
Abstract
“Nature has provided, in the white corpuscles as you call them—in the phagocytes as we call them—a natural means of devouring and destroying all disease germs. There is at bottom only one genuinely scientific treatment for all diseases, and that is to stimulate the phagocytes.” So opined B.B. in G.B. Shaw's The Doctor's Dilemma1 in a dramatic restatement of a key portion of Ilya Metchnikoff's Nobel Prize address: “Whenever the organism enjoys immunity, the introduction of infectious microbes is followed by the accumulation of mobile cells, of white corpuscles of the blood in particular which absorb the microbes and destroy them. The white corpuscles and the other cells capable of doing this have been designated 'phagocytes,' (i.e., devouring cells) and the whole function that ensures immunity has been given the name of 'phagocytosis'”2. Based on these insights into the foundation of resistance to infectious disease, Metchnikoff was awarded the 1908 Nobel Prize in Physiology or Medicine together with Paul Ehrlich (Fig. 1). Although both were cited for discoveries in immunity, the contributions of the two men seem worlds apart. Ehrlich's studies did not deal with generic responses to infection, but rather with the highly specific nature of antibodies and their relationship to the cells producing them: “As the cell receptor is obviously preformed, and the artificially produced antitoxin only the consequence, i.e. secondary, one can hardly fail to assume that the antitoxin is nothing else but discharged components of the cell, namely receptors discharged in excess”3. But biological systems are just that—systems—and the parts need to work together. And so we arrive, a century later, at an appreciation for just how intimately related these two seemingly disparate aspects of host defense really are.

Photo Researchers, Inc.
Paul Erlich (left) and Ilya Metchnikoff (right), winners of the 1908 Nobel Prize in Physiology or Medicine.
Similar content being viewed by others

Main
Transitioning from the age of specificity
The point-counterpoint of the 1908 dual Nobel award set the stage for the ensuing 100 years of advances in immunology. For most of the twentieth century, it was Ehrlich's focus on the remarkable specificity of immune reactions and the lymphoid components of the hematopoietic system that obsessed the research community. Metchnikoff's phagocytes held a secondary position in the immunological pantheon, being viewed mainly as the handservants of antigen-specific immune factors such as opsonizing antibody or activating cytokines like interferon (IFN)-γ. Even Shaw's character held this latter notion when one looks more closely at his text: “Find the germ of the disease; prepare from it a suitable anti-toxin; inject it three times a day quarter of an hour before meals; and what is the result? The phagocytes are stimulated”1. This is a formulation that gives clear pride of place to antibodies as the controlling elements and phagocytes as the recruited henchmen.
The iconography of journals, meeting posters and books for the last few decades of the twentieth century makes apparent the preeminence of antigen-related recognition in immunological thinking. Beginning in the 1970s, the polypeptide chain organization of antibodies4,5,6 was a ubiquitous image. In the late 1980s, this was replaced by the newly determined structure of the major histocompatibility complex (MHC) class I molecule7. Each of these images encapsulated several major accomplishments in immunological research. The basis for the exquisite chemical specificity of the antitoxins studied by Ehrlich was revealed by crystallographic studies of antigen-antibody complexes. The origin of the immense binding-site diversity of such proteins was uncovered by molecular studies documenting the remarkable role of somatic gene segment recombination in producing the mature antibody molecule8, a process also accounting for the diversity of T-cell antigen receptors9,10. The enigmatic nature of genetic control of immune responses at the T-cell level (IR genes11) and of the phenomenon of 'MHC-restricted antigen responses'12,13 was explained conjointly by (i) the roles of MHC class I and class II membrane proteins in presenting antigenic fragments to clonally distributed receptors on T lymphocytes14,15 and (ii) the impact of natural MHC molecule polymorphism on the particular peptides that bound well to each allelic product16,17. The biochemical differences in the peptide binding strategies of MHC class I versus class II molecules18 provided a strikingly clear picture of how these two subsets of highly related proteins evolved for optimized capture of distinct peptides in different intracellular compartments19.
Although the adaptive (antigen-specific, lymphocyte-based) response remains a substantial focus of immunological research, the past decade has seen a renewal of interest in the other (innate) components of host defense. Phagocytes as effectors per se have not been the main subjects of investigation, but myeloid cells have certainly come to the fore as key players in the guise of dendritic cells (DCs), which are thought to orchestrate the extent and quality of antigen-specific immune responses. An entirely new appreciation has emerged of natural killer (NK) cells, previously given short shrift because of their lack of clonally distributed receptors. The analysis of molecules other than immunoglobulins or T-cell receptors that mediate pathogen recognition and response has assumed a major position in the research portfolio. These new directions in understanding the role of innate immunity in host defense join studies on regulatory processes controlling the quantitative and qualitative features of adaptive immune responses as some of the most significant advances in immunology over the past decade.
Here I summarize the progress made in these and other key areas of investigation. Because up-to-date, expert reviews are available on each topic discussed, it is not my intent to dwell on the details. Rather, I focus on those overarching aspects of the work that were unexpected and how the findings changed immunological thinking, ending with some speculations about the future of the field.
Innate immunity to the fore: immunologists' 'dirty little secret'
Over the past decade, new insights have emerged into the enzymology of gene recombination (the cooperation of the RAG 1 and 2 proteins with broadly expressed DNA repair enzymes in mediating immunoglobulin and T cell receptor gene segment rearrangement20,21) and somatic hypermutation or class switching of immunoglobulins (the central role of AID22). Evidence has been provided for a useful rather than harmful role of self-recognition in many aspects of T cell function23,24,25, and the molecular basis of ligand discrimination by T cells has been substantially clarified26,27. Yet even with all these advances dealing with central issues in antigen-specific host defense, the dominant driving force in immunology in the recent past appears to be the field's new appreciation of the importance of the innate immune system, especially its essential role in orchestrating adaptive responses.
Many attribute this new weltanschauung to what was at first an underappreciated but remarkably insightful 1989 article by the late Charles Janeway, Jr. In “Approaching the asymptote? Evolution and revolution in immunology”, he laid out a general hypothesis for why adjuvants (immunologists' 'dirty little secret') were needed to get effective immune responses28. Janeway's argument was that components of the innate system, especially antigen-presenting cells such as DCs, required the microbial stimuli contained in these empirically developed concoctions to become activated and acquire the capacity to induce productive responses from antigen-specific lymphocytes. Without such activation by recognition of infection, Janeway suggested that the adaptive immune system ignored or even became tolerant to the antigens presented by the 'quiescent' DC. He specifically proposed that evolutionarily conserved features of infectious organisms (pathogen-associated molecular patterns or PAMPs) were detected by the immune system through a set of specialized receptors (which he termed pattern-recognition receptors or PRRs), an especially prescient aspect of this hypothesis. Matzinger's competing 'Danger Model'29 argued against Janeway's focus on microbial signals. This hypothesis instead suggested that endogenous mediators produced by damaged or stressed cells held the key to effective immunity. These contrasting views elicited vigorous debate and prompted many investigators to turn their attention to this issue at the bench.
The bell 'tolls' for immunologists
Janeway's article and the competing Matzinger proposal helped focus immunological thinking on the role of antigen-unspecific signals in guiding immune function, but they did not provide a clear path for investigators to follow in translating these concepts into mechanistic understanding. It was a later achievement of Janeway and his associate Medzhitov, together with the critical findings of Beutler and others, that really ignited the revolution in this regard. In 1997, the first mammalian homolog of the gene that encodes Drosophila Toll protein (Toll-like receptor or Tlr) was cloned and shown to activate the NF-κB pathway that is a key feature of inflammatory signaling30. The subsequent evidence that the defect in the endotoxin (lipopolysaccharide) response of two strains of mice could be attributed to mutations in a specific member of the Tlr family (Tlr4)31 provided the crucial missing link between Tlr and in vivo mammalian responses to microbial products.
Toll is a Drosophila receptor critical to this insect's resistance to fungal infections32,33. This molecule has regions of homology to the IL-1 receptor of mammals and signals through an adapter (DmMyD88) to cause the activation of an NF-κB homolog. This transcription factor in turn promotes the production of drosomycin, an antifungal peptide that has a microbicidal role similar to that of members of the mammalian defensin family34. Thus, for Drosophila, which lacks an adaptive immune system, Toll acts as a central regulator of a major limb of its pathogen defense system. A distinct pathway first identified by its dependence on the imd gene is responsible for signals that induce production of antibacterial peptides that protect the insect against Gram-negative invaders33. The latter observations thus define a complementary limb of innate immunity to the one that involves Toll and illustrate how recognition of distinct pathogen products by individual receptors evokes a qualitatively appropriate host response.
The same theme has now emerged from studies in mammals, with specific signals generated upon binding of particular components of infectious agents to distinct receptors directing the quality of the ensuing immune response. A family of Tlr with at least 11 members has been identified, with each member responding to a different spectrum of ligands35. It remains unresolved whether mammalian Tlrs directly bind these stimulatory molecules36 or whether they act indirectly through other host proteins as seen in the activation of Drosophila Toll32,33. Tlrs show a complex pattern of tissue- and cell-specific expression. This distribution is believed to reflect the predilection of certain infectious agents to enter through or colonize specific tissues. There is presumably a match between preferred sites of infection, the nature of the molecular entities characteristic of the responsible organism, the Tlrs expressed by the tissue and associated immune cells and the character of the effector response that ensues35,37,38,39,40,41.
One issue of substantial debate in this field is whether the original Janeway concept of PAMPs and PRRs is correct. Two objections to this hypothesis have been raised: first, that signals from nonpathogenic infectious agents are recognized by the same set of receptors as those dealing with pathogens, and second, that specific molecules and not 'patterns' are actually recognized42. In a strict sense these objections are correct, but a focus on the underlying thinking suggests a more generous view. Janeway proposed that the innate system distinguished “infectious non-self” from “non-infectious self”28,43, and thus the actual concept was more global than the 'pathogen' part of the PAMP catchphrase implies. As to molecules versus patterns, the hypothesis stated that recognition of biochemical features broadly characteristic of the invader and not the host would best reveal infection. This has proven to be correct—Tlrs focus on aspects of prokaryotic, viral or parasitic structure or replicative biology generically distinct from those of the host. But it is also true that many (though not all) of the recognition events are specific for particular molecular species within these categories rather than for the 'molecular pattern' per se. A middle-of-the-road position seems best here.
Dendritic cells as primary translators of the infectious agent—Tlr discourse
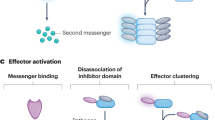
Since their discovery, analysis of Tlrs has proceeded in three directions: (i) the dissection of the molecules (MyD88, TRIF, TIRAP, IRAK, TRAFs and others) involved in transducing Tlr signals within cells35,44,45; (ii) the cataloging of genes activated by such signaling46,47,48; and (iii) the analysis of biological responses to Tlr signaling, including systemic toxicity during sepsis49,50 and control of hematopoietic cell, especially DC, differentiation39,40,41. Although not diminishing the substantial accomplishments of the first two, many consider some of the most exciting advances to have occurred in this last arena. Thirty years after their modern description51, DCs have emerged as the nexus for translating signals from innate recognition into cues guiding adaptive immune function52. Distinct DC subsets have been identified53,54,55,56 and the selective expression of particular members of the Tlr family by each of these subsets has been revealed57,58,59 (Fig. 2a).

(a) Representative subsets of human and mouse DCs are illustrated together with the Tlrs expressed and the location of each receptor (plasma membrane or endosome). The major ligands for each Tlr are also indicated. ?, no direct evidence for signaling by this ligand-receptor combination. +/−, low versus no expression in different studies. Adapted from refs. 40,41,293. DN, CD4−CD8α−. (b) Different Tlr signals direct DC differentiation along distinct pathways, which in turn results in polarization of antigen-specific T cells toward the TH1 versus TH2 lineages. See refs. 40,41,68,69,80,294,295.
One key aspect of Tlr function in DCs involves polarization of effector CD4+ T cells. The T helper type 1 (TH1)–T helper type 2 (TH2) paradigm of effector CD4+ T-cell responses arose from in vitro studies60,61,62. It was given physiological relevance by evidence that the extent of protection or nature of tissue pathology seen upon infection with agents such as Leishmania major or Schistosoma mansoni depended on which of the two types of effector response predominated63,64, as well as by molecular studies that identified distinct transcription factors promoting TH1 IFN-γ responses (T-bet65) or TH2 IL-4 production (GATA-3; refs. 66,67). The more recent discovery that DCs exposed to particular Tlr ligands selectively drive adaptive responses along one or the other of these pathways provided a mechanistic framework for these existing functional observations, linking antigen-independent recognition of the type of invading organism to the class of antigen-specific response that ensued39,40,41,68,69. For example, exposure of mouse DCs to Escherichia coli lipopolysaccharide results in signaling through Tlr4 that induces IL-12 secretion and enhancement of an IFN-γ-focused TH1 response. Conversely, interaction of the same mouse DC population with Porphyromonas gingivalis lipopolysaccharide triggers Tlr2 to promote a TH2 response through as yet undefined molecular events68. Thus, instead of the view espoused by Shaw's character in which antigen-specific proteins controlled the activity of phagocytes, many now see adaptive immunity as beholden to a subset of the latter (DCs) for guidance on how to best assist the host in resisting infections (Fig. 2b).
Although these results have given us a framework for one major aspect of Tlr function, many questions remain. Why is Tlr9 not only expressed by DCs but also by B lymphocytes, where it seems primarily to pose a risk of autoimmunity through signals induced upon binding to host DNA70,71? Why are several Tlrs only functional within endosomal compartments35,72? Is phagocytic uptake of bacteria or of apoptotic, infected cells with associated viral single-stranded or double-stranded RNA central to proper activation of DCs? What happens when two different Tlrs cosignal a given DC? We must also reappraise the Danger Model29 as evidence accumulates for endogenous ligands of Tlrs—albeit with some caution, given the possible role of lipopolysaccharide contamination in these results73. Also, Tlrs are not the sole detectors of infectious agents: C-type lectins on DCs seem to serve signaling functions that also guide DC differentiation69,74,75 and protein kinase R–dependent type 1 IFN responses to intracellular viral double-stranded RNA76 can influence DC maturation and activity77, in addition to serving directly as mediators of antiviral defense. Finally, a continuing debate is the extent to which 'plastic' DCs are driven to support one or another direction of T-cell polarization by Tlr signals as just described versus the 'fixed' predilection of particular DC subsets to foster T cell effector differentiation along a particular path, irrespective of Tlr instruction40,78,79,80.
DC subsets: splitters and lumpers
A description of the conjunction of Tlrs and DC would be incomplete without further discussion of DC subsets. One of the most contentious areas of DC biology over the past decade has been the origin and function of distinguishable variants of this cell type. The more monoclonal antibodies tested, the more unique DC types proposed81. Whether DCs with specific surface phenotypes were of lymphoid or myeloid origin was hotly disputed81. This debate has now been muted (though not fully resolved) by evidence that DCs of the same surface phenotype can be derived from either committed lymphoid or myeloid precursors82,83,84. Other studies have provided strong support for the concept of truly distinct DC subsets diverging in parallel from these precursors85,86,87,88.
With these issues receding from the limelight, the central focus of the field has instead turned to the dichotomy between the previously well-recognized DC subsets, now collectively called 'conventional DC' (CDC) and the new players on the team, the plasmacytoid DCs (PDCs) (Fig. 2a). PDCs were only recently recognized as the previously identified major producers of type 1 IFN following viral infection89,90,91,92. Further study has shown PDCs to have a complex physiology that includes differing from CDCs in genetic control of MHC class II expression93 and rapidly shutting off high-rate type 1 IFN production after Tlr activation, possibly changing their capacity to promote TH1 versus TH2 development during this process94,95. The tissue distribution, maturation and migration pattern, and cell interaction behavior of PDCs are all much less well defined at present in comparison to CDCs96. These two DC subsets also show little overlap in Tlr expression in human cells57,58 and examining the consequences of this disparate pattern of expression is made difficult by the fact that mouse and human differ in this regard, with many mouse but not human CDCs showing high expression of Tlr9 (refs. 57,58). This Tlr responds to CpG-rich DNA sequences97,98, which are being vigorously pursued as possible adjuvants in oligonucleotide form99 and whose presence in bacterial plasmid DNA is considered to impart this material with its immunogenic properties in mice100,101. Finally, PDCs have been suggested as major players in several human diseases102, although most of the results are correlative at present. One especially intriguing example in this regard is a report that PDCs have a key role in limiting the TH2-type inflammation responsible for atopic asthma-like airway disease in mice103.
The other side of the DC coin
If the idea that infectious products regulate the quality of adaptive immune effector responses through effects on DCs is the yin, then the yang of DC biology is the new focus on these cells as critical players in maintaining self-tolerance and avoiding autoimmunity when such microbial signals are absent104,105,106. The thymus is well recognized as the major site for elimination of overtly autoreactive T cells107. Indeed, one of the more unexpected findings of recent years is the role of autoimmune regulator protein AIRE in promoting thymic expression of what are traditionally considered to be tissue-specific antigens, resulting in deletion of the corresponding autoreactive thymocytes108,109,110,111,112,113. The lack of such AIRE-directed 'ectopic' antigen expression can result in autoimmunity111,114,115 and a very recent mouse study showing a gene dose effect of AIRE in susceptibility to diabetes116 suggests that subtle quantitative variations in expression among AIRE alleles may explain the linkage of this locus to human autoimmune disease117. Even with proper AIRE activity, however, potentially autoaggressive cells clearly escape to the periphery. Nonetheless, serious illness from self-reactivity is uncommon, presumably as a result of post-thymic mechanisms that keep such reactivity in check.
Accumulating evidence now points to DCs as important elements in this peripheral control process. The implication of data from several model systems118,119,120,121,122,123 is that the absence of Tlr signaling of DCs in noninfected hosts, together with continuous extrathymic presentation of self antigens by these cells124,125,126, may foster anergy or apoptosis of many of the self-reactive T cells that escape thymic elimination. This is apparently a result of inadequate expression of costimulatory (e.g., CD80, CD86) or viability-promoting (e.g., OX40L, 4.1-BBL) molecules by the nonactivated DC. This 'quiet' state of DC may also promote the expansion or functionality of regulatory or suppressor T cells127,128,129, about which much more will be said below.
Seen from this perspective, the absence of an adjuvant during antigen exposure not only leads to an ineffective host response, but can actively limit subsequent responses to the same antigen. This has important implications for vaccine trials. Inadequate innate stimulation upon vaccination may not only fail to engender protective responses, but may make the individual more susceptible to future infection or to a tumor by actively tolerizing the lymphocytes able to recognize the administered antigens. Conversely, there are good reasons to believe that defects in the capacity of 'resting' (adjuvant-naive) DCs to inactivate self-specific T cells may contribute to autoimmunity130.
The new understanding of the preeminent role of DCs in T-cell activation has combined with breakthroughs in generating and manipulating these otherwise rare cells131,132,133,134 to promote testing of DCs as vehicles for human vaccination135,136,137, with therapeutic cancer immunization as the primary focus. Whether patient-specific therapy with DCs will become a mainstay in the clinic even if it proves successful in specific cases is unclear, but the lessons learned from these studies may aid in designing more tractable ways of stimulating DCs in situ through such receptors. Methods of selective antigen targeting to DCs in vivo have been explored in animal models121,138,139,140 and these targeting approaches are likely to be combined in the future with activating agents that target Tlrs or CD40 to ensure that the delivered antigen is presented in an immunogenic manner and pushes T-cell differentiation toward the desired effector class.
Seeing what is missing—NK cell recognition
Tlrs are not the only poster children of innate recognition. The existence of lymphocytes lacking the unique clonal antigen specificity of T or B cells but showing contact-dependent cytotoxic activity against many tumor or virally infected cells (NK cells) has been appreciated for decades141,142,143 and the first of the receptors involved in regulating NK cell effector function were identified and cloned just over 10 years ago144,145,146,147,148,149. Since then, the diversity of such receptors in mouse and humans (including the presence of both C-type lectin and immunoglobulin-based structures150,151,152,153), the complex, nonclonal pattern of expression of these receptors along with the modulation of their expression to achieve self-tolerance154, the distinction between inhibitory and activating receptors155,156,157, and the ligands158 as well as the structure159 of many of these receptors have been uncovered (Fig. 3a).

(a) Expression of inhibitory and activating NK receptors on human and mouse lymphocyte subsets. See refs. 150,152,154,296,297. Not all possible receptors and expression patterns reported have been illustrated. (b) Loss of NK inhibitory receptor function upon downregulation of normally expressed MHC class I molecule expression is detection of 'missing self'. Induction of activating signals upon NKG2D recognition of MICA/B or UBLP molecules upregulated following infection of human cells is one mechanism of detection of 'stressed/infected self'. MICA/B, MHC class I–related protein A/B; ULBP, UL16 binding proteins. See refs. 152,158,168. (c) Loss of NK inhibitory receptor function upon downregulation of normally expressed MHC class I molecule expression is detection of 'missing self'. Induction of activating signals upon NKG2D recognition of MULT-1, H60 or Rae1 molecules upregulated following infection of mouse cells is one mechanism of detection of 'stressed/infected self'. MULT1, mouse ULBP-like transcript; Rae1, retinoic acid early transcript 1. See refs. 152,158,168. ITIM, immunoreceptor tyrosine-based inhibitory motif; Ig, immunoglobin; DAP, DNAX activating protein; ITAM, immunoreceptor tyrosine-based activation motif; PI3K, phosphotidyl inositol-3-kinase; CD3ζ, ζ chain associated with the CD3-T cell receptor; FcRγ, v chain of the immunoglobin Fc receptor.
Among the most striking conceptual advances that these new results have fostered is the notion that NK cells act very much like mirror images of T lymphocytes160. The importance to antipathogen responses of CD8+ T cells recognizing foreign peptides has been made clear by the evolutionary selection of infectious agents (especially viruses) that possess multiple molecular mechanisms for thwarting MHC class I antigen presentation161,162,163. The delineation of the molecular basis for such effects, involving interference with peptide transport (e.g., ICP47 of Herpes simplex inactivating transporters associated with antigen processing164,165), loading (e.g., human cytomegalovirus–induced inactivation of tapasin166), or display (e.g., human cytomegalovirus–US11-mediated degradation of MHC class I heavy chains167), represents a major accomplishment of the past decade. What we now understand is that pathogen evasion of CD8+ T-cell responses through interference with MHC class I expression and presentation of pathogen-derived peptides reciprocally contributes to the activation of NK cells that mediate many of the same effector functions (cytolysis, IFN-γ secretion).
The 'missing self' model of NK cell activation168 first proposed that NK cell effector function is typically kept in check by inhibitory receptors that are specific for host MHC class I molecules bound to self-peptides169. It has become clear that this model is correct and that when a pathogen interferes with normal MHC class I expression or peptide loading, diminished signaling by the inhibitory receptors contributes to NK cell activation and attack of the infected cell. Loss of inhibitory signals resulting from a reduction in surface MHC class I expression is not the sole mechanism responsible for NK cell activation, however. In some cases activating rather than inhibitory receptors directly bind viral proteins (for example, Ly49H of the mouse recognizes the mouse cytomegalovirus gene product m157 (ref. 170), acting as a nonclonal receptor for antigen), but escape mutations can eliminate effective NK receptor binding in this circumstance171. This risk of escape is obviated by NK recognition of the infected cells through the NKG2D receptor, which binds to a variety of host proteins induced by infection (these ligands include Mult-1, H60 and the Rae1 family in mouse and MICA, MICB and the ULBP family in humans152,158,172,173,174,175). Engagement of stimulatory NK receptors by these 'stress-induced' molecules leads to effector function, including killing of the host cell. Thus, NK cells respond to disruption of normal cell physiology upon infection both by sensing the absence of constitutive self in the form of peptide–MHC class I complexes and the presence of abnormal self in the form of these stress-induced molecules (Fig. 3b,c). This stands in marked contrast to the biology of conventional CD8+ T cells, which respond to new displays of foreign peptides bound to MHC molecules and utilize many central (thymic) and peripheral mechanisms to help minimize just such overt self-reactivity.
The ability of host proteins expressed by stressed cells to activate NK cells may be of particular importance in cancer. Many transformed cells express ligands for activating NK receptors152,176,177,178. Beyond explaining some of the evidence for a role of NK cells in immune surveillance of transformed cells, these data indicate that screening for activating NK receptor ligand expression coupled with treatments to facilitate the effector function of NK cells may prove therapeutically useful. The limiting factor in the success of approaches based on NKG2D recognition may be the propensity of tumor cells to secrete or shed high levels of ligands for this receptor179,180. Such materials can then act as decoys that prevent direct recognition of the tumor cell or promote loss of NK cell reactivity through tonic signaling and/or cognate receptor downregulation. This interfering effect may also apply to the costimulatory function of the NKG2D on human CD8+ T cells that constitutively express this activating receptor along with the TCR152,178.
Lastly, increasing attention has been paid to a transitional cell type between NK cells and conventional CD4+ and CD8+ T lymphocyte populations. These NK T cells arise in the thymus and express antigen receptors formed by gene rearrangement, but the receptors produced are nearly monomorphic, using just a few V, D and J segments and are devoid of the typical junctional diversity that has such a crucial role in the fine specificity of conventional T cells181,182. They also express many of the same inhibitory receptors present on NK cells and encoded by nonrearranging genes, hence the NK T designation. Similar cell populations exist in mice and humans. They are selected in the mouse based on receptor interaction with the MHC class I–like CD1d molecule181, which has a highly hydrophobic ligand-binding pocket suited to interaction with lipid ligands183. Until recently, the sea sponge lipid α-galactosyl ceramide was the best known strongly activating ligand for NK T cells184, but a sphingolipid (isoglobotrihexosylceramide or iGb3) has very recently been found to represent a natural stimulus for CD1d-restricted NK T cells185. Previous work had shown that high doses of lipopolysaccharide can elicit IFN-γ in a CD1d-dependent manner186, raising the possibility of a linkage between self-antigen upregulation, NK T cell activation and Tlr signaling by the same infectious stimuli that also promote adaptive immune responses. NK T cells produce effector cytokines within minutes to hours of initial stimulation181,187,188,189 as do NK cells189,190, and both populations have been found to influence the function of DCs191 and conventional T cells responding to the same infection. Thus, a complex web of connections among Tlr ligands, DCs, NK cells, NK T cells and conventional T cells has been shown, but who is saying what to whom and for what purpose is not yet clear.
From innate to adaptive: rules and regulations
Although innate immunity has been a dominant research theme of late, adaptive immunity has not been ignored. The intersection of innate and adaptive responses through the effects of microbial signals on DCs and the resulting regulation of antigen-specific responses has already been noted. Active regulation of adaptive immune effector function has been a favorite topic of immunologists for many decades, but the conclusions reached by older studies (particularly those involving 'suppressor cells and factors'), had lost credence among most investigators192. In a striking turn-about, there is now an exponentially increasing amount of research dedicated to studies of the role of negative regulation in enforcing effective self-tolerance, in controlling potentially pathologic responses to infectious agents, or in maintaining active immune memory. The primary focus is on 'natural' regulatory T cells (Treg)193,194, lymphocytes characterized by expression of CD25 (the IL-2Rα chain)195 along with the transcription factor FoxP3 (refs. 196–198), and that appear to constitute a unique lineage of cells positively selected along with conventional pre-effector CD4+ and CD8+ T cells in the thymus193,194,199 (Fig. 4). A defect in the function of CD25+CD4+ T regulatory cells resulting from FoxP3 deficiency leads to the 'scurfy' autoimmune phenotype in mice198,200 and to the human autoimmune syndrome IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked)201, emphasizing just one aspect of the clinical relevance of the wealth of basic studies focused on these cells.

A role for Treg cells seems to have been reported in virtually every imaginable type of immune response193,194,199. The CD25+ Treg subset has been reported to decline in number and/or activity in NOD mice, heralding the onset of autoimmune diabetes202, though other studies disagree with this conclusion203. Their elimination substantially augments antitumor immunity in several mouse models204. A variety of organ-specific autoimmunity diseases arise in mice if CD25+ Treg cells are absent and some form of activating signal (such as lymphopenia) is provided193,194. Treg cells can suppress allograft responses205 and temper antiviral immunity206. In the case of the resistant B6 mouse strain, their activity prevents sterilizing immunity to Leishmania major, which in turn appears necessary for maintaining cell-mediated resistance to reinfection by this parasite207. Although the lion's share of attention has gone to the natural Treg cells, other studies suggest that actively induced suppressive T cells generated by antigen exposure in the context of cosignals such as IL-10 (ref. 208) or vitamin D receptor activation209 may also have significant roles in modulating immunity in many of the same circumstances.
Although the origin and MHC-based positive selection of natural Treg cells in the thymus is well established, much remains unknown about these cells (Fig. 4). Are there specific signals beyond those of the T cell receptor involved in driving thymocytes to adopt this cell fate? Did these cells evolve specifically to control potential autoimmunity and are their effects on responses to infectious agents a mere byproduct of this primary function, or is the latter an essential feature of their activity? What specific signals beyond IL-2 (ref. 210) maintain their number or promote their expansion in vivo, how do immature versus mature DCs fit into the picture, and what are the contributions of contact-mediated211,212 and cytokine-dependent (IL-10, TGF-β213,214,215) mechanisms to their in vivo function in different situations?
One especially salient issue is how the system avoids inappropriate interference by Treg cells with effector responses to infections. Tlr signaling may contribute to resistance to Treg cell suppression, possibly by promoting DC production of cytokines such as IL-6 that allow conventional T cells to resist the otherwise dampening effects of the suppressor population216. Such a model would allow Treg cells to dominate in the noninflammatory steady state and block autoreactive responses, while allowing effector development in response to pathogens to proceed unimpeded. But self-reactive cells are still present in a host during an infection and these cells should presumably be activated if Tlr-induced soluble mediators blocked all Treg cell function. If we are to manipulate Treg cells for clinical purposes, the field needs to gain a deeper understanding of how their function is controlled so that unwanted responses are held in check without preventing needed effector activities.
Other advances in the field of immunoregulation involve the contributions of a growing collection of cosignaling receptors and their ligands in establishing the qualitative and quantitative nature of immune responses. The expanded CD28-B7 family can serve as a prime example217,218. Upon binding to CD80 and CD86 on antigen-presenting cells, CD28 augments but CTLA-4 suppresses T-cell activation217,218,219,220. Expression of CD28 and CTLA-4 are regulated quite differently, with the former showing constitutive surface localization and the latter appearing on the plasma membrane in proportion to the strength of T cell receptor signals221. CD80 and CD86 do not bind equivalently to these two receptors, giving each ligand a distinct role to perform in balancing the competing biological effects of stimulation and inhibition222. PD-1 and PD-L1/2 are other members of this same family with various alternative names that can have both activating217,218,223 and inhibitory217,218,224,225,226 roles depending on cellular context. Multiple other activating and inhibitory members of this family have been discovered more recently226,227 A key concept that has emerged from investigation of these various cosignaling proteins is the critical role of sequential, properly timed interactions between particular receptor-ligand pairs in the guidance of the developing immune response217,218,226. Experiments that do not take these temporal features into account are likely to confound our attempts to develop a robust understanding of how these molecules act and how we can manipulate them for clinical purposes.
Death be not proud
Not only does immune regulation function by actively controlling the behavior of living cells, it also acts by determining which lymphocytes will die. Apoptotic cell death is now appreciated as a key element in maintaining immune homeostasis and preventing the emergence of lymphomas or the development of autoimmunity228. Precursor T or B lymphocytes are removed in large numbers from the maturing pool as a result of death in response to high-level self-recognition in the thymus107,229,230 or bone marrow and spleen231,232, respectively. Mature T cells engaging immature or nonactivated DCs displaying self-antigens in secondary lymphoid tissues such as lymph nodes and spleen are also purged from the repertoire106 and a similar fate awaits conventional mature B cells that bind antigen without concomitant T-cell help or that alter their antigen receptor specificity during somatic hypermutation in germinal centers so as to either lose foreign antigen specificity or gain autoreactivity233. Likewise, only a fraction of the large cohort of antigen-specific T cells produced by clonal expansion in response to infection is permitted to survive as memory cells234,235.
The study of regulated cell death in lymphoid cells has resulted in many key contributions to our basic cell biological understanding of apoptotic mechanisms228. As just one example with clinical relevance, individuals with previously unexplained autoimmunity and lymphoproliferation (autoimmune lymphoproliferative syndrome or ALPS) were studied for defects in the molecules known from mouse models to yield similar pathology. It rapidly became clear that a large subset of these patients had defects in Fas, a known death receptor236. The surprising genetic finding was that disease was dominant, an unexpected result that ultimately led to a new understanding of tumor necrosis factor (TNF) family receptor biochemistry characterized by preassembly of the trimeric receptors before ligand binding237.
The TNF family of proteins includes members that prevent death of a crucial subset of activated lymphocytes that go on to form the memory pool238. Antigen-specific recall responses are hallmarks of adaptive immunity and their analysis has been an area of substantial ferment over the past few years. Subsets of phenotypically and functionally distinct memory T cells have been described in mice and humans. The two major subtypes most widely recognized are those cells with the capacity for secondary lymphoid recirculation ('central memory cells' expressing CCR7 and CD62L) and those with tissue-homing preference and capable of immediate effector function upon stimulation ('peripheral effector memory cells' lacking these two essential molecules for lymphoid tissue entry)235,239. Candidates identified over the past few years as the key ligands and receptors involved in promoting the production of such long-lived memory T cells include OX40-OX40L for CD4+ T cells and 4.1BB–4.1BB-L in concert with CD27-CD70 for CD8+ T cells, with expression of all of these TNF family ligands in some measure dependent on CD40L-CD40 signaling between activated CD4+ T cells and mature DCs238. This relationship between CD40 signaling and expression of prosurvival TNF family members may provide a partial explanation for the recent reports indicating that primary CD8+ effector responses can be quite CD4+ T cell–independent but that long-term, functional cell-mediated immune memory generally requires antigen-specific, CD40L-dependent CD4+ T-cell helper function240,241,242,243,244,245. This new information is of critical importance to vaccine development that aims at inducing robust, long-lived CD8+ T-cell priming, for therapeutic immunization in hosts with chronic viral infections (e.g., HIV or hepatitis C virus), or following adoptive immunotherapy for cancer using CD8+ cytotoxic effectors.
Location, location, location
A unique feature of most hematopoietic cells as compared to other cells in an adult organism is their lack of a fixed tissue location. But the function of the various cells comprising the system is not fully autonomous and interactions are necessary both to initiate and effectuate responses. To accomplish this, immune cells must be at the right place at the right time. One of the principal accomplishments of the past decade has been unraveling the codes involving multiple chemokine ligands and receptors that together have a critical role in guiding immune cell movement and localization246,247,248,249,250. The molecules and mechanisms that control entry of T and B cells into lymph nodes for initial antigen-dependent activation (primarily, but not exclusively, involving CCR7-CCL21 binding247,248,251,252), as well as those regulating movement within that site for the cell interactions that underlie effective memory cell generation or humoral immune responses (for example, the balance of B cell CXCR5 and CCR7 function247,253,254), have been determined. Some of the participants in guiding activated cells out of lymphoid tissue (SIP1 and sphingosine-1-phosphate255) and into parenchymal sites for effector function246,248,252 have been identified. How tissue-homing preferences are imprinted on activated T cells is being unraveled, especially the role of the local DC population in fostering the return of stimulated lymphocytes to the tissue from which antigen was acquired256,257,258. Not unexpectedly, various infectious agents have utilized these same critical guidance elements for their own purposes, ranging from acting as sites of viral259 or plasmodial260 cell entry, to manipulating host immune function261.
Better tools for tracking immune responses
The past decade has seen some remarkable technological developments that enhance immunololgists' ability to ask probing questions. The development of soluble multimers of peptide-MHC molecule ligands of the T cell receptor allowed the direct enumeration of antigen-specific T cells for the first time262,263,264, with a major impact on our ability to track and quantify responses to infectious agents and candidate vaccines in animals and humans. High resolution static265 and time-lapse266,267 in vitro microscopy of T cell–antigen-presenting cell pairs documented discrete protein-enriched subdomains in the contacting membranes, leading to an explosion of studies on the organization of the aptly named 'immunological synapse'268,269,270. Techniques allowing assembly of confocal fluorescent images from an entire mouse provided information on the whole-body distribution of immune cells at various stages of an immune response271. Most recently, new live-cell imaging methods have allowed the incredibly dynamic movement of lymphocytes and DCs as well as the duration of interaction of these cells to be visualized for the first time within intact lymphoid structures272,273,274,275,276,277. It is still early, but the latter technique offers the prospect of linking a detailed picture of the in situ behavior of immune cells to the overall response of the organism, potentially providing the information required for eventual modeling of cellular events underlying normal and pathologic immune responses.
Translation
This introductory overview would not be complete without mention of the successes of translational and clinical immunology. Monoclonal antibodies finally were shown to be the powerful therapeutic agents that many had hoped278. In autoimmune disease, tumor therapy, transplantation and anti-infection prophylaxis, unmodified as well as toxin-, drug- or radionuclide-conjugated humanized antibodies have proven their clinical worth. Gene therapy for immunodeficiency was accomplished279, though not without evidence of the potential of this methodology for severe side effects280,281. A host of tumor-associated and tumor-specific antigens were cloned and moved from the bench to the bedside as components in experimental therapeutic cancer vaccines282,283,284. Although to date the results have been mixed, occasional successes suggest the potential for effective use exists if better patient preselection methods, more reproducible protocols and combination treatment regimes that incorporate interventions to augment immune function can be developed. In this last regard, the translation of basic advances in immunoregulation holds substantial promise, through such strategies as elimination of natural or induced Treg cell function285 or the use of antibody specific for CTLA-4, which has already shown its ability to strikingly facilitate antitumor responses, although with the risk of accompanying autoimmune pathology286,287,288.
Epilogue
“Let me not to the marriage of true minds
Admit impediments. Love is not love
Which alters when it alteration finds,
Or bends with the remover to remove:
O no! it is an ever-fixed mark
That looks on tempests and is never shaken;”
W. Shakespeare, Sonnet 116
Although I have emphasized the striking shift in emphasis characterizing immunological research over the past decade, with its increasing focus on innate as opposed to adaptive aspects of system behavior, both parts are necessary to a functional whole. The bard's lines thus aptly describe the steadfast nature of a true immunologist, who even in the face of this new direction does not abandon the long-held view that antigen specificity is a defining feature of mammalian immune function. Different infectious agents may predominate in causing morbidity when innate effector functions are lacking as compared to when adaptive defects are present, but in the end, host survival over the long term depends on the integrated activity of the two. Indeed, if any overarching theme has emerged from the flood of information produced by the last 10 years of study, it is the extensive crosstalk among all the components of the immune system. Particular microbial stimuli activate subsets of innate cells that differentiate and produce secondary mediators; these in turn guide antigen-reactive lymphocytes along the differentiation pathway most relevant for host protection against that particular infectious agent. In addition to their direct attack on infected cells, the activated lymphocytes also produce mediators (both antigen-specific and unspecific) that enhance the protective capacity of nonlymphoid cells. The targets include the phagocytes of Metchnikoff that, through mechanisms such as uptake of antibody-coated organisms and IFN-γ-mediated augmentation of the production of microbicidal molecules like nitric oxide and reactive oxygen species, provide the proximate effector functions that combat the infectious agent.
Has the burst of new knowledge about innate immune function and innate-adaptive crosstalk brought us close to the end in terms of conceptual advances? I suspect that despite the cataloging of nearly all genes in mice and humans, we are still far from knowing which ones contribute to immune activity, much less how they do so. Likewise, many of the cells whose functions currently consume the research efforts of immunologists are numerically minor populations; it would hardly be surprising if other small subsets with similarly important roles, especially in regional responses, came to light in the future.
Nevertheless, it is also true that the basic outline of host defense I have summarized here has been known for decades—barrier function is supported by ready-to-go innate defenses that are followed temporally by the activities of clonally expanded adaptive effector cells whose products also enhance innate mechanisms. So what is really new and where are we going in the future? The devil has been in the details—the substantive advances of the past decade have been to identify major players in pathogen recognition (Tlrs as the prime example), develop an understanding of how specific innate recognition strategies promote the needed quality of immune response (DC subsets and DC differentiation plasticity guiding effector T-cell polarity), show the multilayered nature of the defense strategy so that the rapid evolutionary capacity of microbes does not leave gaping holes in host resistance (consider the reciprocal nature of activating signals for CD8+ T cells and NK cells with respect to host MHC class I expression), reveal the mechanisms by which the right cell arrives at the right place to become activated or to be a useful effector (chemokines, chemokine receptors, selective adhesion molecule expression) and uncover the extensive system of regulatory components that not only help guide the response in the proper direction, but also limit the pathologic consequences of inappropriate immune activity (inhibitory cosignaling receptors like CTLA-4, suppressive Treg cells, death-promoting signals like Fas).
One major problem posed by all this new information is that the extensive feedback and crossregulatory activities documented in the past several years can be expected to yield very nonlinear, even counterintuitive system behavior289,290. Our natural tendency is to think in analog terms about a response (put in graded stimuli, get correspondingly graded responses). But complex systems often display all-or-none behavior, with inputs below a threshold being inadequate to elicit any downstream output and a signal just above that level giving the full response the system can provide. The consequence is that small differences in both the nature of input used and the state of the individual host's immune system can lead to markedly different outcomes, consistent with the finding that with some vaccines, a subset of individuals develops robust cell-mediated responses while others show none.
The lesson is that we must go beyond the qualitative nature of most current analysis of immune behavior and become much more quantitative. We need to understand that a few percentage points of variation in cell cycle rate or survival fraction among cells in an exponentially expanding population can produce outcome differences of more than two orders of magnitude within a week's time, given the rate at which lymphocytes multiply291. We need to appreciate that even a very modest level of inhibition by Treg cells, CTLA-4 or IL-10 that reduces the expansion rate or differentiation frequency of proliferating effector T cells can make all the difference between health and substantial autoimmune pathology, in part by preventing the positive feedback stimulation through self-antigen released from damaged cells that pushes a response beyond a 'fail-safe' threshold.
Acquiring the needed quantitative information, especially in humans, is a major challenge, as is the proper utilization of such information in the context of the ever more intricate networks of interactions that we understand to comprise the immune system. Just as 'rediscovery' of innate immunity and immune regulation were dominant features of the past decade of immunological research, development of new tools for detecting and measuring immune responses, especially in situ, and for predictive analysis of complex system behavior, are features that must eventually characterize the field in the future. The question is when this next era will arrive. This last question has been more eloquently posed that I can manage and so I end with the words of Sydney Brenner: “In one way, you could say all the genetic and molecular biological work of the last 60 years could be considered a long interlude...We have come full circle—back to the problems left behind unsolved. How does a wounded organism regenerate exactly the same structure it had before? How does the egg form the organism? In the next 25 years, we are going to have to teach biologists another language...I don't know what it's called yet; nobody knows”292. I only hope immunologists are among the first to find out.
References
Shaw, G.B. The Doctor's Dilemma http://www.webbooks.com/Classics/Nonfiction/Drama/Shaw/Doctor/Home.htm
Metchnikoff, I.I. On the Present State of the Question of Immunity in Infectious Diseases. (1908). http://nobelprize.org/medicine/laureates/1908/mechnikov-lecture.html
Ehrlich, P. Partial Cell Functions (lecture, 1908). in Nobel Lectures, Physiology or Medicine 1901–1921. (Elsevier Publishing Company, Amsterdam, 1967).
Edelman, G.M. et al. The covalent structure of an entire γG immunoglobulin molecule. Proc. Natl. Acad. Sci. USA 63, 78–85 (1969).
Davies, D.R., Padlan, E.A. & Segal, D.M. Three-dimensional structure of immunoglobulins. Annu. Rev. Biochem. 44, 639–667 (1975).
Amzel, L.M. & Poljak, R.J. Three-dimensional structure of immunoglobulins. Annu. Rev. Biochem. 48, 961–997 (1979).
Bjorkman, P.J. et al. Structure of the human class I histocompatibility antigen, HLA-A2. Nature 329, 506–512 (1987).
Brack, C., Hirama, M., Lenhard-Schuller, R. & Tonegawa, S. A complete immunoglobulin gene is created by somatic recombination. Cell 15, 1–14 (1978).
Hedrick, S.M., Cohen, D.I., Nielsen, E.A. & Davis, M.M. Isolation of cDNA clones encoding T cell-specific membrane-associated proteins. Nature 308, 149–153 (1984).
Yanagi, Y. et al. A human T cell-specific cDNA clone encodes a protein having extensive homology to immunoglobulin chains. Nature 308, 145–149 (1984).
Benacerraf, B. & McDevitt, H.O. Histocompatibility-linked immune response genes. Science 175, 273–279 (1972).
Rosenthal, A.S. & Shevach, E.M. Function of macrophages in antigen recognition by guinea pig T lymphocytes. I. Requirement for histocompatible macrophages and lymphocytes. J. Exp. Med. 138, 1194–1212 (1973).
Zinkernagel, R.M. & Doherty, P.C. Restriction of in vitro T cell–mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. Nature 248, 701–702 (1974).
Townsend, A.R., Gotch, F.M. & Davey, J. Cytotoxic T cells recognize fragments of the influenza nucleoprotein. Cell 42, 457–467 (1985).
Babbitt, B.P., Allen, P.M., Matsueda, G., Haber, E. & Unanue, E.R. Binding of immunogenic peptides to Ia histocompatibility molecules. Nature 317, 359–361 (1985).
Heber-Katz, E., Hansburg, D. & Schwartz, R.H. The Ia molecule of the antigen-presenting cell plays a critical role in immune response gene regulation of T cell activation. J. Mol. Cell. Immunol. 1, 3–18 (1983).
Buus, S., Sette, A., Colon, S.M., Miles, C. & Grey, H.M. The relation between major histocompatibility complex (MHC) restriction and the capacity of Ia to bind immunogenic peptides. Science 235, 1353–1358 (1987).
Stern, L.J. & Wiley, D.C. Antigenic peptide binding by class I and class II histocompatibility proteins. Structure 2, 245–251 (1994).
Germain, R.N. MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell 76, 287–299 (1994).
Hesslein, D.G. & Schatz, D.G. Factors and forces controlling V(D)J recombination. Adv. Immunol. 78, 169–232 (2001).
Jung, D. & Alt, F.W. Unraveling V(D)J recombination; insights into gene regulation. Cell 116, 299–311 (2004).
Honjo, T., Muramatsu, M. & Fagarasan, S. AID: how does it aid antibody diversity? Immunity 20, 659–668 (2004).
Dorfman, J.R. & Germain, R.N. MHC-dependent survival of naive T cells? A complicated answer to a simple question. Microbes Infect. 4, 547–554 (2002).
Wulfing, C. et al. Costimulation and endogenous MHC ligands contribute to T cell recognition. Nat. Immunol. 3, 42–47 (2002).
Stefanova, I., Dorfman, J.R. & Germain, R.N. Self-recognition promotes the foreign antigen sensitivity of naive T lymphocytes. Nature 420, 429–434 (2002).
Germain, R.N. & Stefanova, I. The dynamics of T cell receptor signaling: complex orchestration and the key roles of tempo and cooperation. Annu. Rev. Immunol. 17, 467–522 (1999).
Krummel, M., Wulfing, C., Sumen, C. & Davis, M.M. Thirty-six views of T-cell recognition. Phil. Trans. R. Soc. Lond. B Biol. Sci. 355, 1071–1076 (2000).
Janeway, C.A., Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 1, 1–13 (1989).
Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 12, 991–1045 (1994).
Medzhitov, R., Preston-Hurlburt, P. & Janeway, C.A., Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388, 394–397 (1997).
Poltorak, A. et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088 (1998).
Hoffmann, J.A. The immune response of Drosophila. Nature 426, 33–38 (2003).
Ferrandon, D., Imler, J.L. & Hoffmann, J.A. Sensing infection in Drosophila: Toll and beyond. Semin. Immunol. 16, 43–53 (2004).
Ganz, T. Defensins: antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 3, 710–720 (2003).
Takeda, K., Kaisho, T. & Akira, S. Toll-like receptors. Annu. Rev. Immunol. 21, 335–376 (2003).
Rutz, M. et al. Toll-like receptor 9 binds single-stranded CpG-DNA in a sequence- and pH-dependent manner. Eur. J. Immunol. 34, 2541–2550 (2004).
Beutler, B., Hoebe, K., Du, X. & Ulevitch, R.J. How we detect microbes and respond to them: the Toll-like receptors and their transducers. J. Leukoc. Biol. 74, 479–485 (2003).
Janssens, S. & Beyaert, R. Role of Toll-like receptors in pathogen recognition. Clin. Microbiol. Rev. 16, 637–646 (2003).
Kapsenberg, M.L. Dendritic-cell control of pathogen-driven T-cell polarization. Nat. Rev. Immunol. 3, 984–993 (2003).
Reis e Sousa, C. Toll-like receptors and dendritic cells: for whom the bug tolls. Semin. Immunol. 16, 27–34 (2004).
Iwasaki, A. & Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 5, 987–995 (2004).
Beutler, B. Not “molecular patterns” but molecules. Immunity 19, 155–156 (2003).
Medzhitov, R. & Janeway, C.A., Jr. Innate immunity. N. Engl. J. Med. 343, 338–344 (2000).
Barton, G.M. & Medzhitov, R. Toll-like receptor signaling pathways. Science 300, 1524–1525 (2003).
Beutler, B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature 430, 257–263 (2004).
Vogel, S.N., Fitzgerald, K.A. & Fenton, M.J. TLRs: differential adapter utilization by toll-like receptors mediates TLR-specific patterns of gene expression. Mol. Interv. 3, 466–477 (2003).
Beutler, B., Hoebe, K. & Shamel, L. Forward genetic dissection of afferent immunity: the role of TIR adapter proteins in innate and adaptive immune responses. C. R. Biol. 327, 571–580 (2004).
Weighardt, H. et al. Identification of a TLR4- and TRIF-dependent activation program of dendritic cells. Eur. J. Immunol. 34, 558–564 (2004).
Beutler, B. & Poltorak, A. Sepsis and evolution of the innate immune response. Crit. Care Med. 29, S2–6; discussion S6–7 (2001).
Cristofaro, P. & Opal, S.M. The Toll-like receptors and their role in septic shock. Expert Opin. Ther. Targets 7, 603–612 (2003).
Steinman, R.M. & Cohn, Z.A. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J. Exp. Med. 137, 1142–1162 (1973).
Banchereau, J. & Steinman, R.M. Dendritic cells and the control of immunity. Nature 392, 245–252 (1998).
Liu, Y.J. Dendritic cell subsets and lineages, and their functions in innate and adaptive immunity. Cell 106, 259–262 (2001).
Fazekas De St Groth, B. et al. Experimental models linking dendritic cell lineage, phenotype and function. Immunol. Cell Biol. 80, 469–476 (2002).
Ardavin, C. Origin, precursors and differentiation of mouse dendritic cells. Nat. Rev. Immunol. 3, 582–590 (2003).
Wilson, H.L. & O'Neill, H.C. Murine dendritic cell development: difficulties associated with subset analysis. Immunol. Cell Biol. 81, 239–246 (2003).
Kadowaki, N. et al. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J. Exp. Med. 194, 863–869 (2001).
Jarrossay, D., Napolitani, G., Colonna, M., Sallusto, F. & Lanzavecchia, A. Specialization and complementarity in microbial molecule recognition by human myeloid and plasmacytoid dendritic cells. Eur. J. Immunol. 31, 3388–3393 (2001).
Edwards, A.D. et al. Toll-like receptor expression in murine DC subsets: lack of TLR7 expression by CD8α+ DC correlates with unresponsiveness to imidazoquinolines. Eur. J. Immunol. 33, 827–833 (2003).
Mosmann, T.R., Cherwinski, H., Bond, M.W., Giedlin, M.A. & Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 136, 2348–2357 (1986).
Mosmann, T.R. & Coffman, R.L. Heterogeneity of cytokine secretion patterns and functions of helper T cells. Adv. Immunol. 46, 111–147 (1989).
Seder, R.A. & Paul, W.E. Acquisition of lymphokine-producing phenotype by CD4+ T cells. Annu. Rev. Immunol. 12, 635–673 (1994).
Sacks, D. & Noben-Trauth, N. The immunology of susceptibility and resistance to Leishmania major in mice. Nat. Rev. Immunol. 2, 845–858 (2002).
Wynn, T.A. Fibrotic disease and the TH1/TH2 paradigm. Nat. Rev. Immunol. 4, 583–594 (2004).
Szabo, S.J. et al. A novel transcription factor, T-bet, directs TH1 lineage commitment. Cell 100, 655–669 (2000).
Zheng, W. & Flavell, R.A. The transcription factor GATA-3 is necessary and sufficient for TH2 cytokine gene expression in CD4 T cells. Cell 89, 587–596 (1997).
Ouyang, W. et al. Inhibition of TH1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity 9, 745–755 (1998).
Pulendran, B. et al. Lipopolysaccharides from distinct pathogens induce different classes of immune responses in vivo. J. Immunol. 167, 5067–5076 (2001).
Pulendran, B. Modulating vaccine responses with dendritic cells and Toll-like receptors. Immunol. Rev. 199, 227–250 (2004).
Leadbetter, E.A. et al. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature 416, 603–607 (2002).
Marshak-Rothstein, A., Busconi, L., Rifkin, I.R. & Viglianti, G.A. The stimulation of Toll-like receptors by nuclear antigens: a link between apoptosis and autoimmunity. Rheum. Dis. Clin. North Am. 30, 559–74, ix (2004).
Ahmad-Nejad, P. et al. Bacterial CpG-DNA and lipopolysaccharides activate Toll-like receptors at distinct cellular compartments. Eur. J. Immunol. 32, 1958–1968 (2002).
Tsan, M.F. & Gao, B. Endogenous ligands of Toll-like receptors. J. Leukoc. Biol. 76, 514–519 (2004).
Geijtenbeek, T.B., Engering, A. & Van Kooyk, Y. DC-SIGN, a C-type lectin on dendritic cells that unveils many aspects of dendritic cell biology. J. Leukoc. Biol. 71, 921–931 (2002).
Gordon, S. Pattern recognition receptors: doubling up for the innate immune response. Cell 111, 927–930 (2002).
Diebold, S.S. et al. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature 424, 324–328 (2003).
Santini, S.M. et al. Type I interferon as a powerful adjuvant for monocyte-derived dendritic cell development and activity in vitro and in Hu-PBL-SCID mice. J. Exp. Med. 191, 1777–1788 (2000).
Reid, S.D., Penna, G. & Adorini, L. The control of T cell responses by dendritic cell subsets. Curr. Opin. Immunol. 12, 114–121 (2000).
Liu, Y.J., Kanzler, H., Soumelis, V. & Gilliet, M. Dendritic cell lineage, plasticity and cross-regulation. Nat. Immunol. 2, 585–589 (2001).
Pulendran, B. Modulating TH1/TH2 responses with microbes, dendritic cells, and pathogen recognition receptors. Immunol. Res. 29, 187–196 (2004).
Lotze, M.T. & Thomson, A.W. (eds.). Dendritic Cells: Biology and Clinical Applications (Academic, San Diego, 1999).
Traver, D. et al. Development of CD8α-positive dendritic cells from a common myeloid progenitor. Science 290, 2152–2154 (2000).
Manz, M.G., Traver, D., Miyamoto, T., Weissman, I.L. & Akashi, K. Dendritic cell potentials of early lymphoid and myeloid progenitors. Blood 97, 3333–3341 (2001).
Wu, L. et al. Development of thymic and splenic dendritic cell populations from different hemopoietic precursors. Blood 98, 3376–3382 (2001).
Kamath, A.T. et al. The development, maturation, and turnover rate of mouse spleen dendritic cell populations. J. Immunol. 165, 6762–6770 (2000).
Naik, S., Vremec, D., Wu, L., O'Keeffe, M. & Shortman, K. CD8α+ mouse spleen dendritic cells do not originate from the CD8α - dendritic cell subset. Blood 102, 601–604 (2003).
Tsujimura, H. et al. ICSBP/IRF-8 retrovirus transduction rescues dendritic cell development in vitro. Blood 101, 961–969 (2003).
Aliberti, J. et al. Essential role for ICSBP in the in vivo development of murine CD8α+ dendritic cells. Blood 101, 305–310 (2003).
Siegal, F.P. et al. The nature of the principal type 1 interferon-producing cells in human blood. Science 284, 1835–1837 (1999).
Kadowaki, N., Antonenko, S., Lau, J.Y. & Liu, Y.J. Natural interferon α/β -producing cells link innate and adaptive immunity. J. Exp. Med. 192, 219–226 (2000).
Asselin-Paturel, C. et al. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat. Immunol. 2, 1144–1150 (2001).
Dalod, M. et al. Dendritic cell responses to early murine cytomegalovirus infection: subset functional specialization and differential regulation by interferon alpha/beta. J. Exp. Med. 197, 885–898 (2003).
LeibundGut-Landmann. S., Waldburger, J.M., Reis e Sousa, C., Acha-Orbea, H. & Reith, W. MHC class II expression is differentially regulated in plasmacytoid and conventional dendritic cells. Nat. Immunol. 5, 899–908 (2004).
Rissoan, M.C. et al. Reciprocal control of T helper cell and dendritic cell differentiation. Science 283, 1183–1186 (1999).
Langenkamp, A., Messi, M., Lanzavecchia, A. & Sallusto, F. Kinetics of dendritic cell activation: impact on priming of TH1, TH2 and nonpolarized T cells. Nat. Immunol. 1, 311–316 (2000).
Penna, G., Vulcano, M., Sozzani, S. & Adorini, L. Differential migration behavior and chemokine production by myeloid and plasmacytoid dendritic cells. Hum. Immunol. 63, 1164–1171 (2002).
Hemmi, H. et al. A Toll-like receptor recognizes bacterial DNA. Nature 408, 740–745 (2000).
Hemmi, H., Kaisho, T., Takeda, K. & Akira, S. The roles of Toll-like receptor 9, MyD88, and DNA-dependent protein kinase catalytic subunit in the effects of two distinct CpG DNAs on dendritic cell subsets. J. Immunol. 170, 3059–3064 (2003).
Krieg, A.M. & Davis, H.L. Enhancing vaccines with immune stimulatory CpG DNA. Curr. Opin. Mol. Ther. 3, 15–24 (2001).
Krieg, A.M., Yi, A.K., Schorr, J. & Davis, H.L. The role of CpG dinucleotides in DNA vaccines. Trends Microbiol. 6, 23–27 (1998).
Pisetsky, D.S. Mechanisms of immune stimulation by bacterial DNA. Springer Semin. Immunopathol. 22, 21–33 (2000).
Jahnsen, F.L., Farkas, L., Lund-Johansen, F. & Brandtzaeg, P. Involvement of plasmacytoid dendritic cells in human diseases. Hum. Immunol. 63, 1201–1205 (2002).
de Heer, H.J. et al. Essential role of lung plasmacytoid dendritic cells in preventing asthmatic reactions to harmless inhaled antigen. J. Exp. Med. 200, 89–98 (2004).
Steinman, R.M. & Nussenzweig, M.C. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc. Natl. Acad. Sci. USA 99, 351–358 (2002).
Lu, L. & Thomson, A.W. Manipulation of dendritic cells for tolerance induction in transplantation and autoimmune disease. Transplantation 73, S19–S22 (2002).
Steinman, R.M., Hawiger, D. & Nussenzweig, M.C. Tolerogenic dendritic cells. Annu. Rev. Immunol. 21, 685–711 (2003).
von Boehmer, H., Teh, H.S. & Kisielow, P. The thymus selects the useful, neglects the useless and destroys the harmful. Immunol. Today 10, 57–61 (1989).
Heath, V.L., Moore, N.C., Parnell, S.M. & Mason, D.W. Intrathymic expression of genes involved in organ specific autoimmune disease. J. Autoimmun. 11, 309–318 (1998).
Heino, M. et al. Autoimmune regulator is expressed in the cells regulating immune tolerance in thymus medulla. Biochem. Biophys. Res. Commun. 257, 821–825 (1999).
Derbinski, J., Schulte, A., Kyewski, B. & Klein, L. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat. Immunol. 2, 1032–1039 (2001).
Anderson, M.S. et al. Projection of an immunological self shadow within the thymus by the aire protein. Science 298, 1395–1401 (2002).
Liston, A., Lesage, S., Wilson, J., Peltonen, L. & Goodnow, C.C. Aire regulates negative selection of organ-specific T cells. Nat. Immunol. 4, 350–354 (2003).
Kyewski, B. & Derbinski, J. Self-representation in the thymus: an extended view. Nat. Rev. Immunol. 4, 688–698 (2004).
Scott, H.S. et al. Common mutations in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients of different origins. Mol. Endocrinol. 12, 1112–1119 (1998).
Rosatelli, M.C. et al. A common mutation in Sardinian autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients. Hum. Genet. 103, 428–434 (1998).
Liston, A. et al. Gene dosage–limiting role of Aire in thymic expression, clonal deletion, and organ-specific autoimmunity. J. Exp. Med. 200, 1015–1026 (2004).
Hornum, L. & Markholst, H. New autoimmune genes and the pathogenesis of type 1 diabetes. Curr. Diab. Rep. 4, 135–142 (2004).
Kurts, C., Kosaka, H., Carbone, F.R., Miller, J.F. & Heath, W.R. Class I-restricted cross-presentation of exogenous self-antigens leads to deletion of autoreactive CD8+ T cells. J. Exp. Med. 186, 239–245 (1997).
Steinman, R.M., Turley, S., Mellman, I. & Inaba, K. The induction of tolerance by dendritic cells that have captured apoptotic cells. J. Exp. Med. 191, 411–416 (2000).
Heath, W.R. & Carbone, F.R. Cross-presentation, dendritic cells, tolerance and immunity. Annu. Rev. Immunol. 19, 47–64 (2001).
Hawiger, D. et al. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 194, 769–779 (2001).
Bonifaz, L. et al. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J. Exp. Med. 196, 1627–1638 (2002).
Legge, K.L. et al. On the role of dendritic cells in peripheral T cell tolerance and modulation of autoimmunity. J. Exp. Med. 196, 217–227 (2002).
Stockinger, B. & Hausmann, B. Functional recognition of in vivo processed self antigen. Int. Immunol. 6, 247–254 (1994).
Huang, F.P. et al. A discrete subpopulation of dendritic cells transports apoptotic intestinal epithelial cells to T cell areas of mesenteric lymph nodes. J. Exp. Med. 191, 435–444 (2000).
Scheinecker, C., McHugh, R., Shevach, E.M. & Germain, R.N. Constitutive presentation of a natural tissue autoantigen exclusively by dendritic cells in the draining lymph node. J. Exp. Med. 196, 1079–1090 (2002).
Min, W.P. et al. Inhibitory feedback loop between tolerogenic dendritic cells and regulatory T cells in transplant tolerance. J. Immunol. 170, 1304–1312 (2003).
Yamazaki, S. et al. Direct expansion of functional CD25+ CD4+ regulatory T cells by antigen-processing dendritic cells. J. Exp. Med. 198, 235–247 (2003).
Fisson, S. et al. Continuous activation of autoreactive CD4+ CD25+ regulatory T cells in the steady state. J. Exp. Med. 198, 737–746 (2003).
Steinman, R.M. et al. Dendritic cell function in vivo during the steady state: a role in peripheral tolerance. Ann NY Acad Sci 987, 15–25 (2003).
Inaba, K. et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 176, 1693–1702 (1992).
Maraskovsky, E. et al. Dramatic numerical increase of functionally mature dendritic cells in FLT3 ligand-treated mice. Adv. Exp. Med. Biol. 417, 33–40 (1997).
Young, J.W. Dendritic cells: expansion and differentiation with hematopoietic growth factors. Curr. Opin. Hematol. 6, 135–144 (1999).
Zou, G.M. & Tam, Y.K. Cytokines in the generation and maturation of dendritic cells: recent advances. Eur. Cytokine Netw. 13, 186–199 (2002).
Steinman, R.M. & Dhodapkar, M. Active immunization against cancer with dendritic cells: the near future. Int. J. Cancer 94, 459–473 (2001).
Schuler, G., Schuler-Thurner, B. & Steinman, R.M. The use of dendritic cells in cancer immunotherapy. Curr. Opin. Immunol. 15, 138–147 (2003).
Figdor, C.G., de Vries, I.J., Lesterhuis, W.J. & Melief, C.J. Dendritic cell immunotherapy: mapping the way. Nat. Med. 10, 475–480 (2004).
Carayanniotis, G., Skea, D.L., Luscher, M.A. & Barber, B.H. Adjuvant-independent immunization by immunotargeting antigens to MHC and non-MHC determinants in vivo. Mol. Immunol. 28, 261–267 (1991).
Finkelman, F.D., Lees, A., Birnbaum, R., Gause, W.C. & Morris, S.C. Dendritic cells can present antigen in vivo in a tolerogenic or immunogenic fashion. J. Immunol. 157, 1406–1414 (1996).
van Bergen, J. et al. Get into the groove! Targeting antigens to MHC class II. Immunol. Rev. 172, 87–96 (1999).
Kiessling, R., Klein, E. & Wigzell, H. Natural killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur. J. Immunol. 5, 112–117 (1975).
Kiessling, R. & Haller, O. Natural killer cells in the mouse: an alternative immune surveillance mechanism? Contemp. Top. Immunobiol. 8, 171–201 (1978).
Herberman, R.B. & Holden, H.T. Natural cell-mediated immunity. Adv. Cancer Res. 27, 305–377 (1978).
Karlhofer, F.M., Ribaudo, R.K. & Yokoyama, W.M. MHC class I alloantigen specificity of Ly-49+ IL-2-activated natural killer cells. Nature 358, 66–70 (1992).
Moretta, A. et al. P58 molecules as putative receptors for major histocompatibility complex (MHC) class I molecules in human natural killer (NK) cells. Anti-p58 antibodies reconstitute lysis of MHC class I-protected cells in NK clones displaying different specificities. J. Exp. Med. 178, 597–604 (1993).
Litwin, V., Gumperz, J., Parham, P., Phillips, J.H. & Lanier, L.L. NKB1: a natural killer cell receptor involved in the recognition of polymorphic HLA-B molecules. J. Exp. Med. 180, 537–543 (1994).
Daniels, B.F., Karlhofer, F.M., Seaman, W.E. & Yokoyama, W.M. A natural killer cell receptor specific for a major histocompatibility complex class I molecule. J. Exp. Med. 180, 687–692 (1994).
Brennan, J., Mager, D., Jefferies, W. & Takei, F. Expression of different members of the Ly-49 gene family defines distinct natural killer cell subsets and cell adhesion properties. J. Exp. Med. 180, 2287–2295 (1994).
Wagtmann, N. et al. Molecular clones of the p58 NK cell receptor reveal immunoglobulin-related molecules with diversity in both the extra- and intracellular domains. Immunity 2, 439–449 (1995).
Lanier, L.L. NK cell receptors. Annu. Rev. Immunol. 16, 359–393 (1998).
Yokoyama, W.M. & Plougastel, B.F. Immune functions encoded by the natural killer gene complex. Nat. Rev. Immunol. 3, 304–316 (2003).
Raulet, D.H. Roles of the NKG2D immunoreceptor and its ligands. Nat. Rev. Immunol. 3, 781–790 (2003).
Bottino, C., Moretta, L., Pende, D., Vitale, M. & Moretta, A. Learning how to discriminate between friends and enemies, a lesson from Natural Killer cells. Mol. Immunol. 41, 569–575 (2004).
Raulet, D.H., Vance, R.E. & McMahon, C.W. Regulation of the natural killer cell receptor repertoire. Annu. Rev. Immunol. 19, 291–330 (2001).
Bakker, A.B., Wu, J., Phillips, J.H. & Lanier, L.L. NK cell activation: distinct stimulatory pathways counterbalancing inhibitory signals. Hum. Immunol. 61, 18–27 (2000).
Campbell, K.S. & Colonna, M. Human natural killer cell receptors and signal transduction. Int. Rev. Immunol. 20, 333–370 (2001).
Lanier, L.L. Natural killer cell receptor signaling. Curr. Opin. Immunol. 15, 308–314 (2003).
Cerwenka, A. & Lanier, L.L. Ligands for natural killer cell receptors: redundancy or specificity. Immunol. Rev. 181, 158–169 (2001).
Radaev, S. & Sun, P.D. Structure and function of natural killer cell surface receptors. Annu. Rev. Biophys. Biomol. Struct. 32, 93–114 (2003).
Versteeg, R. NK cells and T cells: mirror images? Immunol. Today 13, 244–247 (1992).
Farrell, H.E. & Davis-Poynter, N.J. From sabotage to camouflage: viral evasion of cytotoxic T lymphocyte and natural killer cell-mediated immunity. Semin. Cell Dev. Biol. 9, 369–378 (1998).
Lorenzo, M.E., Ploegh, H.L. & Tirabassi, R.S. Viral immune evasion strategies and the underlying cell biology. Semin. Immunol. 13, 1–9 (2001).
Basta, S. & Bennink, J.R. A survival game of hide and seek: cytomegaloviruses and MHC class I antigen presentation pathways. Viral Immunol. 16, 231–242 (2003).
York, I.A. et al. A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell 77, 525–535 (1994).
Fruh, K. et al. A viral inhibitor of peptide transporters for antigen presentation. Nature 375, 415–418 (1995).
Park, B. et al. Human cytomegalovirus inhibits tapasin-dependent peptide loading and optimization of the MHC class I peptide cargo for immune evasion. Immunity 20, 71–85 (2004).
Wiertz, E.J. et al. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 84, 769–779 (1996).
Ljunggren, H.G. & Karre, K. In search of the 'missing self': MHC molecules and NK cell recognition. Immunol. Today 11, 237–244 (1990).
Brennan, J., Mahon, G., Mager, D.L., Jefferies, W.A. & Takei, F. Recognition of class I major histocompatibility complex molecules by Ly-49: specificities and domain interactions. J. Exp. Med. 183, 1553–1559 (1996).
Smith, H.R. et al. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc. Natl. Acad. Sci. USA 99, 8826–8831 (2002).
Voigt, V. et al. Murine cytomegalovirus m157 mutation and variation leads to immune evasion of natural killer cells. Proc. Natl. Acad. Sci. USA 100, 13483–13488 (2003).
Bauer, S. et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 285, 727–729 (1999).
Diefenbach, A., Jamieson, A.M., Liu, S.D., Shastri, N. & Raulet, D.H. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat. Immunol. 1, 119–126 (2000).
Sutherland, C.L., Chalupny, N.J. & Cosman, D. The UL16-binding proteins, a novel family of MHC class I-related ligands for NKG2D, activate natural killer cell functions. Immunol. Rev. 181, 185–192 (2001).
Carayannopoulos, L.N., Naidenko, O.V., Fremont, D.H. & Yokoyama, W.M. Cutting edge: murine UL16-binding protein-like transcript 1: a newly described transcript encoding a high-affinity ligand for murine NKG2D. J. Immunol. 169, 4079–4083 (2002).
Long, E.O. Tumor cell recognition by natural killer cells. Semin. Cancer Biol. 12, 57–61 (2002).
Diefenbach, A. & Raulet, D.H. The innate immune response to tumors and its role in the induction of T-cell immunity. Immunol. Rev. 188, 9–21 (2002).
Cerwenka, A. & Lanier, L.L. NKG2D ligands: unconventional MHC class I-like molecules exploited by viruses and cancer. Tissue Antigens 61, 335–343 (2003).
Groh, V., Wu, J., Yee, C. & Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 419, 734–738 (2002).
Salih, H.R. et al. Functional expression and release of ligands for the activating immunoreceptor NKG2D in leukemia. Blood 102, 1389–1396 (2003).
Bendelac, A., Rivera, M.N., Park, S.H. & Roark, J.H. Mouse CD1-specific NK1 T cells: development, specificity, and function. Annu. Rev. Immunol. 15, 535–562 (1997).
Taniguchi, M. & Nakayama, T. Recognition and function of Vα14 NKT cells. Semin. Immunol. 12, 543–550 (2000).
Zeng, Z. et al. Crystal structure of mouse CD1: An MHC-like fold with a large hydrophobic binding groove. Science 277, 339–345 (1997).
Kawano, T. et al. CD1d-restricted and TCR-mediated activation of Vα14 NKT cells by glycosylceramides. Science 278, 1626–1629 (1997).
Zhou, D. et al. Lysosomal glycosphingolipid recognition by NKT cells. Science, published online 11 November 2004 (doi: 10.1126/science.1103440)
Brigl, M., Bry, L., Kent, S.C., Gumperz, J.E. & Brenner, M.B. Mechanism of CD1d-restricted natural killer T cell activation during microbial infection. Nat. Immunol. 4, 1230–1237 (2003).
Teixeira, H.C. & Kaufmann, S.H. Role of NK1.1+ cells in experimental listeriosis. NK1+ cells are early IFN-gamma producers but impair resistance to Listeria monocytogenes infection. J. Immunol. 152, 1873–1882 (1994).
Yoshimoto, T., Bendelac, A., Watson, C., Hu-Li, J. & Paul, W.E. Role of NK1.1+ T cells in a TH2 response and in immunoglobulin E production. Science 270, 1845–1847 (1995).
Biron, C.A. & Brossay, L. NK cells and NKT cells in innate defense against viral infections. Curr. Opin. Immunol. 13, 458–464 (2001).
Biron, C.A., Nguyen, K.B., Pien, G.C., Cousens, L.P. & Salazar-Mather, T.P. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu. Rev. Immunol. 17, 189–220 (1999).
Steinman, R.M. Some interfaces of dendritic cell biology. APMIS 111, 675–697 (2003).
Green, D.R. & Webb, D.R. Saying the 'S' word in public. Immunol. Today 14, 523–525 (1993).
Shevach, E.M. Regulatory T cells in autoimmmunity*. Annu. Rev. Immunol. 18, 423–449 (2000).
Hori, S., Takahashi, T. & Sakaguchi, S. Control of autoimmunity by naturally arising regulatory CD4+ T cells. Adv. Immunol. 81, 331–371 (2003).
Sakaguchi, S., Sakaguchi, N., Asano, M., Itoh, M. & Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor α-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 155, 1151–1164 (1995).
Hori, S., Nomura, T. & Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061 (2003).
Fontenot, J.D., Gavin, M.A. & Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 4, 330–336 (2003).
Khattri, R., Cox, T., Yasayko, S.A. & Ramsdell, F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 4, 337–342 (2003).
Fehervari, Z. & Sakaguchi, S. Development and function of CD25+CD4+ regulatory T cells. Curr. Opin. Immunol. 16, 203–208 (2004).
Ramsdell, F. Foxp3 and natural regulatory T cells: key to a cell lineage? Immunity 19, 165–168 (2003).
Gambineri, E., Torgerson, T.R. & Ochs, H.D. Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked inheritance (IPEX), a syndrome of systemic autoimmunity caused by mutations of FOXP3, a critical regulator of T-cell homeostasis. Curr. Opin. Rheumatol. 15, 430–435 (2003).
Chatenoud, L., Salomon, B. & Bluestone, J.A. Suppressor T cells–they're back and critical for regulation of autoimmunity! Immunol. Rev. 182, 149–163 (2001).
Berzins, S.P., Venanzi, E.S., Benoist, C. & Mathis, D. T-cell compartments of prediabetic NOD mice. Diabetes 52, 327–334 (2003).
Sakaguchi, S. et al. Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol. Rev. 182, 18–32 (2001).
Karim, M., Bushell, A.R. & Wood, K.J. Regulatory T cells in transplantation. Curr. Opin. Immunol. 14, 584–591 (2002).
Rouse, B.T. & Suvas, S. Regulatory cells and infectious agents: detentes cordiale and contraire. J. Immunol. 173, 2211–2215 (2004).
Belkaid, Y., Piccirillo, C.A., Mendez, S., Shevach, E.M. & Sacks, D.L. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature 420, 502–507 (2002).
Roncarolo, M.G., Bacchetta, R., Bordignon, C., Narula, S. & Levings, M.K. Type 1 T regulatory cells. Immunol. Rev. 182, 68–79 (2001).
Barrat, F.J. et al. In vitro generation of interleukin 10-producing regulatory CD4+ T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (TH1)- and TH2-inducing cytokines. J. Exp. Med. 195, 603–616 (2002).
Malek, T.R. The main function of IL-2 is to promote the development of T regulatory cells. J. Leukoc. Biol. 74, 961–965 (2003).
Piccirillo, C.A. & Shevach, E.M. Cutting edge: control of CD8+ T cell activation by CD4+CD25+ immunoregulatory cells. J. Immunol. 167, 1137–1140 (2001).
Thornton, A.M. & Shevach, E.M. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J. Immunol. 164, 183–190 (2000).
Maloy, K.J. et al. CD4+CD25+ T(R) cells suppress innate immune pathology through cytokine-dependent mechanisms. J. Exp. Med. 197, 111–119 (2003).
Nakamura, K., Kitani, A. & Strober, W. Cell contact-dependent immunosuppression by CD4+CD25+ regulatory T cells is mediated by cell surface-bound transforming growth factor β. J. Exp. Med. 194, 629–644 (2001).
Nakamura, K. et al. TGF-β 1 plays an important role in the mechanism of CD4+CD25+ regulatory T cell activity in both humans and mice. J. Immunol. 172, 834–842 (2004).
Pasare, C. & Medzhitov, R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science 299, 1033–1036 (2003).
Sharpe, A.H. & Freeman, G.J. The B7–CD28 superfamily. Nat. Rev. Immunol. 2, 116–126 (2002).
Carreno, B.M. & Collins, M. The B7 family of ligands and its receptors: new pathways for costimulation and inhibition of immune responses. Annu. Rev. Immunol. 20, 29–53 (2002).
Alegre, M.L., Frauwirth, K.A. & Thompson, C.B. T-cell regulation by CD28 and CTLA-4. Nat. Rev. Immunol. 1, 220–228 (2001).
Chambers, C.A., Kuhns, M.S., Egen, J.G. & Allison, J.P. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 19, 565–594 (2001).
Egen, J.G. & Allison, J.P. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity 16, 23–35 (2002).
Pentcheva-Hoang, T., Egen, J.G., Wojnoonski, K. & Allison, J.P. B7–1 and B7–2 selectively recruit CTLA-4 and CD28 to the immunological synapse. Immunity 21, 401–413 (2004).
Tseng, S.Y. et al. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J. Exp. Med. 193, 839–846 (2001).
Nishimura, H., Nose, M., Hiai, H., Minato, N. & Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 11, 141–151 (1999).
Latchman, Y. et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2, 261–268 (2001).
Chen, L. Co-inhibitory molecules of the B7–CD28 family in the control of T-cell immunity. Nat. Rev. Immunol. 4, 336–347 (2004).
Dong, C., Nurieva, R.I. & Prasad, D.V. Immune regulation by novel costimulatory molecules. Immunol. Res. 28, 39–48 (2003).
Newton, K. & Strasser, A. Cell death control in lymphocytes. Adv. Immunol. 76, 179–226 (2000).
Surh, C.D. & Sprent, J. T-cell apoptosis detected in situ during positive and negative selection in the thymus. Nature 372, 100–103 (1994).
Palmer, E. Negative selection–clearing out the bad apples from the T-cell repertoire. Nat. Rev. Immunol. 3, 383–391 (2003).
Defrance, T., Casamayor-Palleja, M. & Krammer, P.H. The life and death of a B cell. Adv. Cancer Res. 86, 195–225 (2002).
Jankovic, M., Casellas, R., Yannoutsos, N., Wardemann, H. & Nussenzweig, M.C. RAGs and regulation of autoantibodies. Annu. Rev. Immunol. 22, 485–501 (2004).
MacLennan, I.C. Germinal centers. Annu. Rev. Immunol. 12, 117–139 (1994).
Ahmed, R. & Gray, D. Immunological memory and protective immunity: understanding their relation. Science 272, 54–60 (1996).
Seder, R.A. & Ahmed, R. Similarities and differences in CD4+ and CD8+ effector and memory T cell generation. Nat. Immunol. 4, 835–842 (2003).
Fisher, G.H. et al. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell 81, 935–946 (1995).
Chan, F.K. et al. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science 288, 2351–2354 (2000).
Croft, M. Costimulatory members of the TNFR family: keys to effective T-cell immunity? Nat. Rev. Immunol. 3, 609–620 (2003).
Sallusto, F., Lenig, D., Forster, R., Lipp, M. & Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401, 708–712 (1999).
Bourgeois, C., Rocha, B. & Tanchot, C. A role for CD40 expression on CD8+ T cells in the generation of CD8+ T cell memory. Science 297, 2060–2063 (2002).
Janssen, E.M. et al. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 421, 852–856 (2003).
Sun, J.C. & Bevan, M.J. Cutting edge: long-lived CD8 memory and protective immunity in the absence of CD40 expression on CD8 T cells. J. Immunol. 172, 3385–3389 (2004).
Sun, J.C. & Bevan, M.J. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science 300, 339–342 (2003).
Shedlock, D.J. & Shen, H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science 300, 337–339 (2003).
Bevan, M.J. Helping the CD8+ T-cell response. Nat. Rev. Immunol. 4, 595–602 (2004).
Butcher, E.C. & Picker, L.J. Lymphocyte homing and homeostasis. Science 272, 60–66 (1996).
Cyster, J.G. Chemokines and cell migration in secondary lymphoid organs. Science 286, 2098–2102 (1999).
von Andrian, U.H. & Mackay, C.R. T-cell function and migration. Two sides of the same coin. N. Engl. J. Med. 343, 1020–1034 (2000).
Mackay, C.R. Chemokines: immunology's high impact factors. Nat. Immunol. 2, 95–101 (2001).
Murphy, P.M. Chemokines. in Fundamental Immunology (ed. Paul, W.E.) 801–840 (Lippincott, Williams, and Wilkins, Philadelphia, 2003).
Forster, R. et al. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell 99, 23–33 (1999).
Sallusto, F., Mackay, C.R. & Lanzavecchia, A. The role of chemokine receptors in primary, effector, and memory immune responses. Annu. Rev. Immunol. 18, 593–620 (2000).
Forster, R. et al. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell 87, 1037–1047 (1996).
Ansel, K.M. et al. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature 406, 309–314 (2000).
Cyster, J.G. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu. Rev. Immunol. published online 11 October 2004 (doi: 10.1146/annurev.immunol.23.021704.115628)
Mora, J.R. et al. Selective imprinting of gut-homing T cells by Peyer's patch dendritic cells. Nature 424, 88–93 (2003).
Kim, C.H., Nagata, K. & Butcher, E.C. Dendritic cells support sequential reprogramming of chemoattractant receptor profiles during naive to effector T cell differentiation. J. Immunol. 171, 152–158 (2003).
Iwata, M. et al. Retinoic Acid imprints gut-homing specificity on T cells. Immunity 21, 527–538 (2004).
Berger, E.A., Murphy, P.M. & Farber, J.M. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu. Rev. Immunol. 17, 657–700 (1999).
Horuk, R. et al. A receptor for the malarial parasite Plasmodium vivax: the erythrocyte chemokine receptor. Science 261, 1182–1184 (1993).
Murphy, P.M. Viral exploitation and subversion of the immune system through chemokine mimicry. Nat. Immunol. 2, 116–122 (2001).
Altman, J.D. et al. Phenotypic analysis of antigen-specific T lymphocytes. Science 274, 94–96 (1996).
Reijonen, H. & Kwok, W.W. Use of HLA class II tetramers in tracking antigen-specific T cells and mapping T-cell epitopes. Methods 29, 282–288 (2003).
Meidenbauer, N., Hoffmann, T.K. & Donnenberg, A.D. Direct visualization of antigen-specific T cells using peptide-MHC-class I tetrameric complexes. Methods 31, 160–171 (2003).
Monks, C.R., Freiberg, B.A., Kupfer, H., Sciaky, N. & Kupfer, A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature 395, 82–86 (1998).
Wulfing, C., Sjaastad, M.D. & Davis, M.M. Visualizing the dynamics of T cell activation: intracellular adhesion molecule 1 migrates rapidly to the T cell/B cell interface and acts to sustain calcium levels. Proc. Natl. Acad. Sci. USA 95, 6302–6307 (1998).
Grakoui, A. et al. The immunological synapse: a molecular machine controlling T cell activation. Science 285, 221–227 (1999).
Bromley, S.K. et al. The immunological synapse. Annu. Rev. Immunol. 19, 375–396 (2001).
Kupfer, A. & Kupfer, H. Imaging immune cell interactions and functions: SMACs and the Immunological Synapse. Semin. Immunol. 15, 295–300 (2003).
Davis, M.M. et al. Dynamics of cell surface molecules during T cell recognition. Annu. Rev. Biochem. 72, 717–742 (2003).
Reinhardt, R.L., Khoruts, A., Merica, R., Zell, T. & Jenkins, M.K. Visualizing the generation of memory CD4 T cells in the whole body. Nature 410, 101–105 (2001).
Stoll, S., Delon, J., Brotz, T.M. & Germain, R.N. Dynamic imaging of T cell-dendritic cell interactions in lymph nodes. Science 296, 1873–1876 (2002).
Cahalan, M.D., Parker, I., Wei, S.H. & Miller, M.J. Two-photon tissue imaging: seeing the immune system in a fresh light. Nat. Rev. Immunol. 2, 872–880 (2002).
Miller, M.J., Wei, S.H., Parker, I. & Cahalan, M.D. Two-photon imaging of lymphocyte motility and antigen response in intact lymph node. Science 296, 1869–1873 (2002).
Bousso, P., Bhakta, N.R., Lewis, R.S. & Robey, E. Dynamics of thymocyte-stromal cell interactions visualized by two-photon microscopy. Science 296, 1876–1880 (2002).
Bousso, P. & Robey, E. Dynamics of CD8+ T cell priming by dendritic cells in intact lymph nodes. Nat. Immunol. 4, 579–585 (2003).
Mempel, T.R., Henrickson, S.E. & Von Andrian, U.H. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature 427, 154–159 (2004).
Waldmann, T.A. Immunotherapy: past, present and future. Nat. Med. 9, 269–277 (2003).
Hacein-Bey-Abina, S. et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N. Engl. J. Med. 346, 1185–1193 (2002).
Hacein-Bey-Abina, S. et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 302, 415–419 (2003).
Chinen, J. & Puck, J.M. Successes and risks of gene therapy in primary immunodeficiencies. J. Allergy. Clin. Immunol. 113, 595–603; quiz 604 (2004).
Van Der Bruggen, P. et al. Tumor-specific shared antigenic peptides recognized by human T cells. Immunol. Rev. 188, 51–64 (2002).
Pardoll, D. Does the immune system see tumors as foreign or self? Annu. Rev. Immunol. 21, 807–839 (2003).
Rosenberg, S.A. Shedding light on immunotherapy for cancer. N. Engl. J. Med. 350, 1461–1463 (2004).
Wang, H.Y. et al. Tumor-specific human CD4+ regulatory T cells and their ligands: implications for immunotherapy. Immunity 20, 107–118 (2004).
Egen, J.G., Kuhns, M.S. & Allison, J.P. CTLA-4: new insights into its biological function and use in tumor immunotherapy. Nat. Immunol. 3, 611–618 (2002).
Hodi, F.S. et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc. Natl. Acad. Sci. USA 100, 4712–4717 (2003).
Phan, G.Q. et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc. Natl. Acad. Sci. USA 100, 8372–8377 (2003).
Herzenberg, L.A., Black, S.J. & Herzenberg, L.A. Regulatory circuits and antibody responses. Eur. J. Immunol. 10, 1–11 (1980).
Germain, R.N. The art of the probable: system control in the adaptive immune system. Science 293, 240–245 (2001).
Gett, A.V. & Hodgkin, P.D. A cellular calculus for signal integration by T cells. Nat. Immunol. 1, 239–244 (2000).
Brenner, S. End of the interlude? Nat. Biotechnol. 22, 1191 (2004).
Amsen, D. et al. Instruction of distinct CD4 T helper cell fates by different notch ligands on antigen-presenting cells. Cell 117, 515–526 (2004).
Maekawa, Y. et al. d1-Notch3 interactions bias the functional differentiation of activated CD4+ T cells. Immunity 19, 549–559 (2003).
Akira, S. & Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 4, 499–511 (2004).
Vivier, E. & Anfossi, N. Inhibitory NK-cell receptors on T cells: witness of the past, actors of the future. Nat. Rev. Immunol. 4, 190–198 (2004).
Bottino, C., Moretta, L., Pende, D., Vitale, M. & Moretta, A. Learning how to discriminate between friends and enemies, a lesson from Natural Killer cells. Mol. Immunol. 41, 569–575 (2004).
Grossman, W.J. et al. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity 21, 589–601 (2004).
Acknowledgements
I am grateful to my many colleagues who willingly suggested their “Top Ten” recent immunology research advances. However, not all the important discoveries of the last 10 years can be dealt with in this limited space and it is inevitable that there will be disagreement with what I have chosen to emphasize. The responsibility for these selections is mine. To those who see either major omissions or excess attention to one topic or another, or who find the citations imperfect, I apologize in advance, offering only the excuse that what I have written represents a good faith attempt to highlight the principal accomplishments of the field during this period and to give as much credit as possible within the limits of this article.
Author information
Authors and Affiliations
Ethics declarations
Competing interests
The author declares no competing financial interests.
Rights and permissions
About this article
Cite this article
Germain, R. An innately interesting decade of research in immunology. Nat Med 10, 1307–1320 (2004). https://doi.org/10.1038/nm1159
Published:
Issue Date:
DOI: https://doi.org/10.1038/nm1159
This article is cited by
-
Immune optimization inspired artificial natural killer cell earthquake prediction method
The Journal of Supercomputing (2022)
-
Molecular Characterization and Expression Analysis of ftr01, ftr42, and ftr58 in Zebrafish (Danio rerio)
Virologica Sinica (2019)
-
Systems biology applied to vaccine and immunotherapy development
BMC Systems Biology (2011)
