
Abstract
Automated annotation of protein function is challenging. As the number of sequenced genomes rapidly grows, the overwhelming majority of protein products can only be annotated computationally. If computational predictions are to be relied upon, it is crucial that the accuracy of these methods be high. Here we report the results from the first large-scale community-based critical assessment of protein function annotation (CAFA) experiment. Fifty-four methods representing the state of the art for protein function prediction were evaluated on a target set of 866 proteins from 11 organisms. Two findings stand out: (i) today's best protein function prediction algorithms substantially outperform widely used first-generation methods, with large gains on all types of targets; and (ii) although the top methods perform well enough to guide experiments, there is considerable need for improvement of currently available tools.
Similar content being viewed by others


Main
The accurate annotation of protein function is key to understanding life at the molecular level and has great biomedical and pharmaceutical implications. However, with its inherent difficulty and expense, experimental characterization of function cannot scale up to accommodate the vast amount of sequence data already available1. The computational annotation of protein function has therefore emerged as a problem at the forefront of computational and molecular biology.
Many solutions have been proposed in the last four decades2,3,4,5,6,7,8,9,10, yet the task of computational functional inference in a laboratory often relies on traditional approaches such as identifying domains or finding Basic Local Alignment Search Tool (BLAST)11 hits among proteins with experimentally determined function. Recently, the availability of genomic-level sequence information for thousands of species, coupled with massive high-throughput experimental data, has created new opportunities for function prediction. A large number of methods have been proposed to exploit these data, including function prediction from amino acid sequence12,13,14,15,16, inferred evolutionary relationships and genomic context17,18,19,20,21, protein-protein interaction networks22,23,24,25, protein structure data26,27,28, microarrays29 or a combination of data types30,31,32,33,34. An unbiased evaluation of these different methods can provide insight into their ability to characterize proteins functionally and can guide biological experiments. So far, however, a comprehensive assessment incorporating a large and diverse set of target sequences has not been conducted because of practical difficulties in providing an accurately annotated target set.
In this report, we present the results of the first CAFA experiment, a worldwide effort aimed at analyzing and evaluating protein function prediction methods. Although protein function can be described in multiple ways, we focus on classification schemes provided by the Gene Ontology (GO) Consortium35. Over the course of 15 months, 30 teams associated with 23 research groups participated in the effort, testing 54 function annotation algorithms. Short descriptions of published methods and detailed descriptions of unpublished methods can be found in the Supplementary Note. These methods were evaluated on a target set of 866 protein sequences from 11 species.
Results
Protein function is a concept that can have different interpretations in different biological contexts. Generally, it describes biochemical, cellular and phenotypic aspects of the molecular events that involve the protein, including how the protein interacts with the environment (such as with small compounds or pathogens). From the various classification schemes developed to standardize descriptions of protein function, we chose the “Molecular Function” and “Biological Process” categories from GO. Each category in GO is a hierarchical set of terms and relationships among them that capture functional information; such a system facilitates computation, and its outputs can be interpreted by humans. GO's consistency across species and its widespread adoption make it suitable for large-scale computational studies. In CAFA, given a new protein sequence, the task of a protein function prediction method is to provide a set of terms in GO along with the confidence scores associated with each term.
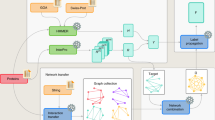
The experiment was organized as follows. A set of 48,298 proteins lacking experimentally validated functional annotation was provided to the community 4 months before the submission deadline for predictions (Fig. 1). Proteins were annotated by the predicting groups, and these annotations were submitted to the assessors. After the submission deadline, GO experimental annotations for those sequences were allowed to accumulate over a period of 11 months. Methods were then evaluated on 866 targets from 11 species that had accumulated functional annotations during the waiting period (Supplementary Table 1). The Swiss-Prot database36 was selected as the gold standard because of its relatively high reliability37.

(a) Timeline for the CAFA experiment. (b) Number of target sequences per organism. The graph shows the number of target sequences for each of the ontologies (Molecular Function and Biological Process) as well as the total number of targets, obtained as a union between sequences in the two ontologies. Of 866 proteins, 531 had Molecular Function annotations and 587 had Biological Process annotations. (c) Distribution of target sequences in each ontology according to the number of leaf terms available for each protein sequence. For example, in the Molecular Function category, 79% of proteins had one leaf term, 16% had two leaf terms, and so on. A term is considered a leaf term for a particular target if no other GO term associated with that sequence is its descendant.
The selection of proteins was ineluctably biased owing to experimenter and annotator choice during the evaluation time frame. Thus, the set of targets was first analyzed to establish that it was representative of those sequences experimentally annotated before the submission deadline. In terms of organismal representation, the eukaryotic targets provided reasonable coverage of taxa (Fig. 1). In contrast, the set of prokaryotic targets was heavily biased toward Escherichia coli K-12. The distribution of terms over the target sequences was representative of the annotations in Swiss-Prot (data not shown); however, we note that in the Molecular Function category a large fraction of target sequences (38%) were associated with “protein binding” as their most specific term. The distribution of term depths over all targets is shown in Supplementary Figure 1 for both ontologies.
Overall predictor performance
The quality of protein function prediction can be measured in different ways that reflect differing motivations for understanding function. In some cases, imprecise experimental characterization means that it is not entirely clear whether a prediction is correct. For CAFA, we principally report a simple metric, the maximum F-measure (Fmax; Online Methods), which considers predictions across the full spectrum from high to low sensitivity. This approach, however, has limitations, such as penalization of specific predictions (see Discussion). We note that the choice of evaluation metric differentially affects different prediction methods, depending on their application objectives.
Top predictor performance, based on maximum F-measure and calculated over all targets, is shown in Figure 2 (precision-recall curves are shown in Supplementary Fig. 2; the performance evaluation for the Molecular Function ontology when proteins annotated with only the “protein binding” term were included is shown in Supplementary Fig. 3). All methods were compared with two baseline tools: (i) BLAST, in which all GO terms of an experimentally annotated sequence (template) from Swiss-Prot were transferred to the target sequence such that the scores equaled pairwise sequence identity between the template and the target (terms with multiple hits retained the highest score), and (ii) a naive method (Naive), in which each GO term for each target was scored with the relative frequency of this term in Swiss-Prot over all annotated proteins (Online Methods). We also evaluated the quality of position-specific iterated (PSI)-BLAST predictions, but we found that it did not provide any advantage over BLAST: specifically, Fmax(PSI-BLAST) = Fmax(BLAST) = 0.38 for Molecular Function; Fmax(PSI-BLAST) = 0.24 and Fmax(BLAST) = 0.26 for Biological Process. We believe that the improved ability of PSI-BLAST to identify remote homologs has been canceled out by its reranking of close hits.

(a,b) The maximum F-measure for the top-performing methods for Molecular Function ontology (a) and Biological Process ontology (b). All panels show the top ten participating methods in each category as well as the BLAST and Naive baseline methods. Note that 33 models outperformed BLAST in the Molecular Function category, whereas 26 models outperformed BLAST in the Biological Process category (cutoff scores below which methods were excluded from the panels were 0.468 and 0.300 for the Molecular Function and Biological Process categories, respectively). In the Molecular Function category, proteins with “protein binding” as their only leaf term were excluded from the analysis because the protein binding term was not considered informative (results that include those proteins are presented in Supplementary Fig. 3). A perfect predictor would be characterized with Fmax = 1. Confidence intervals (95%) were determined using bootstrapping with n = 10,000 iterations on the set of target sequences. For cases in which a principal investigator participated in multiple teams, only the results of the best-scoring method are presented.
We observed a substantial performance difference in the ability to predict the two GO categories (Molecular Function versus Biological Process). This can be partly explained by the topological differences between the ontologies (respectively: number of terms, 8,728 and 18,982; branching factor, 5.9 and 6.4; maximum depth, 11 and 10; number of leaf terms, 7,003 and 8,125). However, more fundamentally, terms in the Biological Process ontology were associated with a more abstract level of function. Such terms were less likely to be predictable solely from amino acid sequence, which was the data source used by most methods in this experiment and may critically depend on the cellular and organismal context.
Predictor performance on categories of targets
We divided the target sequences into a variety of different categories to compare predictor performance across each category. The first division was between easy and difficult targets. A target was considered easy if it had a 60% or higher sequence identity with any experimentally annotated protein. We manually chose the threshold of 60% after plotting the distribution of sequence identities between targets and annotated proteins (Supplementary Fig. 4). This resulted in 188 easy and 343 difficult targets in the Molecular Function category and 247 easy and 340 difficult targets in the Biological Process category. Supplementary Figure 5 shows the precision-recall curves for both categories. Perhaps unsurprisingly, whereas BLAST outperformed Naive in the easy target category, their performance was similar for the difficult targets. However, because of the similar performance among top-ranked predictors over easy and difficult targets, the sequence identity–based classification of targets does not seem to accurately reflect the uncertainty associated with a protein's true function (except for with BLAST). This may be because the methods can compensate for the differences in sequence similarity of the best hit by using multiple sequence hits as well as other data sources.
Next we compared prediction performance on eukaryotic versus prokaryotic targets (Supplementary Fig. 6). Performance was generally similar in the Molecular Function category, but in the Biological Process category we observed high prediction accuracy for prokaryotic targets. We believe this is because most prokaryotic targets came from E. coli, for which reliable experimental data are available, whereas the data for eukaryotic targets came from sources with highly variable coverage and quality. It is important to note that the particular calculation of precision and recall (Online Methods) adversely affected methods that predicted on only eukaryotic targets (BMRF, ConFunc, GOstruct and Tian Lab) and resulted in lower overall performance for these methods. Detailed results for eukaryotic and prokaryotic targets, as well as several individual organisms, are shown in Supplementary Figures 6 and 7.
Finally we separated targets into sequences containing a single domain versus sequences containing multiple protein domains, with domains defined according to Pfam-A classification38 (targets without any Pfam-A hits were grouped together with single-domain proteins). Multidomain proteins were generally longer; however, they were not associated with more functional terms than single-domain proteins. By analyzing the performance of the top ten methods in each category, we found that although the overall accuracy was higher on single-domain proteins, results were significant in only the Molecular Function category and for eukaryotic targets (P = 1.4 × 10−5, n = 10, paired t-test; Fig. 3). Though generally expected, the higher performance on single-domain proteins further emphasizes the need for developing methods that can optimally combine sequence information from multiple domains along with other information to produce a relatively small set of predicted terms.

(a) Distribution of target proteins with respect to the number of Pfam domains they contain. (b) Performance evaluation in the Molecular Function category. Each of the ten top-performing methods showed higher accuracy (higher Fmax) on single-domain proteins. Confidence intervals (95%) were determined using bootstrapping with n = 10,000 iterations on the set of target sequences.
Predictor performance on functional terms
We assessed the ability of methods to predict individual GO terms by calculating the area under the receiver operating characteristic (ROC) curve (AUC; Online Methods). To more confidently assess the performance in predicting individual terms, we considered only terms for which at least 15 targets were annotated. Average AUC values were then calculated from the five top-performing models in each ontology, excluding those models that provide only single-score predictions.
Using the above criteria, we were able to calculate average AUC values for 28 Molecular Function and 223 Biological Process terms (Supplementary Table 2). We found a clear distinction between the average AUC of Molecular Function terms generally associated with catalytic and transporter activity and those associated with binding. In general, the prediction of terms associated with binding showed lower AUC values, even though proteins were biased toward being annotated with binding terms. Among the Biological Process terms, we found, as expected, low AUC values associated with less specific terms such as “locomotion”, “cellular process” and “response to stress.” We also found that prediction of terms associated with “cell adhesion”, “metabolic process”, “transcription” and “regulation of gene expression” showed high performance. We tested whether a high predictor AUC value on individual terms was due to high levels of sequence similarity among sequences experimentally annotated with those terms, and we found a moderate level of correlation (data not shown).
Case study
Here we illustrate some challenges associated with computational protein function prediction. We provide a detailed analysis of the human mitochondrial polynucleotide phosphorylase 1 (hPNPase, encoded by PNPT1), a large (783-amino-acid) protein with seven Pfam domains (Fig. 4a). Human PNPase is characterized by several experimentally determined functions, which makes it an attractive target with which to evaluate the performance of prediction methods. hPNPase belongs to a family of exoribonucleases, which hydrolyze single-stranded RNA in the 3′-to-5′ direction. In complex with other components of the mitochondrial degradasome, hPNPase mediates the translocation of small RNAs into the mitochondrial matrix39. It is also proposed to be involved in several biological processes including cell-cycle arrest40, cellular senescence and response to oxidative stress41.

(a) Domain architecture of human PNPT1 gene according to the Pfam classification. For each domain, the numbers of different leaf terms (for the Molecular Function and Biological Process categories) associated with any protein in Swiss-Prot database containing this domain are shown. (b) Molecular Function terms (six of which are leaves) associated with the human PNPT1 gene in Swiss-Prot as of December 2011. Colored circles represent the predicted terms for three representative methods as well as two baseline methods. The prediction threshold for each method was selected to correspond to the point in the precision-recall space that provides the maximum F-measure. J (blue), Jones-UCL; O (magenta), Team Orengo; d (navy blue), dcGO; B (green), BLAST; N (brown), Naive. Dashed lines indicate the presence of other terms between the source and destination nodes.
Owing to its involvement in several molecular functions and biological processes, the comprehensive and accurate listing of functions of hPNPase is a challenging task. Furthermore, though PNPase is prevalent in bacteria and eukarya, it has accumulated several lineage-specific functions. Specifically, whereas bacterial and chloroplast PNPase have demonstrated exoRNase and polyadenylation activities, hPNPase functions predominantly as an RNA importer39, showing exoRNase activity only in vitro42. Finally, hPNPase is a mitochondrial protein found in the intermembrane matrix. Taken together with its involvement in the rRNA import process, this suggests the need to predict the cellular compartment as part of a comprehensive understanding of function.
Figure 4b shows the experimental GO-term annotation of hPNPase as well as the terms predicted by a representative set of the ten top-performing methods. Within the Molecular Function terms, none of the methods predicted poly(U) or poly(G) RNA binding43 or microRNA binding. However, most methods that did predict function correctly predicted 3′-to-5′ exoRNase activity and polyribonucleotide nucleotidyltransferase activity. It should be noted that poly(U) and poly(G) binding and microRNA binding are uncommon throughout the PNPase lineage. This may be the reason why none of the programs predicted these terms.
In the Biological Process category, the most prominent function of hPNPase in the literature is the import of nuclear 5S rRNA into the mitochondrion39; indeed, it is hypothesized that this is the reason for hPNPase's location in the intermembrane matrix. However, this function, along with other important terms, such as cellular senescence, was not predicted by any of the top-performing methods at the optimal threshold levels. Generally, the Biological Process predictions were highly nonspecific for most models. In sum, the multidomain architecture of hPNPase, its pleiotropy and the different functions it assumes in different taxa all contribute to the challenge of correctly predicting hPNPase function.
Discussion
Protein function is difficult to predict for several reasons. First, function is studied from various aspects and at multiple levels: for example, it describes the biochemical events involving the protein and also how each protein affects pathways, cells, tissues and the entire organism. Second, protein function and its experimental characterization are context dependent: a particular experiment is unlikely to determine a protein's entire functional repertoire under all conditions (such as temperature, pH or the presence of interacting partners). Third, proteins are often multifunctional44 and promiscuous45; in fact, of the experimentally annotated proteins in Swiss-Prot, 30% have more than one leaf term in the Molecular Function ontology, as do 60% in the Biological Process ontology16. Fourth, in addition to being incomplete, available functional annotations are error prone because of experiment interpretation or curation issues37,46. Finally, current efforts largely map protein function to gene names, thus confounding the functions of potentially diverse isoforms. Despite these challenges, the CAFA experiment revealed progress in automated function annotation over the past decade.
Top algorithms are useful and outperform BLAST considerably.
The first generation of function prediction methods performed a simple function transfer via pairwise sequence similarity: that is, the most similar annotated hit was used as the basis of function prediction47. Several studies have been aimed at characterizing performance of these methods3,16,48. The CAFA experiment provides evidence that the best algorithms universally outperform simple functional transfer. The experiment also showed that BLAST is largely ineffective at predicting functional terms related to the Biological Process ontology. This is possibly due to homologs assuming different biological roles in different tissues and organisms49.
Principles underlying best methods.
The methods evaluated in CAFA used a variety of biological and computational concepts. Most methods used sequence alignments with an underlying hypothesis that sequence similarity is correlated with functional similarity. Recent studies have shown that this correlation is weak when applied to pairs of proteins16 and that domain assignments alone are not sufficient to resolve function50. Therefore, the main challenge for the alignment-based methods was to devise ways of combining multiple hits or identified domains into a single prediction score. More than half the methods used data beyond sequence similarity, such as types of evolutionary relationships, protein structure, protein-protein interactions or gene expression data. The challenge for these methods was finding ways to integrate disparate data sources and properly handle incomplete and noisy data. For example, the protein-protein interaction network for yeast is nearly complete (although noisy), whereas the sets of available interactions for Arabidopsis thaliana and Xenopus laevis are rather sparse (but less noisy, given a smaller fraction of high-throughput data). Finally, some methods used literature mining, which could also be related to the task of retrieving the correct function rather than predicting it from the set of textual descriptions about a protein. As information retrieval is still a challenging research problem, it was useful to evaluate performance accuracy of the methods that exploited literature searching.
On the computational side, most methods used machine learning principles: that is, they typically found combinations of sequence-based or other features that correlated with a specific function in a training set of experimentally annotated proteins. Although these methods automate the task of learning and inference, they also require experience in selecting classification models (for example, a support vector machine), learning parameters, features or the training data that would result in good performance. In addition, the sets of rules according to which these methods score new proteins may be difficult to interpret. Despite the added layer of complexity, machine learning generally played a positive role in increasing prediction accuracy. Thus, it may be expected that top-performing methods in the future will be based on well-founded principles of statistical learning and inference.
With few exceptions, the same methods that performed well for the Molecular Function category also performed well in the Biological Process category; however, their overall performance in the latter category was inferior. We believe that this is because homologs may perform their biochemical roles in different pathways, and prediction methods are less able to discern those differences at this time. Because sequence similarity is less predictive of the biological roles of proteins, a key to improving the prediction of a protein's biological function will be our ability to generate better-quality systems data and to develop computational tools that exploit them.
Evaluation metrics.
The choice of evaluation metrics was another interesting aspect of the experiment. We decided to use simple and easily interpretable metrics (Online Methods), although simple measures based on precision and recall have limitations in this domain. First, such metrics are sensitive to problems related to the nonuniform distribution of proteins over GO terms due to the equal weight given to all terms. Second, proteins are weighted equally regardless of the depth of their experimental annotation: that is, a correct prediction on a protein annotated with a shallow term (and its ancestors) is considered as good as a correct prediction on a protein annotated with a deep term. Third, a method that reports only high-confidence deep annotations for a small number of proteins will be penalized (in terms of recall) compared to a method that annotates all proteins with frequently occurring general terms. Finally, in some cases, it is not clear whether to consider a prediction correct or erroneous; with our current approach, we consider only the experimental annotation and more general predictions to be correct. As such, correct and highly specific predictions will be penalized if the protein has been experimentally annotated only in a more generic way. For those reasons, we encourage the development of a diverse set of metrics to understand better the strengths and weaknesses of function prediction in different application contexts.
Summary.
The CAFA experiment was designed to enable the community to periodically reassess the performance of computational methods as experimental evidence accumulates. In addition, the large set of targets released to the community provided us with prediction scores for most proteins across multiple methods. If the experiment is repeated, we expect to be able to evaluate future methods against those that deposited predictions in the first CAFA experiment and therefore monitor progress in the field over time.
Though the CAFA experiment has seen positive outcomes, it is also clear that there is significant room for the improvement of protein function prediction. In the Molecular Function category, performance may be considered accurate. However, in the Biological Process category, the overall performance of the top-scoring methods was below our expectations. This was true for any subset of targets. Another area in need of improvement is the availability of tools that can easily be used by experimental scientists and that can be maintained and upgraded on a regular basis. As the community moves beyond the initial algorithm development stage, there is a need to provide stand-alone tools (similar to the BLAST package) capable of predicting protein function at several different levels.
Given its significance, its intellectual challenge and the growing need for accurate functional annotations, protein function prediction is likely to remain an active and expanding research field. As the quality of data improves and the number of experimentally annotated proteins grows, we expect that computational prediction will become more accurate. On the basis of the CAFA experiment, it seems that the most powerful methods will be those that will devise principled ways to integrate a variety of experimental evidence and weigh different data appropriately and separately for each functional term. Novel ideas and approaches are necessary as well.
Methods
Experiment design.
The CAFA experiment was conceived in the fall of 2009. The Organizing, Steering and Assessment Committees were designated by March 2010. During the same period a feasibility study was conducted to determine the rate at which experimental annotations accumulated in Swiss-Prot between 2007 and 2010. We concluded that a period of 6 months or more would result in annotations of at least 300–500 proteins, which would be sufficient for statistically reliable comparisons between algorithms. The experiment was announced in July 2010 and subsequently heavily advertised. The set of targets was announced on 15 September 2010 with a prediction submission deadline of 18 January 2011 (Fig. 1).
Predictors were asked to submit predictions for each target along with scores ranging between 0 and 1 that would indicate the strength of the prediction (ideally, posterior probabilities). To reduce the amount of data submitted, we allowed no more than 1,000 term annotations for each target. Prediction algorithms were also associated with keywords from a predetermined set, which were used to provide insight into the types of approaches that performed well. A list of all participating teams, principal investigators and methods is provided in Supplementary Table 3.
Initial comparative evaluation of models was conducted in July 2011 during the Automated Function Prediction (AFP) Special Interest Group (SIG) meeting associated with the ISMB 2011 conference. This study provides the analysis on a set of targets from the Swiss-Prot database from 14 December 2011.
Target proteins.
A set of 48,298 target amino acid sequences was announced in September 2010. Because our feasibility study showed that only a handful of species were steadily accumulating experimental annotations, target proteins were selected from predominantly those species. The targets contained all the sequences in Swiss-Prot from 7 eukaryotic and 11 prokaryotic species that were not associated with any experimental GO terms. A protein was considered experimentally annotated if it was associated with GO terms having EXP, IDA, IMP, IGI, IEP, TAS or IC evidence codes. An additional set of targets was announced consisting of 1,301 enzymes from multiple species and metagenomic studies that were the focus of the Enzyme Function Initiative project51.
18 January 2011 was set as the deadline for the submission of function predictions. To exclude targets that had accumulated annotations before the submission deadline, we obtained annotated proteins from the January version of Swiss-Prot, GO35 and UniProt-GOA52 databases. We refer to those sets of proteins as Swiss-Prot(t0), GO(t0) and GOA(t0), respectively.
We later determined the evaluation set of target proteins by downloading a newer version of the Swiss-Prot database, denoted as Swiss-Prot(t). The set of target proteins for the CAFA experiment was then selected using the following scheme

Note that this experiment was designed to allow for reassessment of algorithm performance at some later point in time.
Evaluation metrics.
Algorithms were evaluated in two scenarios: (i) protein centric and (ii) term centric. These two types of evaluations were chosen to address the following related questions: (i) what is the function of a particular protein? and (ii) what are the proteins associated with a particular functional term?
1. Protein-centric metrics. The main evaluation metric in CAFA was the precision-recall curve. For a given target protein i and some decision threshold t ∈ [0, 1], the precision and recall were calculated as

and

where f is a functional term in the ontology, Ti is a set of experimentally determined (true) nodes for protein i, and Pi(t) is a set of predicted terms for protein i with score greater than or equal to t. Note that f ranges over the entire ontology (separately for Molecular Function and Biological Process), excluding the root. Function I(·) is the standard indicator function. For a fixed threshold t, a point in the precision-recall space is then created by averaging precision and recall across targets. Precision at threshold t is calculated as

where m(t) is the number of proteins on which at least one prediction was made above threshold t. On the other hand, recall is calculated over all n proteins in a target set, i.e.,

regardless of the prediction threshold. The maximum ratio between m(t) and n (over all thresholds t) is referred to as the prediction coverage. If a particular algorithm outputs only a fixed score (for example, 1), its performance will be described by a single point in the precision-recall space instead of by a curve.
For submissions with unpropagated functional annotations, the organizers recursively propagated all scores toward the root of the ontology such that each parent term received the highest score among its children. The annotations were propagated regardless of the type of relationship between terms. We note that it may be useful to associate different weights with different ontological terms and therefore reward algorithms that are better at predicting more difficult or less frequent terms. However, for simplicity, in our main evaluation, each term was associated with an equal weight of 1 (weighted precision-recall curves are shown in Supplementary Fig. 8).
The main appeal of the precision-recall evaluation stems from its interpretability: if, for a particular threshold, a method has a precision of 0.7 at a recall of 0.5, this indicates that on average 70% of the predicted terms will be correct and that about 50% of the true annotations will be revealed for a previously unseen protein. On the other hand, a limitation of this evaluation method is that the terms are not independent because of ontological relationships, and the unequal level of specificity of functional terms at the same depth in the ontology was not taken into account.
To provide a single number for comparisons between methods, we calculated the F-measure (a harmonic mean between precision and recall) for each threshold and calculated its maximum value over all thresholds. More specifically, we used

2. Term-centric metrics. For each functional term f, we calculated the area under the ROC curve (AUC) using a sliding threshold approach. The ROC curve is a plot of sensitivity (or recall) for a given false positive rate (or 1 − specificity). The sensitivity and specificity for a particular functional term f and threshold t were calculated as

and

where Pi(t) is the set of predicted terms for protein i with a score greater than or equal to threshold t, and Ti is the set of true terms for protein i. Once the sensitivity and specificity for a particular functional term were determined over all proteins for different values of the prediction threshold, the AUC was calculated using the trapezoid rule. The AUC has a useful probabilistic interpretation: given a randomly selected protein associated with functional term f and a randomly selected protein not associated with f, the AUC is the probability that the former protein will receive a higher score than the latter protein53.
Baseline methods.
In addition to the methods implemented by the community, we used two additional methods as baselines. The first such method is based on BLAST11 hits to the database of proteins with experimentally annotated functions (roughly 37,000 proteins). The score for a particular term was calculated as the maximum sequence identity between the target protein and any protein experimentally annotated with that term. More specifically, if a particular protein was hit with the local sequence identity 75%, all its functional terms were transferred to the target sequence with the score of 0.75. If a term was hit with multiple sequence identity scores, the highest one was retained. BLAST was selected as a baseline method because of its ubiquitous use. We note that the same method was tested using the BLAST bit scores, which resulted in slightly better performance. In addition to BLAST, we also tested PSI-BLAST11, in which the profiles were created using the most recent “nr” database and −j 3 −h 0.0001 parameters. These profiles were then searched against a database of experimentally annotated proteins with E-values used to rank the hits. The second baseline method, referred to as Naive, used the prior probability of each term in the database of experimentally annotated proteins as the prediction score for that term. If a term “protein binding” occurs with relative frequency 0.25, each target protein was associated with score 0.25 for that term. Thus, the Naive method assigned the same predictions to all targets.
References
Liolios, K. et al. The Genomes On Line Database (GOLD) in 2009: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 38, D346–D354 (2010).
Bork, P. et al. Predicting function: from genes to genomes and back. J. Mol. Biol. 283, 707–725 (1998).
Rost, B., Liu, J., Nair, R., Wrzeszczynski, K.O. & Ofran, Y. Automatic prediction of protein function. Cell Mol. Life Sci. 60, 2637–2650 (2003).
Watson, J.D., Laskowski, R.A. & Thornton, J.M. Predicting protein function from sequence and structural data. Curr. Opin. Struct. Biol. 15, 275–284 (2005).
Friedberg, I. Automated protein function prediction—the genomic challenge. Brief. Bioinform. 7, 225–242 (2006).
Sharan, R., Ulitsky, I. & Shamir, R. Network-based prediction of protein function. Mol. Syst. Biol. 3, 88 (2007).
Lee, D., Redfern, O. & Orengo, C. Predicting protein function from sequence and structure. Nat. Rev. Mol. Cell Biol. 8, 995–1005 (2007).
Punta, M. & Ofran, Y. The rough guide to in silico function prediction, or how to use sequence and structure information to predict protein function. PLoS Comput. Biol. 4, e1000160 (2008).
Rentzsch, R. & Orengo, C.A. Protein function prediction—the power of multiplicity. Trends Biotechnol. 27, 210–219 (2009).
Xin, F. & Radivojac, P. Computational methods for identification of functional residues in protein structures. Curr. Protein Pept. Sci. 12, 456–469 (2011).
Altschul, S.F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Jensen, L.J. et al. Prediction of human protein function from post-translational modifications and localization features. J. Mol. Biol. 319, 1257–1265 (2002).
Wass, M.N. & Sternberg, M.J. ConFunc—functional annotation in the twilight zone. Bioinformatics 24, 798–806 (2008).
Martin, D.M., Berriman, M. & Barton, G.J. GOtcha: a new method for prediction of protein function assessed by the annotation of seven genomes. BMC Bioinformatics 5, 178 (2004).
Hawkins, T., Luban, S. & Kihara, D. Enhanced automated function prediction using distantly related sequences and contextual association by PFP. Protein Sci. 15, 1550–1556 (2006).
Clark, W.T. & Radivojac, P. Analysis of protein function and its prediction from amino acid sequence. Proteins 79, 2086–2096 (2011).
Pellegrini, M., Marcotte, E.M., Thompson, M.J., Eisenberg, D. & Yeates, T.O. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc. Natl. Acad. Sci. USA 96, 4285–4288 (1999).
Marcotte, E.M. et al. Detecting protein function and protein-protein interactions from genome sequences. Science 285, 751–753 (1999).
Enault, F., Suhre, K. & Claverie, J.M. Phydbac “Gene Function Predictor”: a gene annotation tool based on genomic context analysis. BMC Bioinformatics 6, 247 (2005).
Engelhardt, B.E., Jordan, M.I., Muratore, K.E. & Brenner, S.E. Protein molecular function prediction by Bayesian phylogenomics. PLoS Comput. Biol. 1, e45 (2005).
Gaudet, P., Livstone, M.S., Lewis, S.E. & Thomas, P.D. Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium. Brief. Bioinform. 12, 449–462 (2011).
Deng, M., Zhang, K., Mehta, S., Chen, T. & Sun, F. Prediction of protein function using protein-protein interaction data. J. Comput. Biol. 10, 947–960 (2003).
Letovsky, S. & Kasif, S. Predicting protein function from protein/protein interaction data: a probabilistic approach. Bioinformatics 19 (suppl. 1), i197–i204 (2003).
Vazquez, A., Flammini, A., Maritan, A. & Vespignani, A. Global protein function prediction from protein-protein interaction networks. Nat. Biotechnol. 21, 697–700 (2003).
Nabieva, E., Jim, K., Agarwal, A., Chazelle, B. & Singh, M. Whole-proteome prediction of protein function via graph-theoretic analysis of interaction maps. Bioinformatics 21 (suppl. 1), i302–i310 (2005).
Pazos, F. & Sternberg, M.J. Automated prediction of protein function and detection of functional sites from structure. Proc. Natl. Acad. Sci. USA 101, 14754–14759 (2004).
Pal, D. & Eisenberg, D. Inference of protein function from protein structure. Structure 13, 121–130 (2005).
Laskowski, R.A., Watson, J.D. & Thornton, J.M. Protein function prediction using local 3D templates. J. Mol. Biol. 351, 614–626 (2005).
Huttenhower, C., Hibbs, M., Myers, C. & Troyanskaya, O.G. A scalable method for integration and functional analysis of multiple microarray datasets. Bioinformatics 22, 2890–2897 (2006).
Troyanskaya, O.G., Dolinski, K., Owen, A.B., Altman, R.B. & Botstein, D. A Bayesian framework for combining heterogeneous data sources for gene function prediction (in Saccharomyces cerevisiae). Proc. Natl. Acad. Sci. USA 100, 8348–8353 (2003).
Lee, I., Date, S.V., Adai, A.T. & Marcotte, E.M. A probabilistic functional network of yeast genes. Science 306, 1555–1558 (2004).
Costello, J.C. et al. Gene networks in Drosophila melanogaster: integrating experimental data to predict gene function. Genome Biol. 10, R97 (2009).
Kourmpetis, Y.A., van Dijk, A.D., Bink, M.C., van Ham, R.C. & ter Braak, C.J. Bayesian Markov Random Field analysis for protein function prediction based on network data. PLoS ONE 5, e9293 (2010).
Sokolov, A. & Ben-Hur, A. Hierarchical classification of gene ontology terms using the GOstruct method. J. Bioinform. Comput. Biol. 8, 357–376 (2010).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 (2000).
Bairoch, A. et al. The Universal Protein Resource (UniProt). Nucleic Acids Res. 33, D154–D159 (2005).
Schnoes, A.M., Brown, S.D., Dodevski, I. & Babbitt, P.C. Annotation error in public databases: misannotation of molecular function in enzyme superfamilies. PLoS Comput. Biol. 5, e1000605 (2009).
Punta, M. et al. The Pfam protein families database. Nucleic Acids Res. 40, D290–D301 (2012).
Wang, G. et al. PNPASE regulates RNA import into mitochondria. Cell 142, 456–467 (2010).
Sarkar, D. et al. Down-regulation of Myc as a potential target for growth arrest induced by human polynucleotide phosphorylase (hPNPaseold-35) in human melanoma cells. J. Biol. Chem. 278, 24542–24551 (2003).
Wu, J. & Li, Z. Human polynucleotide phosphorylase reduces oxidative RNA damage and protects HeLa cell against oxidative stress. Biochem. Biophys. Res. Commun. 372, 288–292 (2008).
Wang, D.D., Shu, Z., Lieser, S.A., Chen, P.L. & Lee, W.H. Human mitochondrial SUV3 and polynucleotide phosphorylase form a 330-kDa heteropentamer to cooperatively degrade double-stranded RNA with a 3′-to-5′ directionality. J. Biol. Chem. 284, 20812–20821 (2009).
Portnoy, V., Palnizky, G., Yehudai-Resheff, S., Glaser, F. & Schuster, G. Analysis of the human polynucleotide phosphorylase (PNPase) reveals differences in RNA binding and response to phosphate compared to its bacterial and chloroplast counterparts. RNA 14, 297–309 (2008).
Jeffery, C.J. Moonlighting proteins. Trends Biochem. Sci. 24, 8–11 (1999).
Khersonsky, O. & Tawfik, D.S. Enzyme promiscuity: a mechanistic and evolutionary perspective. Annu. Rev. Biochem. 79, 471–505 (2010).
Brenner, S.E. Errors in genome annotation. Trends Genet. 15, 132–133 (1999).
Doolittle, R.F. Of URFS and ORFS: A Primer on How to Analyze Derived Amino Acid Sequences (University Science Books, 1986).
Addou, S., Rentzsch, R., Lee, D. & Orengo, C.A. Domain-based and family-specific sequence identity thresholds increase the levels of reliable protein function transfer. J. Mol. Biol. 387, 416–430 (2009).
Nehrt, N.L., Clark, W.T., Radivojac, P. & Hahn, M.W. Testing the ortholog conjecture with comparative functional genomic data from mammals. PLoS Comput. Biol. 7, e1002073 (2011).
Brown, S.D., Gerlt, J.A., Seffernick, J.L. & Babbitt, P.C. A gold standard set of mechanistically diverse enzyme superfamilies. Genome Biol. 7, R8 (2006).
Gerlt, J.A. et al. The Enzyme Function Initiative. Biochemistry 50, 9950–9962 (2011).
Barrell, D. et al. The GOA database in 2009—an integrated Gene Ontology Annotation resource. Nucleic Acids Res. 37, D396–D403 (2009).
Hanley, J.A. & McNeil, B.J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 143, 29–36 (1982).
Acknowledgements
We gratefully acknowledge I. Landsberg-Halperin for coining the term “CAFA,” T. Theriault for the initial graphical design of Figure 1, G. Schuster for illuminating discussions on hPNPase and A. Facchinetti, R. Velasco, E. Cilia, D.A. Lee, P. Vats, R. Banerjee and A. Bayaskar for their participation in various individual projects. The Automated Function Prediction Special Interest Group meeting at the ISMB 2011 conference was supported by the US National Institutes of Health (NIH) grant R13 HG006079-01A1 (P.R.) and Office of Science (Biological and Environmental Research), US Department of Energy (DOE BER), grant DE-SC0006807TDD (I.F.). Individual projects were partially supported by the following awards: US National Science Foundation (NSF) DBI-0644017 (P.R.), ABI-0965768 (A.B.-H.), DMS0800568 (D. Kihara), CCF-0905536 and DBI-1062455 (O.L.), DBI-0965768 (K.V.) and ABI-1146960 (I.F.); Marie Curie International Outgoing Fellowship PIOF-QA-2009-237751 (S.R.); PRIN 2009 project 009WXT45Y Italian Ministry for University and Research MIUR (R.C.); NIH GM093123 (J.C.), GM075004 and GM097528 (D. Kihara), GM079656 and GM066099 (O.L.), LM00945102 (C.F.), R01 GM071749 (S.E.B.) and LM009722 and HG004028 (S.D.M.); FP7 “Infrastructures” project TransPLANT Award 283496 (A.D.J.v.D.); UK Biotechnology and Biological Sciences Research Council (BBSRC) grant BB/G022771/1 (J.G.), BB/K004131/1 (A.P.) and BB/F020481/1 (M.N.W. and M.J.E.S.); BBSRC (D.T.J.); Marie Curie Intra European Fellowship Award PIEF-GA-2009-237292 (D.T.J.); Department of Information Technology, Government of India (R.J.); EU, BBSRC and NIH Awards (C.O.); Natural Sciences and Engineering Research Council of Canada Discovery Award #298292-2009, Discovery Accelerator Award #380478-2009, Canada Foundation for Innovation New Opportunities Award 10437 and Ontario's Early Researcher Award #ER07-04-085 (H.S.); Netherlands Genomics Initiative (Y.A.I.K. and C.J.F.t.B.); National Information and Communication Technology Australia (K.V.); National Natural Science Foundation of China grants 31071113 and 30971643 (W.T.); DOE BER KP110201 (S.E.B.); and Alexander von Humboldt Foundation (B.R.). P.R. acknowledges the Indiana University high-performance computing resources (NSF grant CNS-0723054). I.F. acknowledges the assistance of the high-performance computing group at Miami University.
Author information
Authors and Affiliations
Contributions
P.R. and I.F. conceived of the CAFA experiment, supervised the project and wrote most of the manuscript. S.D.M. participated in the design of and supervised the method assessment. W.T.C. performed the analysis of feasibility of the experiment and most of the target and performance analysis and contributed to writing. P.R. and W.T.C. designed and produced figures. T.R.O. developed the web interface, including the portal for submission and the storage of predictions. T.R.O. and T.W. verified the assessment code and participated in analysis. A.M.S. designed and performed the analysis of targets. A. Bairoch, M.L., P.C.B., S.E.B., C.O. and B.R. steered the CAFA experiment, provided critical guidance and participated in writing. The remaining authors participated in the experiment, provided writing and data for their methods and contributed comments on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–8, Supplementary Table 3 and Supplementary Note (PDF 2718 kb)
Supplementary Table 1
List of all target sequences and their experimentally determined functional terms. (XLSX 92 kb)
Supplementary Table 2
Area under the ROC curves (AUC) for the functional terms covering at least 15 target sequences. (XLSX 23 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Radivojac, P., Clark, W., Oron, T. et al. A large-scale evaluation of computational protein function prediction. Nat Methods 10, 221–227 (2013). https://doi.org/10.1038/nmeth.2340
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nmeth.2340
This article is cited by
-
Improvements in viral gene annotation using large language models and soft alignments
BMC Bioinformatics (2024)
-
GO2Sum: generating human-readable functional summary of proteins from GO terms
npj Systems Biology and Applications (2024)
-
Discovering functionally important sites in proteins
Nature Communications (2023)
-
Exploring complexity of class-A Beta-lactamase family using physiochemical-based multiplex networks
Scientific Reports (2023)
-
User structural information in priority-based ranking for top-N recommendation
Advances in Computational Intelligence (2023)
