cases exist of inherent resistance of certain fungi to specific antifungals, but mostly resistance is due to induced changes and mutations.
The imidazoles and more modern triazoles (collectively known as the ‘azoles’) constitute the main class of antifungals used in the treatment of infections. Various modifications of the triazole ring have generated a series of antifungals including fluconazole (used mainly in the treatment of Candida infections), and itraconazole, voriconazole, posaconazole, ravuconazole and the recently licenced isavuconazole, which have improved activity against Aspergillus and filamentous fungal species. These compounds have important differences in antifungal potencies, spectrum of activities, bioavailability, drug interactions and potential toxicity. For example, some patients treated with voriconazole suffer from photosensitivity and an elevated risk of skin carcinoma88. Other sterol inhibitors include the allylamines squalene epoxidase inhibitors and phenylmorpholine Erg24 D14 reductase and Erg2 D8-D7 isomerase inhibitors that are used topically against dermatophytic infections for which clinical resistance is low.
Although some fungi such as Candida krusei are inherently azole resistant, multiply triazole-resistant strains are now emerging89,90, as well as strains with cross-resistance to azoles and echinocandins, suggesting the emergence of concerning multidrug-resistant phenotypes in medically important fungi91. A threat from multi-azole-resistant strains of A. fumigatus may have arisen under the selective pressure of agricultural azole fungicides and subsequent transmission of azole-resistant strains to the clinic by spore dispersal92–95. The prevalence of these alleles is increasing in Europe and now in other parts of the world90,96,97. In Candida, mutants harbouring azole resistance have a fitness deficit98; however, multidrug-resistant strains of Aspergillus do not seem to have markedly decreased fitness, implying that they may become stably represented in the environment.
The most recently developed major class of antifungal is the echinocandin antibiotics, of which caspofungin, micafungin and anidulafungin are used clinically. These have similar pharmacokinetic properties, although a new echinocandin (CD101; formerly biofungin) is in clinical trials and has improved stability in vivo and requires less frequent intravenous dosing. Echinocandins are fungicidal against Candida species and fungistatic or fungicidal against Aspergillus, causing hyphal or bud tip lysis, but they are not efficacious against Pneumocystis jiroveci and some other species.
Hsp90-mediated changes in drug tolerance have also been implicated in determining echinocandin sensitivity99. Recently, multidrug azole/echinocandin resistance has been identified in fungi and this is particularly frequent in strains of C. glabrata, which is common in patients with haematological malignancies and solid tumours100,101. These multidrug-resistant strains of C. glabrata can then only be treated with intravenous amphotericin, and since this agent has poor penetration into urine such infections are essentially untreatable.
Outstanding challenges and future prospects
We began by highlighting the similarities and differences between the emergence of drug resistance in prokaryotic and eukaryotic microorganisms. The control of emerging drug resistance in eukaryotic microbial pathogens also has similarities and distinctions from prokaryotic drug resistance, and the challenge for the future is to ensure best practice is employed for both groups. One effective mechanism to control the spread of drug resistance in bacterial pathogens is the application of appropriate antibiotic stewardship, applying the right drug at the right dose, at the right time, for the right duration. This approach operates effectively where there is well-regulated healthcare, effective and rapid screening, a selection of available drugs as contingency, and the necessary education and engagement between the patient and healthcare provider. Moreover, bacterial drug resistance is a global phenomenon in which resistance selected through poor stewardship in one geographical area may be contained by stringent practices in other areas, or combatted by an investment in new pharmaceutical development in wealthy countries. These containment measures are inevitably less effective where primary care is limited or too expensive, education is lacking or where the diseases involved do not have a direct impact in the developed world. As a consequence, the limitation of many eukaryotic pathogens to the poorer parts of the world makes a coordinated response to resistance emergence more difficult to achieve.
The drivers of the emergence of resistance are also more difficult to mitigate for many eukaryotic pathogens. As highlighted earlier, drug provenance and effective delivery is a considerable challenge in the developing world. Delivery is a particular challenge for prospective mass drug administration programmes, in which delivery to a population on a broad or local scale, if incomplete, can counteract its intention to contain the spread of existing resistance in target regions. A further complication in low- and middle-income countries is the effects of co-infection or malnutrition in populations treated with drugs targeting a particular pathogen (discussed previously102). Notably, the pharmacokinetic behaviour of drugs in malnourished individuals may be variable and unpredictable, leading to inadvertent under-dosing, which can drive resistance. When combined with immunosuppression induced by many parasites, or the hospital-induced immunosuppression of patients that become susceptible to fungal infection, drug concentrations that would clear infections in the context of a robust immune system may fall short in its absence. The ecological balance between distinct pathogens in patients with co-infections can also lead to unanticipated consequences, in which the removal of one pathogen can create a niche exploited by a distinct pathogen, or in which the normal interactions between pathogens with each other, and with the immune system, is perturbed with drug pressure. The resistance mechanisms selected in drug-treated populations can also alter pathogen phenotypes with the risk of enhanced virulence.
Although the factors that drive drug resistance are well known, it remains essential to identify when drug resistance arises and to respond rapidly and effectively. As with health care, surveillance is a key challenge for diseases in the developing world, where populations may be inaccessible, reluctant to engage or where treatment failure can have multiple causes beyond the emergence of drug resistance. Moreover, resistance can show considerable variation among populations or in different geographical settings. Here, accurate and rapid detection is critical to understand resistance epidemiology and thereby the best treatment to deliver, but this can be difficult to achieve. Despite this, developments in field polymerase chain reaction (PCR) assays and next-generation sequencing permit the sensitive identification and tracking of emergent resistance, allowing earlier control responses than could be previously achieved. Hence, an integration of improved therapeutic delivery and treatment monitoring are critical control points to reduce the emergence of resistance, in tandem with the discovery of the relevant resistance mechanisms and the search for new drug therapies. These combined approaches span the individual scientific researcher to clinicians, to health agencies, governments and populations, all of which must be well integrated and alert, with effective and rapid communication between different levels to allow appropriate responses to be put into action if needed.
Fortunately, while drug resistance is emerging in many eukaryotic microbial pathogens, new tools and methodologies are being developed to (1) predict resistance mechanisms, (2) identify modes of drug action and potential escape pathways, and (3) understand pathogen biochemistry as a means of discovering possible new therapies. With respect to drug resistance, the advent of cost-effective and rapid genome resequencing allows signatures of selection to be identified103–106, while genome-wide RNA interference screens allow the mapping of resistance pathways107,108, and overexpression libraries109 can assist with drug target deconvolution through selective screens. These genetic tools are complemented by improvements in proteomics such that adaptations accompanying drug resistance can be pinpointed, providing information on resistance mechanisms, and potential diagnostic tools to detect the emergence of resistance110. Combined with the improved sensitivity and resolution of metabolomics analysis111, biochemical pathways can also be mapped in the context of drug exposure, allowing bypass mechanisms to be highlighted, if present. These each provide the essential early warning systems necessary to identify and combat the spread of drug resistance. Furthermore, certain combination therapies might offer novel transmission blocking strategies: very recently, resistance to the antimalarial atovaquone, a component (with proguanil) of the widely used and successful treatment marketed as Malarone, was further characterized. Resistance mutations that appear during the blood stage of infection localize to the mitochondrial protein cytochrome b, one of the few proteins encoded by the highly reduced Plasmodium mitochondrial genome. All atovaquone-resistance mutations examined generate a deficient mitochondrion and a parasite that, while viable in the blood, is incapable of development in the mosquito and thereby cannot be transmitted112. Thus, despite the fact that resistance to atovaquone might arise repeatedly, each incident is isolated. Drugs that target cytochrome b could form part of combination therapies that are self-limiting in terms of spread of drug resistance and may delay any transmission of resistance that arises to the drug it is partnered with.
Concluding remarks
Drug resistance in eukaryotic microorganisms is an increasing global problem that threatens the advances in health care made over the last 50 years. This mirrors the situation for bacterial and viral pathogens but is particularly acute given the abundance of eukaryotic pathogens in the poorest regions of the world. These countries have the least capacity to respond to the emergence of resistance through the development of new drugs, vaccines and diagnostics, while developed countries lack financial incentives to assist. Nonetheless, there are opportunities to respond to this threat owing to the distinct biology of many major eukaryotic pathogens, uncovered through basic research. Furthermore, many eukaryotic microorganisms are arthropod-borne diseases, such that targeting transmission can be a route to pathogen control not available for opportunistic pathogens. This can take the form of transmission-blocking vaccines or drugs targeting Plasmodium113, or the application of vector control measures such as insecticide-impregnated bed nets114, peri-domestic and indoor residual insecticide spraying114,115, tsetse traps116, or improved housing117. Sterile insect release is also a route to limiting the vector population and so restricting disease spread118,119. Eukaryotic microorganisms have also, like some bacterial pathogens, been found to show cooperative and social behaviours to optimize their establishment and transmission in their hosts or vectors120,121. These social responses can control parasite density or the development of transmission stages122–124, such that blocking or mimicking signals for communication or their transduction pathways provides new routes for limiting the impact of the pathogens using strategies that might be less susceptible to the emergence of resistance.
Whether or not new targets or approaches can be identified, there is a real need to optimize the delivery and deployment of drugs. Control of drug quality and distribution, and the supply of cost-effective drugs is crucial. Furthermore, the application of both epidemiological modelling and evolutionary theory to guide drug treatment policies is important in prolonging the lifespan of drugs and thereby maximizing the return on the considerable cost associated with developing and introducing a new drug. Targeted therapy as opposed to mass drug administration is key to limiting the emergence of resistance, or containing resistance when it is detected. This requires an integration of epidemiology, diagnosis, detection and supply chain control, as well as investment in a pipeline of new therapeutics ready to be deployed when resistance inevitably emerges. Only through slowing the emergence of resistance and accelerating new drug discovery will the control successes achieved against eukaryotic microbial pathogens be sustained.
References
Woolhouse, M. & Farrar, J. Policy: an intergovernmental panel on antimicrobial resistance. Nature 509, 555–557 (2014).
Lutje, V., Seixas, J. & Kennedy, A. Chemotherapy for second-stage human African trypanosomiasis. Cochrane. Database. Syst. Rev. 6, CD006201 (2013).
Fernandez, F. M., Green, M. D. & Newton, P. N. Prevalence and detection of counterfeit pharmaceuticals: a mini review. Ind. Eng. Chem. Res. 47, 585–590 (2008).
Nayyar, G. M., Breman, J. G., Newton, P. N. & Herrington, J. Poor-quality antimalarial drugs in southeast Asia and sub-Saharan Africa. Lancet Infect. Dis. 12, 488–496 (2012).
Woolhouse, M., Ward, M., van, B. B. & Farrar, J. Antimicrobial resistance in humans, livestock and the wider environment. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370, 20140083 (2015).
Verweij, P. E., Chowdhary, A., Melchers, W. J. & Meis, J. F. Azole resistance in Aspergillus fumigatus: can we retain the clinical use of mold-active antifungal azoles?. Clin. Infect. Dis. 62, 362–368 (2016).
Lewis, K. Persister cells, dormancy and infectious disease. Nature Rev. Microbiol. 5, 48–56 (2007).
Cohen, N. R., Lobritz, M. A. & Collins, J. J. Microbial persistence and the road to drug resistance. Cell Host. Microbe 13, 632–642 (2013).
LaFleur, M. D., Kumamoto, C. A. & Lewis, K. Candida albicans biofilms produce antifungal-tolerant persister cells. Antimicrob. Agents Chemother. 50, 3839–3846 (2006).
Kucharikova, S., Tournu, H., Lagrou, K., Van, D. P. & Bujdakova, H. Detailed comparison of Candida albicans and Candida glabrata biofilms under different conditions and their susceptibility to caspofungin and anidulafungin. J. Med. Microbiol. 60, 1261–1269 (2011).
LaFleur, M. D., Qi, Q. & Lewis, K. Patients with long-term oral carriage harbor high-persister mutants of Candida albicans. Antimicrob. Agents Chemother. 54, 39–44 (2010).
Kussell, E., Kishony, R., Balaban, N. Q. & Leibler, S. Bacterial persistence: a model of survival in changing environments. Genetics 169, 1807–1814 (2005).
Rovira-Graells, N. et al. Transcriptional variation in the malaria parasite Plasmodium falciparum. Genome Res. 22, 925–938 (2012).
Keren, I., Kaldalu, N., Spoering, A., Wang, Y. & Lewis, K. Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 230, 13–18 (2004).
Pankey, G. A. & Sabath, L. D. Clinical relevance of bacteriostatic versus bactericidal mechanisms of action in the treatment of Gram-positive bacterial infections. Clin. Infect. Dis. 38, 864–870 (2004).
Laufer, M. K., Djimde, A. A. & Plowe, C. V. Monitoring and deterring drug-resistant malaria in the era of combination therapy. Am. J. Trop. Med. Hyg. 77, 160–169 (2007).
Peters, W. The prevention of antimalarial drug resistance. Pharmacol. Ther. 47, 499–508 (1990).
White, N. J. & Olliaro, P. L. Strategies for the prevention of antimalarial drug resistance: rationale for combination chemotherapy for malaria. Parasitol. Today 12, 399–401 (1996).
Priotto, G. et al. Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: a multicentre, randomised, phase III, non-inferiority trial. Lancet 374, 56–64 (2009).
Nosten, F. & White, N. J. Artemisinin-based combination treatment of falciparum malaria. Am. J. Trop. Med. Hyg. 77, 181–192 (2007).
Hotez, P. J., Bottazzi, M. E. & Strych, U. New vaccines for the world's poorest people. Annu. Rev. Med. 67, 405–417 (2015).
Kumar, R. & Engwerda, C. Vaccines to prevent leishmaniasis. Clin. Transl. Immunology 3 e13 (2014).
Cassone, A. Development of vaccines for Candida albicans : fighting a skilled transformer. Nature Rev. Microbiol. 11, 884–891 (2013).
Wellems, T. E. & Plowe, C. V. Chloroquine-resistant malaria. J. Infect. Dis 184, 770–776 (2001).
Wongsrichanalai, C., Pickard, A. L., Wernsdorfer, W. H. & Meshnick, S. R. Epidemiology of drug-resistant malaria. Lancet Infect. Dis. 2, 209–218 (2002).
Awasthi, G. & Das, A. Genetics of chloroquine-resistant malaria: a haplotypic view. Mem. Inst. Oswaldo Cruz 108, 947–961 (2013).
Fidock, D. A. et al. Mutations in the P-falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol. Cell 6, 861–871 (2000).
Pulcini, S. et al. Mutations in the Plasmodium falciparum chloroquine resistance transporter, PfCRT, enlarge the parasite's food vacuole and alter drug sensitivities. Sci. Rep. 5, 14552 (2015).
Noedl, H. et al. Evidence of artemisinin-resistant malaria in western Cambodia. N. Engl. J. Med. 359, 2619–2620 (2008).
Amaratunga, C. et al. Artemisinin-resistant Plasmodium falciparum in Pursat province, western Cambodia: a parasite clearance rate study. Lancet Infect. Dis. 12, 851–858 (2012).
Dondorp, A. M. et al. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 361, 455–467 (2009).
Mok, S. et al. Drug resistance. Population transcriptomics of human malaria parasites reveals the mechanism of artemisinin resistance. Science 347, 431–435 (2015).
Miotto, O. et al. Multiple populations of artemisinin-resistant Plasmodium falciparum in Cambodia. Nature Genet. 45, 648–655 (2013).
Takala-Harrison, S. et al. Genetic loci associated with delayed clearance of Plasmodium falciparum following artemisinin treatment in Southeast Asia. Proc. Natl Acad. Sci. USA 110, 240–245 (2013).
Ariey, F. et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 505, 50–55 (2014).
Ghorbal, M. et al. Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nature Biotechnol. 32, 819–821 (2014).
Straimer, J. et al. Drug resistance. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science 347, 428–431 (2015).
MalariaGEN Plasmodium falciparum Community Project. Genomic epidemiology of artemisinin resistant malaria. Elife 5, e08714 (2016).
Mbengue, A. et al. A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature 520, 683–687 (2015).
Gabryszewski, S. J., Modchang, C., Musset, L., Chookajorn, T. & Fidock, D. A. Combinatorial genetic modeling of pfcrt-mediated drug resistance evolution in Plasmodium falciparum. Mol. Biol. Evol. (2016).
Sisowath, C. et al. In vivo selection of Plasmodium falciparum parasites carrying the chloroquine-susceptible pfcrt K76 allele after treatment with artemether-lumefantrine in Africa. J. Infect. Dis 199, 750–757 (2009).
Venkatesan, M. et al. Polymorphisms in Plasmodium falciparum chloroquine resistance transporter and multidrug resistance 1 genes: parasite risk factors that affect treatment outcomes for P. falciparum malaria after artemether-lumefantrine and artesunate-amodiaquine. Am. J. Trop. Med. Hyg. 91, 833–843 (2014).
Amaratunga, C. et al. Dihydroartemisinin-piperaquine resistance in Plasmodium falciparum malaria in Cambodia: a multisite prospective cohort study. Lancet Infect. Dis. 16, 357–365 (2016).
Franco, J. R., Simarro, P. P., Diarra, A., Ruiz-Postigo, J. A. & Jannin, J. G. The journey towards elimination of gambiense human African trypanosomiasis: not far, nor easy. Parasitology 141, 748–760 (2014).
Fairlamb, A. H., Blackburn, P., Ulrich, P., Chait, B. T. & Cerami, A. Trypanothione: a novel bis(glutathionyl)spermidine cofactor for glutathione reductase in trypanosomatids. Science 227, 1485–1487 (1985).
Fairlamb, A. H., Henderson, G. B. & Cerami, A. Trypanothione is the primary target for arsenical drugs against African trypanosomes. Proc. Natl Acad. Sci. USA 86, 2607–2611 (1989).
Matovu, E. et al. Melarsoprol refractory T.b. gambiense from Omugo, north-western Uganda. Trop. Med. Int. Health 6, 407–411 (2001).
Robays, J. et al. High failure rates of melarsoprol for sleeping sickness, Democratic Republic of Congo. Emerg. Infect. Dis. 14, 966–967 (2008).
Munday, J. C. et al. Trypanosoma brucei aquaglyceroporin 2 is a high-affinity transporter for pentamidine and melaminophenyl arsenic drugs and the main genetic determinant of resistance to these drugs. J. Antimicrob. Chemother. 69, 651–663 (2014).
Song, J. et al. Pentamidine is not a permeant but a nanomolar inhibitor of the Trypanosoma brucei aquaglyceroporin-2. PLoS Pathog. 12 e1005436 (2016).
Urbina, J. A. & Docampo, R. Specific chemotherapy of Chagas disease: controversies and advances. Trends Parasitol. 19, 495–501 (2003).
Molina, I. et al. Randomized trial of posaconazole and benznidazole for chronic Chagas' disease. N. Engl. J. Med. 370, 1899–1908 (2014).
Morillo, C. A. et al. Randomized trial of benznidazole for chronic Chagas' cardiomyopathy. N. Engl. J. Med. 373, 1295–1306 (2015).
Chatelain, E. Chagas disease drug discovery: toward a new era. J. Biomol. Screen. 20, 22–35 (2015).
Urbina, J. A. Recent clinical trials for the etiological treatment of chronic chagas disease: advances, challenges and perspectives. J. Euk. Microbiol. 62, 149–156 (2015).
McGwire, B. S. & Satoskar, A. R. Leishmaniasis: clinical syndromes and treatment. QJM 107, 7–14 (2014).
Sundar, S. & Chakravarty, J. An update on pharmacotherapy for leishmaniasis. Expert Opin. Pharmacother. 16, 237–252 (2015).
World Health Organization. Control of the Leishmaniases Technical Report Series 949 1–186 (World Health Organization, 2010).
Olliaro, P. L. et al. Treatment options for visceral leishmaniasis: a systematic review of clinical studies done in India, 1980–2004. Lancet Infect. Dis. 5, 763–774 (2005).
Perry, M. R. et al. Visceral leishmaniasis and arsenic: an ancient poison contributing to antimonial treatment failure in the Indian subcontinent? PLoS Negl. Trop. Dis. 5 e1227 (2011).
Dey, S. et al. High level arsenite resistance in Leishmania tarentolae is mediated by an active extrusion system. Mol. Biochem. Parasitol. 67, 49–57 (1994).
Perry, M. R., Wyllie, S., Raab, A., Feldmann, J. & Fairlamb, A. H. Chronic exposure to arsenic in drinking water can lead to resistance to antimonial drugs in a mouse model of visceral leishmaniasis. Proc. Natl Acad. Sci. USA 110, 19932–19937 (2013).
Perry, M. R. et al. Arsenic exposure and outcomes of antimonial treatment in visceral leishmaniasis patients in Bihar, India: a retrospective cohort study. PLoS Negl. Trop. Dis. 9 e0003518 (2015).
Shaked-Mishan, P., Ulrich, N., Ephros, M. & Zilberstein, D. Novel intracellular Sb-V reducing activity correlates with antimony susceptibility in Leishmania donovani. J. Biol. Chem. 276, 3971–3976 (2001).
Decuypere, S. et al. Gene expression analysis of the mechanism of natural Sb(V) resistance in Leishmania donovani isolates from Nepal. Antimicrob. Agents Chemother. 49, 4616–4621 (2005).
Decuypere, S. et al. Molecular mechanisms of drug resistance in natural Leishmania populations vary with genetic background. PLoS Negl. Trop. Dis. 6 e1514 (2012).
Brotherton, M. C. et al. Proteomic and genomic analyses of antimony resistant Leishmania infantum mutant. J. Parasitol. Res. 8 e81899 (2013).
Biyani, N., Singh, A. K., Mandal, S., Chawla, B. & Madhubala, R. Differential expression of proteins in antimony-susceptible and -resistant isolates of Leishmania donovani. Mol. Biochem. Parasitol. 179, 91–99 (2011).
Fyfe, P. K., Westrop, G. D., Silva, A. M., Coombs, G. H. & Hunter, W. N. Leishmania TDR1 structure, a unique trimeric glutathione transferase capable of deglutathionylation and antimonial prodrug activation. Proc. Natl Acad. Sci. USA 109, 11693–11698 (2012).
Zhou, Y., Messier, N., Ouellette, M., Rosen, B. P. & Mukhopadhyay, R. Leishmania major LmACR2 is a pentavalent antimony reductase that confers sensitivity to the drug Pentostam. J. Biol. Chem. 279, 37445–37451 (2004).
Plourde, M. et al. Generation of an aquaglyceroporin AQP1 null mutant in Leishmania major. Mol. Biochem. Parasitol. 201, 108–111 (2015).
Marquis, N., Gourbal, B., Rosen, B. P., Mukhopadhyay, R. & Ouellette, M. Modulation in aquaglyceroporin AQP1 gene transcript levels in drug-resistant Leishmania. Mol. Microbiol. 57, 1690–1699 (2005).
Gourbal, B. et al. Drug uptake and modulation of drug resistance in Leishmania by an aquaglyceroporin. J. Biol. Chem. 279, 31010–31017 (2004).
Mandal, S., Maharjan, M., Singh, S., Chatterjee, M. & Madhubala, R. Assessing aquaglyceroporin gene status and expression profile in antimony-susceptible and -resistant clinical isolates of Leishmania donovani from India. J. Antimicrob. Chemother. 65, 496–507 (2010).
Maharjan, M., Singh, S., Chatterjee, M. & Madhubala, R. Role of aquaglyceroporin (AQP1) gene and drug uptake in antimony-resistant clinical isolates of Leishmania donovani. Am. J. Trop. Med. Hyg. 79, 69–75 (2008).
Rogers, M. B. et al. Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res. 21, 2129–2142 (2011).
Sterkers, Y., Crobu, L., Lachaud, L., Pages, M. & Bastien, P. Parasexuality and mosaic aneuploidy in Leishmania: alternative genetics. Trends Parasitol. 30, 429–435 (2014).
Rojo, D. et al. A multiplatform metabolomic approach to the basis of antimonial action and resistance in Leishmania infantum. J. Parasitol. Res. 10 e0130675 (2015).
Berg, M. et al. Metabolic adaptations of Leishmania donovani in relation to differentiation, drug resistance, and drug pressure. Mol. Microbiol. 90, 428–442 (2013).
Dey, S., Ouellette, M., Lightbody, J., Papadopoulou, B. & Rosen, B. P. An ATP-dependent As(III)-glutathione transport system in membrane vesicles of Leishmania tarentolae. Proc. Natl Acad. Sci. USA 93, 2192–2197 (1996).
Manzano, J. I., Garcia-Hernandez, R., Castanys, S. & Gamarro, F. A new ABC half-transporter in Leishmania major is involved in resistance to antimony. Antimicrob. Agents Chemother. 57, 3719–3730 (2013).
Sundar, S. et al. Efficacy of miltefosine in the treatment of visceral leishmaniasis in India after a decade of use. Clin. Infect. Dis. 55, 543–550 (2012).
Rijal, S. et al. Increasing failure of miltefosine in the treatment of Kala-azar in Nepal and the potential role of parasite drug resistance, reinfection, or noncompliance. Clin. Infect. Dis. 56, 1530–1538 (2013).
Seifert, K. et al. Characterisation of Leishmania donovani promastigotes resistant to hexadecylphosphocholine (miltefosine). Int. J. Antimicrob. Ag. 22, 380–387 (2003).
Kulshrestha, A., Sharma, V., Singh, R. & Salotra, P. Comparative transcript expression analysis of miltefosine-sensitive and miltefosine-resistant Leishmania donovani. Parasitol. Res. 113, 1171–1184 (2014).
Perlin, D. S., Shor, E. & Zhao, Y. Update on antifungal drug resistance. Curr. Clin. Microbiol. Rep. 2, 84–95 (2015).
Denning, D. W. & Bromley, M. J. Infectious disease. How to bolster the antifungal pipeline. Science 347, 1414–1416 (2015).
Epaulard, O. et al. A multistep voriconazole-related phototoxic pathway may lead to skin carcinoma: results from a French nationwide study. Clin. Infect. Dis. 57 e182–e188 (2013).
Shor, E. & Perlin, D. S. Coping with stress and the emergence of multidrug resistance in fungi. PLoS Pathog. 11, e1004668 (2015).
Kidd, S. E., Goeman, E., Meis, J. F., Slavin, M. A. & Verweij, P. E. Multi-triazole-resistant Aspergillus fumigatus infections in Australia. Mycoses 58, 350–355 (2015).
van der Linden, J. W. et al. Prospective multicenter international surveillance of azole resistance in Aspergillus fumigatus. Emerg. Infect. Dis 21, 1041–1044 (2015).
Wang, E. et al. The ever-evolving landscape of candidaemia in patients with acute leukaemia: non-susceptibility to caspofungin and multidrug resistance are associated with increased mortality. J. Antimicrob. Chemother. 70, 2362–2368 (2015).
Chowdhary, A., Kathuria, S., Xu, J. & Meis, J. F. Emergence of azole-resistant Aspergillus fumigatus strains due to agricultural azole use creates an increasing threat to human health. PLoS Pathog. 9, e1003633 (2013).
Vermeulen, E., Lagrou, K. & Verweij, P. E. Azole resistance in Aspergillus fumigatus: a growing public health concern. Curr. Opin. Infect. Dis 26, 493–500 (2013).
Snelders, E. et al. Triazole fungicides can induce cross-resistance to medical triazoles in Aspergillus fumigatus. J. Parasitol. Res. 7, e31801 (2012).
Snelders, E., Melchers, W. J. & Verweij, P. E. Azole resistance in Aspergillus fumigatus: a new challenge in the management of invasive aspergillosis?. Future. Microbiol. 6, 335–347 (2011).
Abdolrasouli, A. et al. Genomic context of azole resistance mutations in Aspergillus fumigatus determined using whole-genome sequencing. mBio 6, e00536 (2015).
Katiyar, S. K. et al. Fks1 and Fks2 are functionally redundant but differentially regulated in Candida glabrata: implications for echinocandin resistance. Antimicrob. Agents Chemother. 56, 6304–6309 (2012).
Singh, S. D. et al. Hsp90 governs echinocandin resistance in the pathogenic yeast Candida albicans via calcineurin. PLoS Pathog. 5, e1000532 (2009).
Pfaller, M. A. & Diekema, D. J. Progress in antifungal susceptibility testing of Candida spp. by use of Clinical and Laboratory Standards Institute broth microdilution methods, 2010 to 2012. J. Clin. Microbiol. 50, 2846–2856 (2012).
Alexander, B. D. et al. Increasing echinocandin resistance in Candida glabrata: clinical failure correlates with presence of FKS mutations and elevated minimum inhibitory concentrations. Clin. Infect. Dis. 56, 1724–1732 (2013).
Denoeud-Ndam, L. et al. A multi-center, open-label trial to compare the efficacy and pharmacokinetics of Artemether-Lumefantrine in children with severe acute malnutrition versus children without severe acute malnutrition: study protocol for the MAL-NUT study. BMC Infect. Dis. 15, 228 (2015).
Kinga, M. K. et al. Quantitative genome re-sequencing defines multiple mutations conferring chloroquine resistance in rodent malaria. BMC Genomics 13, 106 (2012).
Manske, M. et al. Analysis of Plasmodium falciparum diversity in natural infections by deep sequencing. Nature 487, 375–379 (2012).
Cheeseman, I. H. et al. A major genome region underlying artemisinin resistance in malaria. Science 336, 79–82 (2012).
Yuan, J. et al. Chemical genomic profiling for antimalarial therapies, response signatures, and molecular targets. Science 333, 724–729 (2011).
Glover, L. et al. Genome-scale RNAi screens for high-throughput phenotyping in bloodstream-form African trypanosomes. Nature Protoc. 10, 106–133 (2015).
Alsford, S. et al. High-throughput decoding of antitrypanosomal drug efficacy and resistance. Nature 482, 232–236 (2012).
Begolo, D., Erben, E. & Clayton, C. Drug target identification using a trypanosome overexpression library. Antimicrob. Agents Chemother. 58, 6260–6264 (2014).
Cowman, A. F. Functional analysis of drug resistance in Plasmodium falciparum in the post-genomic era. Int. J. Parasitol. 31, 871–878 (2001).
Vincent, I. M. & Barrett, M. P. Metabolomic-based strategies for anti-parasite drug discovery. J. Biomol. Screen. 20, 44–55 (2015).
Goodman, C. D. et al. Parasites resistant to the antimalarial atovaquone fail to transmit by mosquitoes. Science 352, 349–353 (2016).
Guttery, D. S., Roques, M., Holder, A. A. & Tewari, R. Commit and transmit: molecular players in Plasmodium sexual development and zygote differentiation. Trends Parasitol. 31, 676–685 (2015).
Hemingway, J. The role of vector control in stopping the transmission of malaria: threats and opportunities. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369, 20130431 (2014).
Dias, J. C. Southern Cone Initiative for the elimination of domestic populations of Triatoma infestans and the interruption of transfusional Chagas disease. Historical aspects, present situation, and perspectives. Mem. Inst. Oswaldo Cruz 102 (suppl. 1), 11–18 (2007).
Welburn, S. C. & Maudlin, I. Priorities for the elimination of sleeping sickness. Adv. Parasitol. 79, 299–337 (2012).
Dias, J. C., Silveira, A. C. & Schofield, C. J. The impact of Chagas disease control in Latin America: a review. Mem. Inst. Oswaldo Cruz 97, 603–612 (2002).
Abd-Alla, A. M. et al. Improving Sterile Insect Technique (SIT) for tsetse flies through research on their symbionts and pathogens. J. Invertebr. Pathol. 112 (suppl.), S2–S10 (2013).
Oliva, C. F. et al. Current status and future challenges for controlling malaria with the sterile insect technique: technical and social perspectives. Acta Trop. 132 (suppl.), S130–S139 (2014).
Dantzler, K. W., Ravel, D. B., Brancucci, N. M. & Marti, M. Ensuring transmission through dynamic host environments: host-pathogen interactions in Plasmodium sexual development. Curr. Opin. Microbiol. 26, 17–23 (2015).
Imhof, S. & Roditi, I. The social life of African trypanosomes. Trends Parasitol. 31, 490–498 (2015).
Mony, B. M. et al. Genome-wide dissection of the quorum sensing signalling pathway in Trypanosoma brucei. Nature 505, 681–685 (2014).
Mantel, P. Y. et al. Malaria-infected erythrocyte-derived microvesicles mediate cellular communication within the parasite population and with the host immune system. Cell Host. Microbe 13, 521–534 (2013).
Regev-Rudzki, N. et al. Cell-cell communication between malaria-infected red blood cells via exosome-like vesicles. Cell 153, 1120–1133 (2013).
World Health Organization. Guidelines for the Treatment of Malaria 3rd edn, 1–316 (World Health Organization, 2015).
World Health Organization. WHO Model List of Essential Medicines 19th edn, 1–51 (World Health Organization, 2015).
World Health Organization. Rapid Advice: Diagnosis, Prevention and Management of Cryptococcal Disease in HIV-Infected Adults, Adolescents and Children 1–37 (World Health Organization, 2011).
Leroux, S. & Ullmann, A. J. Management and diagnostic guidelines for fungal diseases in infectious diseases and clinical microbiology: critical appraisal. Clin. Microbiol. Infect. 19, 1115–1121 (2013).
Kullberg, B. J. & Arendrup, M. C. Invasive candidiasis. N. Engl. J. Med. 373, 1445–1456 (2015).
Ecker, A., Lehane, A. M., Clain, J. & Fidock, D. A. PfCRT and its role in antimalarial drug resistance. Trends Parasitol. 28, 504–514 (2012).
Wootton, J. C. et al. Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum. Nature 418, 320–323 (2002).
Sanchez, C. P., Stein, W. & Lanzer, M. Trans stimulation provides evidence for a drug efflux carrier as the mechanism of chloroquine resistance in Plasmodium falciparum. Biochemistry 42, 9383–9394 (2003).
Sanchez, C. P., Dave, A., Stein, W. D. & Lanzer, M. Transporters as mediators of drug resistance in Plasmodium falciparum. Int. J. Parasitol. 40, 1109–1118 (2010).
Ferdig, M. T. et al. Dissecting the loci of low-level quinine resistance in malaria parasites. Mol. Microbiol. 52, 985–997 (2004).
Valderramos, S. G. et al. Identification of a mutant PfCRT-mediated chloroquine tolerance phenotype in Plasmodium falciparum. PLoS Pathog. 6, e1000887 (2010).
Pelleau, S. et al. Adaptive evolution of malaria parasites in French Guiana: reversal of chloroquine resistance by acquisition of a mutation in pfcrt. Proc. Natl Acad. Sci. USA 112, 11672–11677 (2015).
Wellems, T. E., Walker-Jonah, A. & Panton, L. J. Genetic mapping of the chloroquine-resistance locus on Plasmodium falciparum chromosome 7. Proc. Natl Acad. Sci. USA 88, 3382–3386 (1991).
Duraisingh, M. T. et al. Linkage disequilibrium between two chromosomally distinct loci associated with increased resistance to chloroquine in Plasmodium falciparum. Parasitology 121, 1–7 (2000).
Meshnick, S. R., Taylor, T. E. & Kamchonwongpaisan, S. Artemisinin and the antimalarial endoperoxides — from herbal remedy to targeted chemotherapy. Microbiol. Rev. 60, 301 (1996).
Wang, P., Read, M., Sims, P. F. G. & Hyde, J. E. Sulfadoxine resistance in the human malaria parasite Plasmodium falciparum is determined by mutations in dihydropteroate synthetase and an additional factor associated with folate utilization. Mol. Microbiol. 23, 979–986 (1997).
Heinberg, A. & Kirkman, L. The molecular basis of antifolate resistance in Plasmodium falciparum : looking beyond point mutations. Ann. N. Y. Acad. Sci. 1342, 10–18 (2015).
Cowman, A. F. in Malaria: Parasite Biology, Pathogenesis, and Protection (ed. Sherman, I. W. ) 317–330 (1998).
Rose, G. W., Suh, K. N., Kain, K. C., Le, S. N. & McCarthy, A. E. Atovaquone-proguanil resistance in imported falciparum malaria in a young child. Pediatr. Infect. Dis. J. 27, 567–569 (2008).
Carter, N. S. & Fairlamb, A. H. Arsenical-resistant trypanosomes lack an unusual adenosine transporter. Nature 361, 173–176 (1993).
Maser, P., Sutterlin, C., Kralli, A. & Kaminsky, R. A nucleoside transporter from Trypanosoma brucei involved in drug resistance. Science 285, 242–244 (1999).
Baker, N. et al. Aquaglyceroporin 2 controls susceptibility to melarsoprol and pentamidine in African trypanosomes. Proc. Natl Acad. Sci. USA 109, 10996–11001 (2012).
Graf, F. E. et al. Chimerization at the AQP2-AQP3 locus is the genetic basis of melarsoprol-pentamidine cross-resistance in clinical Trypanosoma brucei gambiense isolates. Int. J. Parasitol. Drugs Drug Resist. 5, 65–68 (2015).
Pyana, P. P. et al. Melarsoprol sensitivity profile of Trypanosoma brucei gambiense isolates from cured and relapsed sleeping sickness patients from the Democratic Republic of the Congo. PLoS Negl. Trop. Dis. 8, e3212 (2014).
Graf, F. E. et al. Aquaporin 2 mutations in Trypanosoma brucei gambiense field isolates correlate with decreased susceptibility to pentamidine and melarsoprol. PLoS Negl. Trop. Dis. 7, e2475 (2013).
Kazibwe, A. J. et al. Genotypic status of the TbAT1/P2 adenosine transporter of Trypanosoma brucei gambiense isolates from Northwestern Uganda following melarsoprol withdrawal. PLoS Negl. Trop. Dis. 3, e523 (2009).
Vincent, I. M. et al. A molecular mechanism for eflornithine resistance in African trypanosomes. PLoS Pathog. 6, e1001204 (2010).
Mathieu, C. et al. Trypanosoma brucei eflornithine transporter AAT6 is a low-affinity low-selective transporter for neutral amino acids. Biochem. J. 463, 9–18 (2014).
Wilkinson, S. R., Taylor, M. C., Horn, D., Kelly, J. M. & Cheeseman, I. A mechanism for cross-resistance to nifurtimox and benznidazole in trypanosomes. Proc. Natl Acad. Sci. USA 105, 5022–5027 (2008).
Hall, B. S., Bot, C. & Wilkinson, S. R. Nifurtimox activation by trypanosomal type I nitroreductases generates cytotoxic nitrile metabolites. J. Biol. Chem. 286, 13088–13095 (2011).
Patterson, S. & Wyllie, S. Nitro drugs for the treatment of trypanosomatid diseases: past, present, and future prospects. Trends Parasitol. 30, 289–298 (2014).
Sokolova, A. Y. et al. Cross-resistance to nitro drugs and implications for treatment of human African trypanosomiasis. Antimicrob. Agents Chemother. 54, 2893–2900 (2010).
Wyllie, S. et al. Nitroheterocyclic drug resistance mechanisms in Trypanosoma brucei. J. Antimicrob. Chemother. 71, 625–634 (2015).
Mejia, A. M. et al. Benznidazole-resistance in Trypanosoma cruzi is a readily acquired trait that can arise independently in a single population. J. Infect. Dis. 206, 220–228 (2012).
Trochine, A., Creek, D. J., Faral-Tello, P., Barrett, M. P. & Robello, C. Benznidazole biotransformation and multiple targets in Trypanosoma cruzi revealed by metabolomics. PLoS Negl. Trop. Dis. 8, e2844 (2014).
Hall, B. S. & Wilkinson, S. R. Activation of benznidazole by trypanosomal type I nitroreductases results in glyoxal formation. Antimicrob. Agents Chemother. 56, 115–123 (2012).
Zingales, B. et al. A novel ABCG-like transporter of Trypanosoma cruzi is involved in natural resistance to benznidazole. Mem. Inst. Oswaldo Cruz 110, 433–444 (2015).
Murta, S. M. F. et al. Deletion of copies of the gene encoding old yellow enzyme (TcOYE), a NAD(P)H flavin oxidoreductase, associates with in vitro-induced benznidazole resistance in Trypanosoma cruzi. Mol. Biochem. Parasitol. 146, 151–162 (2006).
Andrade, H. M. et al. Proteomic analysis of Trypanosoma cruzi resistance to benznidazole. J. Proteome Res. 7, 2357–2367 (2008).
Kubata, B. K. et al. A key role for old yellow enzyme in the metabolism of drugs by Trypanosoma cruzi. J. Exp. Med. 196, 1241–1251 (2002).
Wyllie, S., Cunningham, M. L. & Fairlamb, A. H. Dual action of antimonial drugs on thiol redox metabolism in the human pathogen Leishmania donovani. J. Biol. Chem. 279, 39925–39932 (2004).
Baiocco, P., Colotti, G., Franceschini, S. & Ilari, A. Molecular basis of antimony treatment in leishmaniasis. J. Med. Chem. 52, 2603–2612 (2009).
Wyllie, S. et al. Elevated levels of tryparedoxin peroxidase in antimony unresponsive Leishmania donovani field isolates. Mol. Biochem. Parasitol. 173, 162–164 (2010).
Mukhopadhyay, R. et al. Trypanothione overproduction and resistance to antimonials and arsenicals in Leishmania. Proc. Natl Acad. Sci. USA 93, 10383–10387 (1996).
Wyllie, S., Vickers, T. J. & Fairlamb, A. H. Roles of trypanothione S-transferase and tryparedoxin peroxidase in resistance to antimonials. Antimicrob. Agents Chemother. 52, 1359–1365 (2008).
Callahan, H. L. & Beverley, S. M. Heavy metal resistance: a new role for P-glycoproteins in Leishmania. J. Biol. Chem. 266, 18427–18430 (1991).
Mukherjee, A. et al. Role of ABC transporter MRPA, γ-glutamylcysteine synthetase and ornithine decarboxylase in natural antimony-resistant isolates of Leishmania donovani. J. Antimicrob. Chemother. 59, 204–211 (2007).
El Fadili, K. et al. Role of the ABC transporter MRPA (PGPA) in antimony resistance in Leishmania infantum axenic and intracellular amastigotes. Antimicrob. Agents Chemother. 49, 1988–1993 (2005).
Brochu, C., Haimeur, A. & Ouellette, M. The heat shock protein HSP70 and heat shock cognate protein HSC70 contribute to antimony tolerance in the protozoan parasite Leishmania. Cell Stress Chaperones 9, 294–303 (2004).
Chawla, B., Jhingran, A., Panigrahi, A., Stuart, K. D. & Madhubala, R. Paromomycin affects translation and vesicle-mediated trafficking as revealed by proteomics of paromomycin–susceptible–resistant Leishmania donovani. J. Parasitol. Res. 6, e26660 (2011).
Rakotomanga, M., Saint-Pierre-Chazalet, M. & Loiseau, P. M. Alteration of fatty acid and sterol metabolism in miltefosine-resistant Leishmania donovani promastigotes and consequences for drug-membrane interactions. Antimicrob. Agents Chemother. 49, 2677–2686 (2005).
Rakotomanga, M., Blanc, S., Gaudin, K., Chaminade, P. & Loiseau, P. A. Miltefosine afects lipid metabolism in Leishmania donovani promastigotes. Antimicrob. Agents Chemother. 51, 1425–1430 (2007).
Vincent, I. M. et al. Untargeted metabolomic analysis of miltefosine action inLeishmania infantum reveals changes to the internal lipid metabolism. Int. J. Parasitol. Drugs Drug Resist. 4, 20–27 (2014).
Dorlo, T. P. C., Balasegaram, M., Beijnen, J. H. & de Vries, P. J. Miltefosine: a review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J. Antimicrob. Chemother. 67, 2576–2597 (2012).
Perez-Victoria, F. J., Gamarro, F., Ouellette, M. & Castanys, S. Functional cloning of the miltefosine transporter: a novel P-type phospholipid translocase from Leishmania involved in drug resistance. J. Biol. Chem. 278, 49965–49971 (2003).
Perez-Victoria, F. J., Castanys, S. & Gamarro, F. Leishmania donovani resistance to miltefosine involves a defective inward translocation of the drug. Antimicrob. Agents Chemother. 47, 2397–2403 (2003).
Coelho, A. C. et al. Multiple mutations in heterogeneous miltefosine-resistant Leishmania major population as determined by whole genome sequencing. PLoS Negl. Trop. Dis. 6, e1512 (2012).
Perez-Victoria, F. J., Sanchez-Canete, M. P., Castanys, S. & Gamarro, F. Phospholipid translocation and miltefosine potency require both L. donovani miltefosine transporter and the new protein LdRos3 in Leishmania parasites. J. Biol. Chem. 281, 23766–23775 (2006).
Perez-Victoria, J. M. et al. Alkyl-lysophospholipid resistance in multidrug-resistant Leishmania tropica and chemosensitization by a novel P-glycoprotein-like transporter modulator. Antimicrob. Agents Chemother. 45, 2468–2474 (2001).
Castanys-Munoz, E., Perez-Victoria, J. M., Gamarro, F. & Castanys, S. Characterization of an ABCG-like transporter from the protozoan parasite Leishmania with a role in drug resistance and transbilayer lipid movement. Antimicrob. Agents Chemother. 52, 3573–3579 (2008).
Canuto, G. A. et al. Multi-analytical platform metabolomic approach to study miltefosine mechanism of action and resistance in Leishmania. Anal. Bioanal. Chem. 406, 3459–3476 (2014).
Blum, G. et al. New insight into amphotericin B resistance in Aspergillus terreus. Antimicrob. Agents Chemother. 57, 1583–1588 (2013).
Gray, K. C. et al. Amphotericin primarily kills yeast by simply binding ergosterol. Proc. Natl Acad. Sci. USA 109, 2234–2239 (2012).
Odds, F. C., Brown, A. J. & Gow, N. A. Antifungal agents: mechanisms of action. Trends Microbiol. 11, 272–279 (2003).
Prasad, R. & Rawal, M. K. Efflux pump proteins in antifungal resistance. Front Pharmacol. 5, 202 (2014).
Costa, C., Dias, P. J., Sa-Correia, I. & Teixeira, M. C. MFS multidrug transporters in pathogenic fungi: do they have real clinical impact?. Front Physiol 5, 197 (2014).
Cowen, L. E., Sanglard, D., Howard, S. J., Rogers, P. D. & Perlin, D. S. Mechanisms of antifungal drug resistance. Cold Spring Harb. Perspect. Med. 5, a019752 (2015).
Flowers, S. A. et al. Gain-of-function mutations in UPC2 are a frequent cause of ERG11 upregulation in azole-resistant clinical isolates of Candida albicans. Eukaryot. Cell 11, 1289–1299 (2012).
Cannon, R. D. et al. Efflux-mediated antifungal drug resistance. Clin. Microbiol. Rev. 22, 291–321, Table (2009).
Kwon-Chung, K. J. & Chang, Y. C. Aneuploidy and drug resistance in pathogenic fungi. PLoS Pathog. 8, e1003022 (2012).
Selmecki, A., Gerami-Nejad, M., Paulson, C., Forche, A. & Berman, J. An isochromosome confers drug resistance in vivo by amplification of two genes, ERG11 and TAC1. Mol. Microbiol. 68, 624–641 (2008).
Selmecki, A., Forche, A. & Berman, J. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science 313, 367–370 (2006).
Nishikawa, J. L. et al. Inhibiting fungal multidrug resistance by disrupting an activator–mediator interaction. Nature 530, 485–489 (2016).
Cowen, L. E. The evolution of fungal drug resistance: modulating the trajectory from genotype to phenotype. Nature Rev. Microbiol. 6, 187–198 (2008).
Sun, H. Y. & Singh, N. Characterisation of breakthrough invasive mycoses in echinocandin recipients: an evidence-based review. Int. J. Antimicrob. Agents 35, 211–218 (2010).
Vandeputte, P., Ferrari, S. & Coste, A. T. Antifungal resistance and new strategies to control fungal infections. Int. J. Microbiol. 2012, 713687 (2012).
Acknowledgements
This Review is a collaboration between the Wellcome Trust funded centres of Infectious Disease Research in Scotland (IDRIS). This comprises the Edinburgh University ‘Centre for Immunity, Infection and Evolution’ (Wellcome Trust grant reference 095831), Glasgow University ‘Wellcome Trust Centre for Molecular Parasitology’ (Wellcome Trust grant reference WT104111AIA), Dundee University Wellcome Trust strategic award in ‘Chemical Biology for Target Identification’ (Wellcome Trust grant reference 10502) and the Aberdeen University Wellcome Trust Strategic Award in ‘Medical Mycology and Fungal Immunology’ (Wellcome Trust grant reference 097377). Personal research support involved Wellcome Trust senior investigator awards to K.R.M. (Wellcome Trust grant reference 103740/Z/14/Z) and N.A.R.G. (Wellcome Trust grant reference 101873), and Wellcome Trust Principal Research fellowships to A.H.F. (Wellcome Trust grant reference 079838) and A.P.W. (Wellcome Trust grant reference 107046/Z/15/Z).
Author information
Authors and Affiliations
Contributions
All authors contributed equally to the preparation of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Fairlamb, A., Gow, N., Matthews, K. et al. Drug resistance in eukaryotic microorganisms. Nat Microbiol 1, 16092 (2016). https://doi.org/10.1038/nmicrobiol.2016.92
Published:
DOI: https://doi.org/10.1038/nmicrobiol.2016.92
This article is cited by
-
The importance of antimicrobial resistance in medical mycology
Nature Communications (2022)
-
The power and promise of genetic mapping from Plasmodium falciparum crosses utilizing human liver-chimeric mice
Communications Biology (2021)
-
Aspergillus spp. burden on filtering respiratory protective devices. Is there an occupational health concern?
Air Quality, Atmosphere & Health (2020)
-
Interactions between invasive fungi and symbiotic bacteria
World Journal of Microbiology and Biotechnology (2020)
-
Drug repurposing for antimicrobial discovery
Nature Microbiology (2019)
