
Abstract
The large capacity of vaccinia virus (VAC) for added DNA, cytoplasmic expression and broad host range make it a popular choice for gene delivery, despite the burdensome need for multiple plaque purifications to isolate recombinants. Here we describe how a bacterial artificial chromosome (BAC) containing the entire VAC genome can be engineered in Escherichia coli by homologous recombination using bacteriophage λ–encoded enzymes. The engineered VAC genomes can then be used to produce clonally pure recombinant viruses in mammalian cells without the need for plaque purification.
Similar content being viewed by others
Main
VAC, a member of the poxvirus family, has a linear double-stranded DNA genome of about 200 kilobase pairs (kbp) with covalently closed hairpin ends1. Recombinant forms of VAC are used to synthesize biologically active proteins and investigate structure-function relationships, determine the targets of humoral and cell-mediated immunity, and construct live recombinant vaccines2,3,4. The classic technique for inserting foreign genes is based on homologous recombination between a transfer plasmid and the VAC genome in mammalian or avian cells5. Typically, multiple cycles of plaque purification are necessary, even with selection or screening protocols. Some advantage has been obtained by the use of purified VAC DNA for in vitro or in vivo insertion of genes6,7,8. We previously described cloning the full-length VAC genome as a circular BAC9. Because poxvirus DNA is not infectious, the VAC-BAC is safe for use with minimal precautions. Infectious virus is readily obtained, however, by transfecting the VAC-BAC into mammalian cells in the presence of a distantly related helper avian poxvirus. The helper fowlpox virus (FPV) provides enzyme functions necessary to initiate replication, but does not produce infectious progeny in mammalian cells and no recombination with the VAC genome is detectable7. During rescue and replication, the circular VAC-BAC is linearized and the progeny are indistinguishable from wild-type VAC, except for vector sequences retained in the genome. Our purpose here was to develop methods of genetically engineering the VAC-BAC to facilitate the production of recombinant VAC expression vectors.
We adapted recombinogenic engineering technology, which avoids the use of restriction enzymes or ligases and allows recombination of BACs maintained in E. coli10,11. A mini–λ prophage encoding the red recombination system, composed of 5′ to 3′ exonuclease (Exo), a single-strand DNA binding protein (Bet) and a nuclease inhibitor (Gam), under the control of the temperature-sensitive λ cI857 repressor12,13, was integrated into the chromosome of E. coli DH10B harboring the VAC-BAC to make E. coli DH10B/VAC-BAC/λ. The VAC-BAC appeared stable after four successive overnight cultures of E. coli DH10B/VAC-BAC/λ and after temperature induction of the λ red system for 30–45 min, as judged by restriction endonuclease analysis of DNA from individual clones and by the recovery of infectious VAC following transfection of mammalian cells in the presence of a helper FPV as previously described9. Further evidence for stability will be presented below.
The ability of the λ red system to mediate homologous recombination was tested by incubating E. coli DH10B/VAC-BAC/λ for varying lengths of time at 42 °C to induce expression of the recombination proteins. The cells were then electroporated with linear DNA containing approximately 100 bp of homologous sequences flanking the chloramphenicol acetyltransferase (cat) gene essentially as described13. The number of chloramphenicol-resistant colonies from cells induced for 0, 15, 30 and 45 min was 0, 7, 47 and 49, respectively. Based on these experiments we recommend the following protocol to induce expression of λ recombination genes. First, 50 ml of LB medium containing tetracycline (25 μg/ml) and chloramphenicol (34 μg/ml) is inoculated with 0.5 ml of an overnight culture of E. coli DH10B/VAC-BAC/λ and incubated at 32 °C until the OD600 is approximately 0.6. Then, a 10-ml culture is induced at 42 °C for 30 or 45 min in 50-ml flasks. The cells are chilled on ice for 10 min, collected by centrifugation, washed three times with ice-cold 10% glycerol in water and resuspended in 100 ml of the same solution. The cells maintain viability at −80 °C for at least six weeks.
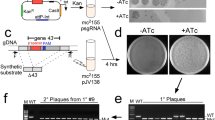
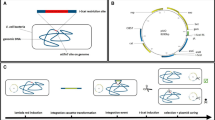
The VAC-BAC is expected to have two main uses: construction of expression vectors and mutagenesis of the VAC genome. The former is of general interest and is the focus of this report. Recombinogenic engineering typically involves two rounds of recombination. In the first round, positive selection is used to isolate clones that have a cassette consisting of adjacent positive and counterselectable genes targeted to the insertion site. In the second round, counterselection is used to isolate clones in which the cassette is replaced with the desired sequence10. This approach is extremely versatile, as it can be used for any kind of modification at any place in a BAC. Counterselection is less efficient than positive selection, however, and many colonies must be screened to isolate the final recombinant. To avoid counterselection, we used the red recombination system to modify the VAC-BAC for one-step antibiotic selection of recombinants. The promoter of the cat gene in the VAC-BAC was replaced by homologous recombination with a zeomycin-resistance gene (zeoR) to form VAC-BAC.zeo (Fig. 1a). The object of the latter manipulation was to make the E. coli sensitive to chloramphenicol, so that subsequent inclusion of the cat promoter in an expression cassette would allow isolation of recombinants because of chloramphenicol resistance. In addition to the cat promoter, the expression cassette contains the gene of interest regulated by a VAC promoter and flanking sequences comprising the start of the cat open reading frame on one side and DNA downstream of zeoR on the other. Homologous recombination results in the deletion of zeoR and the insertion of the cat promoter to restore chloramphenicol resistance and add the gene of interest. To insert a single gene, the insertion cassette is easily built by PCR, as <100 bp of flanking sequence is sufficient for phage-mediated recombination. This would be the preferred method for large-scale projects such as cDNA cloning. For the simultaneous expression of two genes, we constructed a plasmid containing the promoter for the cat gene and two VAC promoters with multiple cloning sites between flanking sequences of 300–400 bp (Fig. 1b). As a test of the system, the genes encoding glucuronidase (GUS) and red fluorescent protein (RFP) were inserted into the transfer plasmid, each controlled by a separate VAC promoter. The expression cassette was amplified by PCR and the purified product was used for electroporation of E. coli DH10B/VAC-BAC.zeo/λ, which had been induced at 42 °C for 30 min. The bacteria were plated on chloramphenicol-containing agar plates and antibiotic-resistant colonies were picked. In the present experiment, four of five colonies examined contained plasmid DNA with sequences encoding GUS and RFP at the targeted site, as determined by PCR using forward and reverse primers (Fig. 1c), and one colony was still contaminated with E. coli containing VAC-BAC.zeo. Restriction enzyme analysis (Fig. 1d) and sequencing of the regions including and flanking the sites of recombination of VAC-BAC.GUS.RFP (data not shown) confirmed the absence of rearrangements, mutations or small deletions accompanying recombination.

(a) Construction of VAC-BAC.zeo. The red recombination system in E. coli DH10B/VAC-BAC/λ was used to replace the cat promoter (Pr) of VAC-BAC with zeoR. The targeting DNA contained zeoR and 50 bp of flanking DNA excluding the cat promoter. Clones were selected for zeomycin resistance and recombination at the correct site was confirmed by PCR and HindIII restriction endonuclease digestion. Symbol '/', inactivation of cat expression. (b) Schematic showing insertion of genes encoding GUS and RFP into the VAC-BAC.zeo plasmid to form VAC-BAC.GUS.RFP. The targeting DNA contained RFP and GUS genes adjacent to two VAC promoters (2vPr), the cat promoter (Pr) and 371–394 bp of flanking DNA. Bacterial colonies were selected by chloramphenicol resistance. (c) PCR screening of VAC-BAC.GUS.RFP plasmids. Five colonies, isolated as in b, were analyzed by PCR using 5′ and 3′ primers outside the recombination site. PCR products of the size expected for zeo and GUS.RFP are indicated by arrowheads on the right. The GeneRuler 1 kbp DNA ladder (Fermentas Life Sciences) is shown on the left with the sizes of selected bands indicated (M). (d) HindIII restriction endonuclease analysis of VAC-BAC.GUS.RFP plasmid. DNA was extracted from bacteria by alkaline lysis method, ethanol precipitated, washed, digested with HindIII endonuclease, analyzed by electrophoresis on a 0.7% agarose gel and stained with ethidium bromide. Arrows on the right show the positions of the two predicted additional bands in the VAC-BAC.GUS.RFP plasmid. The 1 kbp DNA extension ladder (Invitrogen) is shown on the left (M) with the sizes of selected bands indicated.
To further evaluate the stability of the VAC-BAC after induction of the red system, we analyzed the tandem repeat region near the ends of the viral genome. Vaccinia virus contains two blocks of 70 bp repeats separated by a 453-bp unique segment of DNA on each side of the concatemer junction14 (Fig. 2). As a result of recombination, the number of repeats per block naturally varies from 10 to 20 in individual virus isolates15. DdeI fragments of 2,200 and 2,950 bp were detected by Southern blotting of VAC-BAC DNA as previously described14,15, consistent with the presence of two blocks of 12 repeats and two blocks of 16 repeats, respectively (Fig. 2). There was no change in the size of either DdeI fragment after transformation of E. coli with the λ red system, but the size of the larger fragment was reduced by approximately one repeat after insertion of zeoR (Fig. 2). No further change was noted in any of the three clones encoding GUS and RFP (Fig. 2). We attribute the notable DNA stability in E. coli DH10B/VAC-BAC/λ to the low copy number of the VAC-BAC and the short induction periods used for homologous recombination.

(a) The schematic shows four blocks of 70-bp repeats (striped bars), non-repeat regions (filled bars) and DdeI sites in the concatemer junction region of the BAC DNA. The sizes of the non-repeat regions are indicated. Double-headed arrows represent the DdeI fragments seen in b. (b) Southern blotting with 32P-labeled 70-bp repeat sequence DNA. The original VAC-BAC, VAC-BAC/λ, VAC-BAC.zeo and 3 VAC-BAC.GUS.RFP clones were analyzed.
The final step, rescue of the infectious recombinant virus, was carried out by infecting CV-1 cells with FPV and transfecting them with 1 μg of each VAC-BAC.GUS.RFP plasmid clone as previously described9. Cytopathic effects were seen after 3 d at 37 °C and the cells were harvested 2 d later. The recovery of approximately 2 × 105 plaque forming units of infectious recombinant VAC was demonstrated by a plaque assay on BS-C-1 cells, which, like CV-1 cells, are nonpermissive for FPV. All of the plaques contained virus that expressed GUS and RFP as well as GFP, which was present in the original VAC-BAC (Fig. 3a). Virus in control plaques expressed only GFP. The presence of the insert was verified by restriction endonuclease digestion of purified VAC-BAC.GUS.RFP viral DNA (Fig. 3b).

(a) Expression of RFP and GUS. CV-1 cells were infected with FPV and transfected with 1 μg of VAC-BAC.GUS.RFP plasmid and harvested after 5 d. The lysates were diluted and applied to BS-C-1 cells to allow individual virus plaques to form. After 2 d, the plaques were visualized by fluorescence microscopy with appropriate filters to visualize GFP and RFP. The plates were stained with 5-bromo-4-chloro-3-indolyl-β-D-glucuronic acid to detect GUS expression. (b) HindIII restriction endonuclease analysis of DNA from VAC-BAC.GUS.RFP virus. Virus from a single plaque was amplified in BS-C-1 cells and purified by sucrose gradient sedimentation. The virus was digested with proteinase K and the DNA was purified by phenol and chloroform extractions and collected by precipitation with ethanol. DNA from VAC-BAC virus and VAC-BAC.GUS.RFP virus was digested with HindIII restriction endonuclease and analyzed by electrophoresis on a 0.7% agarose gel. The arrow shows the position of the predicted additional band in DNA from VAC-BAC.GUS.RFP virus. The 1-kbp DNA extension ladder is shown on the left (M) with the sizes of selected bands indicated. Note that the second marker band is 20 kbp.
In summary, recombinogenic engineering of VAC-BAC greatly facilitates the construction and isolation of recombinant virus. Genes can be inserted into the VAC-BAC.zeo plasmid by a one-step procedure with positive selection of bacterial colonies. Moreover, the recombinant VAC-BAC plasmids are then used to produce clonally pure recombinant virus without the need for plaque purifications. The simplified procedure may be particularly useful for high-throughput applications and can be adapted to other BAC vectors.
References
Moss, B. in Fields Virology (eds. Knipe, D.M. & Howley, P.M.) 2849–2883 (Lippincott Williams & Wilkins, Philadelphia, 2001).
Moss, B. Proc. Natl. Acad. Sci. USA 93, 11341–11348 (1996).
Mastrangelo, M.J., Eisenlohr, L.C., Gomella, L. & Lattime, E.C. J. Clin. Invest. 105, 1031–1034 (2000).
Paoletti, E. Proc. Natl. Acad. Sci. USA 93, 11349–11353 (1996).
Earl, P.L., Moss, B., Wyatt, L.S. & Carroll, M.W. in Current Protocols in Molecular Biology (eds. Ausubel, F.M. et al.) 16.17.1–16.17.19 (Greene Publishing Associates & Wiley Interscience, New York, 1998).
Merchlinsky, M. & Moss, B. Virology 190, 522–526 (1992).
Scheiflinger, F., Dorner, F. & Falkner, F.G. Proc. Natl. Acad. Sci. USA 89, 9977–9981 (1992).
Smith, E.S. et al. Nat. Med. 7, 967–972 (2001).
Domi, A. & Moss, B. Proc. Natl. Acad. Sci. USA 99, 12415–12420 (2002).
Copeland, N.G., Jenkins, N.A. & Court, D.L. Nat. Rev. Genet. 2, 769–779 (2001).
Britt, W.J., Jarvis, M., Seo, J.Y., Drummond, D. & Nelson, J. J. Virol. 78, 539–543 (2004).
Yu, D. et al. Proc. Natl. Acad. Sci. USA 97, 5978–5983 (2000).
Court, D.L., Sawitzke, J.A. & Thomason, L.C. Annu. Rev. Genet. 36, 361–388 (2002).
Wittek, R. & Moss, B. Cell 21, 277–284 (1980).
Moss, B., Winters, E. & Cooper, N. Proc. Natl. Acad. Sci. USA 78, 1614–1618 (1981).
Acknowledgements
We thank D.L. Court for providing mini-λ DNA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Domi, A., Moss, B. Engineering of a vaccinia virus bacterial artificial chromosome in Escherichia coli by bacteriophage λ–based recombination. Nat Methods 2, 95–97 (2005). https://doi.org/10.1038/nmeth734
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nmeth734
This article is cited by
-
Characterization of virus-mediated immunogenic cancer cell death and the consequences for oncolytic virus-based immunotherapy of cancer
Cell Death & Disease (2020)
-
Recovery of infectious virus from full-length cowpox virus (CPXV) DNA cloned as a bacterial artificial chromosome (BAC)
Veterinary Research (2011)
-
Viruses as vaccine vectors for infectious diseases and cancer
Nature Reviews Microbiology (2010)
