
Abstract
Sepsis, a systemic inflammatory response to infection, commonly progresses to acute lung injury (ALI), an inflammatory lung disease with high morbidity. We postulated that sepsis-associated ALI is initiated by degradation of the pulmonary endothelial glycocalyx, leading to neutrophil adherence and inflammation. Using intravital microscopy, we found that endotoxemia in mice rapidly induced pulmonary microvascular glycocalyx degradation via tumor necrosis factor-α (TNF-α)-dependent mechanisms. Glycocalyx degradation involved the specific loss of heparan sulfate and coincided with activation of endothelial heparanase, a TNF-α–responsive, heparan sulfate–specific glucuronidase. Glycocalyx degradation increased the availability of endothelial surface adhesion molecules to circulating microspheres and contributed to neutrophil adhesion. Heparanase inhibition prevented endotoxemia-associated glycocalyx loss and neutrophil adhesion and, accordingly, attenuated sepsis-induced ALI and mortality in mice. These findings are potentially relevant to human disease, as sepsis-associated respiratory failure in humans was associated with higher plasma heparan sulfate degradation activity; moreover, heparanase content was higher in human lung biopsies showing diffuse alveolar damage than in normal human lung tissue.
Similar content being viewed by others
Main
Sepsis is a common and severe medical condition characterized by a systemic inflammatory response to infection1. The inflammatory consequences of sepsis are particularly apparent within the pulmonary circulation, which is continuously exposed to circulating pathogen-associated molecular patterns (such as the endotoxin lipopolysaccharide (LPS)) capable of triggering innate immunity. Over 40% of individuals with sepsis develop ALI, a syndrome characterized by neutrophilic inflammation and pulmonary vascular hyperpermeability2. The development of ALI markedly worsens patient prognosis, increasing intensive care unit mortality from 11% to 38% in patients with septic shock3. Survivors of ALI are often left with considerable long-term morbidity and increased healthcare expenditures4. Despite this clinical significance, no sepsis-specific treatments prevent the onset of inflammatory lung injury, reflecting an incomplete understanding of septic ALI pathogenesis.
Neutrophil adhesion to the vascular intima is crucial to the initiation of inflammatory tissue injury. Intimal surfaces are lined by the endothelial glycocalyx, an extracellular layer of glycoproteins, proteoglycans and glycosaminoglycans (GAGs)5. In vivo, heparan sulfate and other glycocalyx GAGs are highly hydrated, forming a substantial endothelial surface layer (ESL) that acts as a barrier to circulating cells and large molecules (Fig. 1a)6. Little, however, is known about the influence of the glycocalyx on neutrophil adhesion during inflammation5. Enzymatic degradation of the glycocalyx in isolated guinea pig coronary capillaries, microperfused rat mesenteric venules and microperfused mouse cremasteric venules is associated with increased leukocyte retention; however, the mechanisms underlying this association are uncertain7,8,9,10. Glycocalyx degradation–associated neutrophil retention may be relevant to sepsis-induced organ inflammation, given recent observations of circulating glycocalyx fragments in patients with septic shock11.

(a) Left, graphical representation (not to scale) of the in vivo endothelial glycocalyx, forming a substantial ESL that excludes large molecules (for example, dextrans) from the vessel surface. Right, representative images of mouse subpleural microvessels (MV) using simultaneous differential interference contrast (DIC) and FITC-dextran (FITC) microscopy. Differences in DIC and FITC vascular widths (inclusive and exclusive of the ESL, respectively) reflect ESL thickness (brackets). Scale bars, 10 μm. A, alveolus. (b) Assessment of pulmonary ESL thickness within subpleural microvessels (mean DIC diameter 18.11 ± 1.01 μm, mean FITC diameter 14.17 ± 0.96 μm) of wild-type mice injected with intravenous saline, LPS (20 μg per g body weight) or TNF-α (200 ng) at t = 0 min and imaged at 0, 30, 60 and 90 min. n = 5 mice per group; *P < 0.05 in comparison to other groups. (c) Assessment of pulmonary ESL thickness of TNFR1-deficient Tnfrsf1atm1Imx mice treated at t = 0 min with intravenous saline or LPS (20 μg per g body weight); n = 3 or 4 mice per group. Data are represented as means ± s.e.m.
Our study aimed to elucidate the mechanisms by which glycocalyx loss occurs during sepsis and how this loss allows for neutrophil adhesion within the pulmonary circulation. We hypothesized that sepsis induces activation of heparanase (an endogenous heparan sulfate–specific glucuronidase) within the pulmonary microvasculature, with consequent degradation of the pulmonary endothelial glycocalyx playing a central part in neutrophil adherence and inflammatory lung injury. Our findings may suggest new therapeutic approaches to prevent the onset of inflammatory lung injury during sepsis.
Results
Endotoxemia induces ESL loss via activation of heparanase
Intravital microscopy (in vivo microscopy, IVM) of the ESL provides an ideal means of studying glycocalyx structure during disease states, as the glycocalyx has been found to be aberrant when studied using in vitro preparations and is often damaged during tissue handling and fixation5,12,13. We therefore used closed-chest mouse pulmonary IVM to determine glycocalyx integrity by measuring ESL exclusion of fluorescently labeled, high-molecular-weight (150 kDa) dextrans from vessel surfaces (Fig. 1a and Supplementary Fig. 1). The mean baseline pulmonary microvascular ESL thickness of the wild-type C57BL/6 mice studied (n = 43) was 1.67 ± 0.09 μm (mean ± s.e.m.), substantially greater than the ESL thickness observed in cremasteric (systemic) microvessels (0.67 ± 0.08 μm, n = 9, Supplementary Fig. 2a). In saline-treated mice, pulmonary ESL thickness remained stable throughout 90 min of observation (Fig. 1b). Intravenous injection of LPS (20 μg per g body weight) induced a rapid (<30 min) loss of pulmonary ESL thickness, suggesting sepsis-associated glycocalyx degradation (Fig. 1b). Intravenous injection of TNF-α, a cytokine released early in sepsis, similarly triggered rapid pulmonary ESL loss (Fig. 1b). LPS-induced pulmonary ESL loss was prevented in mice lacking TNFR1, the major TNF-α receptor implicated in sepsis14, suggesting a necessary role for TNF-α in septic glycocalyx degradation (Fig. 1c). Notably, cremasteric ESL thickness was unchanged after LPS injection, suggesting vascular bed heterogeneity in the response to endotoxemia (Supplementary Fig. 2a).
Intravenous injection of heparinase-III, a heparan sulfate–specific bacterial glucuronidase15, rapidly decreased pulmonary ESL thickness in wild-type mice, demonstrating the essential role of glycocalyx heparan sulfate in the maintenance of ESL structure (Fig. 2a). We hypothesized that LPS, via TNF-α–dependent mechanisms, similarly induces the degradation of the pulmonary ESL by rapidly activating pulmonary endothelial heparanase, a mammalian heparinase-III analog constitutively expressed as a 65-kDa proenzyme16. Treatment of mouse lung microvascular endothelial cells (MLMVECs) with TNF-α rapidly (within 30 min) induced cleavage of 65-kDa heparanase to its active 50-kDa isoform (Fig. 2b), coincident with increased heparan sulfate degradation activity within cell lysates (Fig. 2c).

(a) Assessment of pulmonary ESL thickness within subpleural microvessels of wild-type mice treated with heparinase-III or heat-inactivated heparinase-III (1 U) at t = 0. n = 4–6 mice per group; *P < 0.05. (b,c) Expression of active (50 kDa) and inactive (65 kDa) endothelial heparanase (b, representative of three independent experiments) and heparan sulfate degradation activity (c, n = 4 per group) in cultured mouse lung microvascular endothelial cells treated with TNF-α (5 and 50 ng ml−1) or saline control for 30 min; *P < 0.05. (d) Assessment of pulmonary ESL thickness within subpleural microvessels of wild-type mice injected with intravenous vehicle or LPS (20 μg per g body weight) in addition to the heparanase inhibitor heparin (5 U administered intravenously at t = 0); n = 4–6 mice per group. (e) Assessment of pulmonary ESL thickness within subpleural microvessels of wild-type mice injected with intravenous vehicle or LPS (20 μg per g body weight) in addition to NAH (150 μg administered intravenously at t = 0); n = 4 or 5 mice per group. (f) Assessment of pulmonary microvascular ESL thickness in Hpse−/− mice injected with intravenous saline or LPS (20 μg per g body weight); n = 3 mice per group. Data are represented as means ± s.e.m.
In vivo inhibition of heparanase activity with the competitive antagonist heparin completely prevented endotoxemia-induced ESL loss (Fig. 2d) without interfering with LPS activation of Toll-like receptor signaling (Supplementary Fig. 3). This effect of heparin was independent of anticoagulation, as ESL loss was also prevented by the nonanticoagulant heparanase inhibitor N-desulfated/re-N-acetylated heparin (NAH, Fig. 2e) as well as in mice genetically lacking heparanase expression (Hpse−/− mice, Fig. 2f). These findings demonstrate that activation of constitutively expressed endothelial heparanase mediates the rapid thinning of the pulmonary ESL that occurs after LPS exposure.
The pulmonary ESL regulates neutrophil adhesion
To assess in real time the relationship between ESL loss and pulmonary neutrophil recruitment, a key early step in the alveolar inflammation characteristic of ALI, we performed adoptive transfer of GFP-expressing neutrophils into wild-type mice. Circulating GFP+ neutrophils adhered to the pulmonary endothelium within 30 min of intravenous LPS administration (remaining stationary for >15 min thereafter, Fig. 3a and Supplementary Fig. 4), a time course that closely followed ESL loss (Figs. 1b and 2a). Similarly, fluorescent microspheres labeled with antibodies against intercellular adhesion molecule 1 (ICAM-1), an endothelial adhesion molecule implicated in endotoxin-induced pulmonary neutrophil adhesion (Supplementary Fig. 5) and lung injury17, bound to microvascular endothelial surfaces within 45 min of LPS administration (Fig. 3b). These findings were not limited to the subpleural microvasculature, as we noted concordant microsphere adhesion throughout frozen sections of the left lung (Supplementary Fig. 6). The possibility that anti–ICAM-1 microspheres were being captured by neutrophils was excluded, as neutrophil depletion did not prevent microsphere adhesion during endotoxemia (Supplementary Fig. 7). As other adhesion molecules in addition to ICAM-1 participate in neutrophil-endothelial adhesion18, we repeated our experiments with microspheres targeted to vascular cell adhesion molecule 1 (VCAM-1); these similarly adhered to the microvasculature 45 min after LPS administration (Supplementary Fig. 8a). Together, these findings suggest that the rapid neutrophil-endothelial binding that occurs after intravenous LPS may reflect an acute change in the availability of multiple endothelial adhesion molecules to circulating cells.

(a) Adherence of adoptively transferred GFP+ neutrophils within subpleural microvessels before and 30–45 min after intravenous saline, LPS (20 μg per g body weight) or LPS (20 μg per g body weight) with heparin (5 U) in wild-type mice or 30–45 min after intravenous LPS (20 μg per g body weight) in Hpse−/− mice. Representative images reflect changes occurring within a single, serially imaged, low-powered field. Scale bars, 40 μm. n = 3 mice per group; *P < 0.05 compared to saline. PMN, polymorphonuclear leukocytes. (b) Visualization of anti-ICAM-1–coated fluorescent microspheres within wild-type mouse subpleural microvessels, simultaneously imaged by DIC and fluorescence microscopy. Images obtained 45 min after intravenous saline, LPS (20 μg per g body weight), LPS (20 μg per g body weight) with heparin (5 U) or heparinase-III (1 U). Isotype-matched antibody-coated microspheres serve as controls for nonspecific adhesion. Scale bars, 20 μm. n = 3 or 4 mice per group; *P < 0.05 compared to saline. Data are represented as means ± s.e.m.
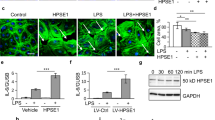
To determine the mechanism by which endothelial adhesion molecule availability rapidly increases during endotoxemia, we subjected wild-type mice and MLMVECs to a short-term (45-min) LPS exposure. This exposure induced an early increase in ICAM-1 mRNA levels (Supplementary Fig. 9a,b); however, the duration of exposure was insufficient to alter ICAM-1 (or VCAM-1) total protein content or cell-surface expression (Supplementary Figs. 8b,c and 9c,d). We suspected that the increased adhesion molecule availability was instead a consequence of glycocalyx degradation, exposing constitutively expressed endothelial surface proteins typically buried within the thick pulmonary ESL10. Indeed, LPS-induced anti–ICAM-1 microsphere adhesion in the lung was attenuated by heparanase inhibition (Fig. 3b and Supplementary Fig. 6). Furthermore, heparinase-III–induced glycocalyx degradation led to anti–ICAM-1 microsphere adhesion (Fig. 3b), albeit in a heterogeneous pattern that may reflect differences between the exogenous administration of heparanase analogs and the endogenous induction of endothelial heparanase. Although heparin and heparinase-III influenced anti–ICAM-1 microsphere adhesion, they did not alter ICAM-1 mRNA levels (Supplementary Fig. 9a,b), further indicating that the rapid increase in ICAM-1 availability occurs independently of any induction of endothelial ICAM-1 expression. Finally, heparin treatment attenuated GFP+ neutrophil adhesion after LPS exposure (Fig. 3a), consistent with previously described effects of heparin on neutrophil-endothelial cell binding in response to LPS19. Similarly, GFP+ (Hpse+/+) neutrophils did not adhere to the pulmonary microvasculature of LPS-treated Hpse−/− mice (Fig. 3a). These findings provide a teleological rationale for LPS-induced heparanase activation: pathogen-associated molecular patterns prompt endothelial cells to cleave the endothelial glycocalyx, preparing the vascular surface for neutrophil adhesion and subsequent inflammation.
Heparanase-mediated ESL loss is necessary for septic ALI
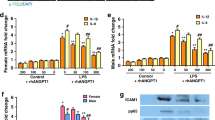
To determine the impact of ESL loss and consequent neutrophil adhesion on the development of septic ALI, we induced ALI in wild-type mice via intraperitoneal LPS injection (40 μg per g body weight)20. We chose this higher dose of LPS to rapidly produce robust lung injury21,22. Six hours after LPS administration, pulmonary endothelial permeability and neutrophil extravasation increased, suggestive of early ALI (Fig. 4a–c). Heparanase expression increased and was localized to the pulmonary endothelium, as demonstrated by association with the endothelial cell-surface marker thrombomodulin (Fig. 4d and Supplementary Fig. 10). Consistent with heparanase activation, small pulmonary arteries of LPS-treated mice (prepared using glycocalyx-preserving fixation techniques) had diminished vascular surface heparan sulfate content in comparison to saline-treated controls (Supplementary Fig. 11). Other glycocalyx components were unchanged, demonstrating the heparan sulfate–specific nature of heparanase activity (Supplementary Fig. 11a). Heparanase inhibitors significantly prevented LPS-induced heparan sulfate loss, endothelial hyperpermeability and neutrophil extravasation, suggesting that heparanase-mediated glycocalyx loss contributes to the later development of septic ALI (Fig. 4a,b and Supplementary Fig. 11b). Similarly, pulmonary endothelial hyperpermeability and neutrophil extravasation 6 h after intraperitoneal LPS administration were attenuated in Hpse−/− mice compared to wild-type controls (Fig. 4a,c). Notably, lung myeloperoxidase activity was suppressed in saline-treated Hpse−/− mice (Fig. 4c) despite normal plasma neutrophil counts (254 ± 53 neutrophils per μl in Hpse−/− mice versus 197 ± 47 neutrophils per μl in wild-type mice, n = 4 per group), suggesting that heparanase influences baseline neutrophil extravasation into healthy mouse lungs. In sum, these findings demonstrate that heparanase activation (with consequent glycocalyx degradation) is necessary to the development of ALI pathophysiology during endotoxemia.

(a) Assessment of pulmonary endothelial permeability (filtration coefficient, Kf) in wild-type or Hpse−/− mice 6 h after intraperitoneal LPS (40 μg per g body weight in 500 μl saline) or saline. Wild-type mice were pretreated with subcutaneous saline (200 μl), heparin (5 U in 200 μl saline) or NAH (150 μg in 200 μl saline) 3 h before LPS. Hpse−/− mice received no pharmacologic pretreatment. n = 5–7 mice per group; *P < 0.05 compared to all other groups. (b,c) Myeloperoxidase (MPO) activity within lung homogenates of wild-type (b) or wild-type and Hpse−/− (c) mice treated as described in a, normalized to saline/saline (b) or saline/wild type (c) control. n = 4–8 mice per group; *P < 0.05 compared to saline control. (d) Representative z-stacked imaging (1-μm increments) of heparanase (red) and the endothelial cell marker thrombomodulin (green) in LPS- or saline-treated wild-type mouse lungs, as described in a. Bottom left, area of positive heparanase immunofluorescence in ten random low-power lung fields, normalized to saline control. Bottom right, percentage of area positive for thrombomodulin immunofluorence that is additionally positive for heparanase, as quantified in ten random low-power fields. Scale bars, 50 μm. n = 5 mice per group; *P < 0.05. Data are represented as means ± s.e.m.
Heparanase in human sepsis and ALI
To determine the relevance of endothelial heparanase activation and consequent heparan sulfate degradation to human sepsis, we collected plasma samples from healthy human donors as well as from three groups of individuals with respiratory failure: those mechanically ventilated for altered mental status (for example, ischemic stroke or alcohol withdrawal), for pneumonia (that is, direct injury to the lung) and those ventilated with a primary diagnosis of nonpulmonary sepsis (that is, potentially indirect injury to the lung) (Supplementary Table 1). Plasma heparan sulfate degradation activity was elevated only in individuals with nonpulmonary sepsis, consistent with endothelial heparanase activation and release into the circulation (Fig. 5a).

(a) Heparan sulfate degradation activity measured in plasma collected from healthy donors and three groups of mechanically ventilated individuals: those with altered mental status–induced respiratory failure, pneumonia- or aspiration-induced respiratory failure or respiratory failure associated with antecedent nonpulmonary sepsis. *P < 0.05 compared to healthy donors. (b) Heparanase immunofluorescence in normal human lung tissue and in lung biopsies with diffuse alveolar damage. *P < 0.05 compared to normal lung tissue. (c) Top, confocal fluorescent images of normal human lung showing minimal heparanase expression. Representative fluorescent images of a lung from a patient with diffuse alveolar damage, with high heparanase expression (red) within capillaries (arrow) and conduit vessels (arrowhead), ascertained by the endothelial marker CD31 (green). Nuclei stained with DAPI. Bottom, H&E staining, with capillaries (arrow) and conduit vessels (arrowhead) noted. Scale bars, 50 μm. Data are represented as means ± s.e.m.
We explored the relevance of heparanase to human inflammatory lung disease using human lung biopsies that showed diffuse alveolar damage, the histologic manifestation of ALI (Supplementary Table 2). These biopsies are infrequently performed, as the diagnosis of ALI during sepsis is typically made on clinical criteria alone. In normal human lung tissue, expression of heparanase was minimal (Fig. 5b,c). In biopsies with diffuse alveolar damage, heparanase immunofluorescence was eightfold higher (Fig. 5b) and was noted around capillaries and conduit vessels (Fig. 5c). These findings, although limited by a small sample size (19 plasma samples, 14 lung samples), suggest that heparanase is active in human sepsis and contributes to inflammatory lung injury.
Heparanase inhibition is protective after sepsis onset
Given that heparan sulfate degradation activity was elevated in human plasma collected a mean of 35.4 h after the onset of sepsis-associated respiratory failure (Supplementary Table 1), we hypothesized that interventions targeting heparanase activity would be lung-protective even if administered after sepsis onset. Accordingly, administration of heparin 3 h after intraperitoneal LPS administration prevented endotoxemia-induced pulmonary endothelial hyperpermeability (Fig. 6a).

(a) Assessment of pulmonary endothelial permeability (Kf) in wild-type mice 6 h after intraperitoneal LPS administration (40 μg per g body weight in 500 μl saline). Mice received saline (200 μl) or heparin (5 U in 200 μl saline) subcutaneously 3 h after LPS administration (3 h before measurement of Kf). n = 3 or 4 per group. *P < 0.05. (b) Pulmonary heparanase expression (red) after CLP in wild-type mice. Images are representative of two or three mice per group. Scale bars, 100 μm. (c) Mouse pulmonary Kf measured 48 h after CLP surgery. Wild-type mice received saline (200 μl) or heparin (5 U in 200 μl saline) subcutaneously 24 h after CLP (24 h before measurement of Kf). n = 3 per group. *P < 0.05. (d) H&E staining of representative (n > 4 per group) lungs of wild-type or Hpse−/− mice 48 h after CLP or sham surgery with ensuing exposure to 60% oxygen (FiO2). Scale bars, 500 μm in low-powered images. High-powered imaging (bottom left; scale bar, 50 μm) details an area of neutrophilic alveolitis. (e) Pulmonary neutrophil infiltration 48 h after CLP and hyperoxia with or without delayed heparin treatment as assessed by histology (top) and myeloperoxidase activity (bottom). Mice were treated with subcutaneous saline (200 μl) or heparin (5 U in 200 μl saline) 24 h after CLP. Scale bars, 100 μm. n = 4 per group; *P < 0.05. (f) Survival of wild-type (n = 14) and Hpse−/− (n = 6) mice exposed to CLP and 60% hyperoxia. Survival of wild-type mice after sham surgery and 60% hyperoxia was 100% (n = 8). Data are represented as means ± s.e.m.
We additionally tested heparin treatment in mice subjected to cecal ligation and puncture (CLP), a clinically relevant model of polymicrobial sepsis. Although CLP alone does not typically cause robust histologic lung injury, it has been demonstrated to induce endothelial and epithelial pathophysiology consistent with ALI23. Within 24 h of CLP, mice appeared systemically ill with piloerection and decreased activity. Pulmonary heparanase expression peaked 48 h after CLP, coincident with an increase in endothelial permeability (Fig. 6b,c and Supplementary Fig. 2c). Notably, we observed no CLP-associated heparanase induction in the cremaster, a tissue with stable ESL thickness during endotoxemia (Supplementary Fig. 2). Delayed heparanase inhibition via a one-time dose of heparin (administered 24 h after CLP) attenuated pulmonary endothelial hyperpermeability (Fig. 6c), suggesting that heparin is a lung-protective intervention even in established sepsis. To augment CLP-induced neutrophilic alveolitis, we repeated CLP experiments in the presence of 60% fraction of inspired oxygen (FiO2), a moderate level of hyperoxia not injurious to normal mice24. In these mice, pulmonary neutrophilic infiltration was apparent 48 h after CLP (Fig. 6d) and was attenuated by delayed heparin therapy (Fig. 6e). Hpse−/− mice were similarly protected from CLP- and hyperoxia-induced alveolitis (Fig. 6d) and experienced no CLP- and hyperoxia-associated mortality (Fig. 6f).
Discussion
Our findings demonstrate the contribution of the endothelial glycocalyx to inflammatory diseases such as ALI (Supplementary Fig. 12). In the presence of circulating pathogen-associated molecular patterns, pulmonary microvascular endothelial cells rapidly activate endogenous stores of heparanase. Activated heparanase cleaves heparan sulfate from the pulmonary endothelial glycocalyx, inducing a rapid thinning of the ESL. This ESL loss exposes previously hidden endothelial surface adhesion molecules including ICAM-1 and VCAM-1, allowing neutrophil recognition of and adhesion to the endothelial surface. These events, which pertain to multiple adhesion molecules along the endothelial surface, may work in concert with modifications to individual adhesion molecules (such as increased expression and adhesiveness25) to control inflammatory cell influx in the setting of ALI. Consequently, inhibition of heparanase activity represents a potential lung-protective therapy during sepsis.
The glycocalyx has been generally understudied, in part because of the aberrancy of in vitro preparations of the ESL and its degradation during tissue handling26. Advancements in IVM have coincided with a greater appreciation of glycocalyx and ESL structure and function in health and disease. We examined ESL thickness by dextran exclusion, an IVM technique that allows measurements to be made from images captured at a single timepoint, thereby negating lung motion artifacts. Our findings, to our knowledge representing the first in vivo observations of the pulmonary ESL, indicate that pulmonary microvascular ESL thickness (1.67 μm) is considerably greater than ESL thickness in systemic microvessels (0.67 μm, consistent with previous reports27). Our measurements of pulmonary ESL thickness are consistent with prior in vitro estimates of glycocalyx thickness (2.8 ± 0.5 μm) using fluorescence correlation spectroscopy of bovine pulmonary microvascular endothelial cells28. The physiological significance of differential ESL size in the systemic and pulmonary microcirculations is uncertain but potentially reflects functional heterogeneity across different vascular beds29.
Enzymatic degradation of glycocalyx heparan sulfate rapidly induced a loss of pulmonary ESL thickness, consistent with the dominant role of this GAG in glycocalyx structure. We found that ESL loss similarly occurred during endotoxemia and was mediated by activation of heparanase, an endogenous heparan sulfate–specific glucuronidase. We did not observe concordant endotoxemic ESL loss in the cremaster muscle, a tissue with low levels of heparanase expression (and limited clinical relevance) during sepsis. Extensively studied in cancer metastasis30, heparanase has also recently been implicated in inflammatory diseases such as ulcerative colitis31 and atherosclerosis32. Our findings indicate that heparanase also contributes to inflammatory lung injury, with pathologic heparan sulfate degradation arising from post-translational heparanase activation (that is, proenzyme cleavage) as well as a delayed increase in enzyme expression. The proinflammatory consequences of heparanase conflict with a previous report suggesting a protective effect of the 65-kDa heparanase proenzyme as a pretreatment in endotoxemic rats33. These seemingly discordant results may be explained by recently described nonenzymatic effects of the heparanase proenzyme34. The resistance of Hpse−/− mice (which lack both activated and inactivated heparanase) to septic lung injury suggests that any potential benefits of the endogenous heparanase proenzyme are outweighed by the untoward enzymatic effects of activated heparanase on the endothelial glycocalyx.
We found that loss of pulmonary ESL thickness exposes endothelial surface adhesion molecules to circulating cells, consistent with a previous study of systemic vessels10. Our work reveals key mechanisms underlying ESL regulation of adhesion molecule availability and their impact on inflammatory lung injury. Although the exposure of adhesion molecules is necessary for neutrophil adhesion, additional mechanisms by which glycocalyx loss could induce neutrophil extravasation may also exist. Transgenic mice with constitutively aberrant (desulfated) or truncated heparan sulfate have decreased neutrophil adherence, extravasation or both35,36. These findings, combined with our data, suggest that the constitutive absence of heparan sulfate is not sufficient for neutrophil adherence; rather, there must be acute degradation of heparan sulfate from the endothelial glycocalyx. Acute heparan sulfate degradation not only would expose underlying adhesion molecules but also could release proinflammatory mediators (such as the chemokine IL-8) typically sequestered within the ESL, facilitating neutrophil adherence and extravasation26.
Pulmonary ESL loss is not an all-or-none phenomenon: a < 0.5-μm-thick glycocalyx remnant remains during endotoxemia. Heparanase may not only decrease ESL thickness but also may increase ESL fluidity, resulting in a more profound effect on barrier function than what is apparent based on measurements of 150-kDa dextran exclusion and perhaps modulating the ability of neutrophils to traverse this remnant ESL. Rigorous testing of this hypothesis will require the development of new in vivo techniques to measure ESL stiffness and structural integrity.
We speculate that heparanase-mediated regulation of pulmonary ESL thickness exists to control neutrophil influx into the lung, an organ continuously exposed to airborne pathogens and their associated molecular patterns. In the presence of these danger signals, endothelial cells react by clearing the intimal surface, inviting an influx of inflammatory cells to address the triggering alveolar pathogen. The large size of the pulmonary ESL could reflect the severe consequences of excessive pulmonary neutrophil influx: a thick ESL could dampen an overexuberant response to localized infection, avoiding lung injury and hypoxemia. In sepsis, high concentrations of circulating danger signals might diffusely activate pulmonary ESL degradation, predisposing for inappropriate neutrophilic inflammation and ALI. Inhibition of heparanase activity could therefore represent a new lung-protective intervention in patients with nonpulmonary sepsis. The effect of ESL protection in the setting of a direct injury to the lung (for example, pneumonia) is less certain: heparanase inhibition might oppose the host response to pulmonary infection and be harmful.
If glycocalyx degradation is necessary for the neutrophilic response to danger signals, then glycocalyx reconstitution should also be important during resolution of inflammation. Little, however, is known regarding the mechanisms underlying reconstitution of a damaged glycocalyx. Recovery of cremasteric ESL thickness occurs 72 h after local (high-dose) TNF-α injection37; it is unclear whether this recovery period is a function of constitutive GAG turnover (with reconstitution occurring after cessation of the inciting inflammatory insult) or reflects an induction of endothelial repair processes.
In conclusion, our data reveal a crucial role for the pulmonary endothelial glycocalyx in the regulation of neutrophil adhesion to the endothelial surface. In the presence of circulating danger signals, endothelial cells initiate rapid, heparanase-mediated degradation of the ESL, leading to the neutrophilic alveolitis characteristic of ALI.
Methods
Reagents.
We purchased LPS (Escherichia coli O55:B5), TNF-α, heparinase-III (Flavobacterium heparinum), 150-kDa dextran (FITC-labeled, TRITC-labeled and unlabeled) and BSA from Sigma. We inactivated heparinase-III by heating at 100 °C for 5 min. We purchased heparin from Moore Medical and NAH from Iduron.
Mice.
The Institutional Animal Care and Use Committee of the University of Colorado approved all mouse protocols. We purchased 8- to 12-week-old male C57BL/6 wild-type, TNFR1 knockout (Tnfrsf1atm1Imx), ubiquitin-GFP (Tg(UBC-GFP)30Scha) and ICAM-1 knockout (Icam1Tm1Jcgr) mice from The Jackson Laboratory. J.P. Li (Uppsula University) generously provided C57BL/6 heparanase knockout (Hpse−/−) mice38.
Humans.
We obtained de-identified lung samples with diffuse alveolar damage (that is, ALI) and noninjured controls from the Department of Pathology archives of the University of Colorado. Between June 2010 and March 2011, we collected plasma samples from mechanically ventilated individuals admitted to the Denver Health Medical Intensive Care Unit as part of a study of ventilator-associated pneumonia (ClinicalTrials.gov NCT00938002). We obtained written, informed consent from patients' proxy decision makers before study inclusion. After the individuals regained decision-making capacity, they were told about their participation and re-consented according to institutional policies. Exclusion criteria included therapeutic anticoagulation. C. Silliman (University of Colorado) generously provided plasma from healthy donors, who had provided written informed consent. The Colorado Multiple Institutions Review Board approved all human protocols and waived the requirement for informed consent for the use of archived, de-identified paraffin-embedded lung samples.
Closed-chest pulmonary intravital (in vivo) microscopy.
We performed IVM as previously described39. We administered experimental drugs, LPS or both at thoracic closure (time = 0 min) and performed IVM using a Nikon LV-150 microscope (CFI 75 LWD 16× objective, numerical aperture 0.8). An image splitter (Photometrics) allowed capture of simultaneous reflected-light DIC and FITC or GFP fluorescent images during IVM. We performed confocal imaging using Nikon A1R or LiveScan SFC microscopes with a CFI 75 Apo LWD 25× objective (numerical aperture 1.1).
Measurement of endothelial surface layer thickness.
We determined ESL thickness by 150-kDa FITC-dextran exclusion (200 μl 6% solution injected at time = 0) as previously described40,41 and as illustrated in Figure 1a. We administered heparin and NAH at doses previously shown to inhibit heparanase in mice42,43. Simultaneously captured DIC and FITC images were randomized, and a blinded observer measured the width of vessels in focus (three to five vessels per mouse per time point) by averaging the length of more than three perpendicular intercepts (NIS Elements). We defined ESL thickness as one-half of the difference between DIC and FITC widths.
In vivo microspheres.
We incubated avidin-coated fluorescent 0.97-μm polystyrene microspheres (Bangs Laboratories) with anti–ICAM-1 (clone YN1/1.7.4), anti–VCAM-1 (clone 429) or isotype-matched (IgG2b eB149/10H5 (control for ICAM-1); IgG2a eBR2a (control for VCAM-1)) biotinylated antibodies for 30 min at room temperature (all antibodies from eBioscience, 1:50 dilution). Microspheres were washed and injected (1 × 108 in 100 μl) into the jugular vein at t = 30 min; intravital images were captured at t = 45 min and quantified with Metamorph (Molecular Devices). Mice were killed and lungs were perfused with RNALater (Ambion) and snap-frozen.
GFP+ neutrophil adhesion.
We isolated neutrophils from UBC-GFP mice, as previously described44. We injected 2 × 107 neutrophils (in 100 μl PBS) into the jugular vein at t = 0 min. An adherent neutrophil was defined as being stationary for 15 min. Experiments were performed after confirming the absence of baseline neutrophil adherence during an initial 15 min of imaging.
Microsphere counting.
After completion of intravital microscopy, left (nonimaged) lungs were flushed with PBS, snap-frozen and sectioned (5 μm). We captured ten random low-power fields (10×) and performed microsphere counts using Metamorph.
Cremaster intravital microscopy.
We performed intravital microscopy of the cremasteric microcirculation in 8- to 12-week-old male C57BL/6 wild-type mice, as previously described45. After completion of the surgical preparation, mice were given 200 μl intravenous 6% 150 kDa FITC-dextran, followed by either intravenous saline (200 μl) or LPS (20 μg per g body weight in 200 μl saline). We performed ESL measurements as described above.
Blood neutrophil counts.
We collected blood from anesthetized mice via tail vein puncture. After erythrocytes were lysed with hypertonic ammonium chloride (Sigma), we measured leukocyte counts using a Coulter counter. We identified neutrophils by flow cytometry, as described below.
Cecal ligation and puncture.
We anesthetized mice with isoflurane. After a 1-cm abdominal incision, the cecum was externalized, ligated (50%) with 4:0 silk sutures and punctured twice with a 22-gauge needle. We then internalized the cecum and closed the incision with 4:0 sutures and glue. We resuscitated mice with 1 ml subcutaneous saline and administered buprenorphine for pain. In delayed heparin treatment experiments, mice received 200 μl saline or 5 U heparin in 200 μl saline subcutaneously 24 h after CLP. In select experiments, we maintained mice at 60% FiO2 in a hyperoxia chamber. At 48 h, mice were killed for lung isolation.
Lung digestion.
Forty-five minutes after intravenous saline (200 μl) or LPS (20 mg per kg body weight in 200 μl saline) administration, mice were killed and lungs harvested. Lungs were not perfusion fixed, ensuring loss of the endothelial glycocalyx (as previously described12), and thereby avoiding potential glycocalyx interference with binding of fluorescent antibodies to the cell surface (for purposes of flow cytometry). After flushing of the pulmonary vasculature with PBS, we minced the lung with a razor and performed digestion for 30 min at 37° using a protease cocktail (1 mg/ml Liberase, Roche), additionally assuring glycocalyx loss via protease digestion46. After 30 min, we added 100 μl of 100 mM EDTA and 1 ml of cold RPMI medium (Sigma) and pipetted the digestate 50 times. We filtered the resultant single-cell suspension through 100-μm mesh, followed by three rounds of washing.
Neutrophil depletion.
R.L. Coffman (DNAX Research Institute) created and generously donated an anti–Gr-1 monoclonal antibody (RB6-8C5) hybridoma47. We purified antibody from the hybridoma culture supernatant using Protein G affinity chromatography, followed by dialysis against PBS and sterilization via 0.2-μm filtration. We measured rat IgG2b concentration by ELISA (SouthernBiotech) according to the manufacturer's instructions. We depleted neutrophils in mice by administering anti-Gr1 antibody (125 μg) intraperitoneally 48 h and 24 h before intravenous microscopy experiments. We injected control mice with 125 μg rat IgG2b isotype antibody (eBioscience, eB149/10H5) at 48 h and 24 h before intravital microscopy. We confirmed neutrophil depletion immediately after completion of intravital microscopy using peripheral blood smears and flow cytometry, as described below. Antibodies for flow cytometry are described below.
Isolated, perfused mouse lung.
We performed lung isolation and perfusion as previously described48. We calculated the filtration coefficient (Kf) as an index of endothelial permeability48. Alternatively, we perfusion-fixed isolated lungs for 2 min with 2% formalin, a technique designed to minimize glycocalyx loss12.
Quantification of neutrophil extravasation.
After measurement of Kf, we flushed and snap-froze isolated lungs for later homogenization. We calculated myeloperoxidase activity using a fluorometric assay (Enzo).
Cell culture.
We isolated mouse lung microvascular endothelial cells and grew them to confluence as previously described49.
Heparan sulfate degradation activity.
We measured heparan sulfate degradation activity in freshly lysed cells or plasma according to the manufacturer's instructions (GenWay).
Protein and mRNA expression.
MLMVECs or harvested lungs were homogenized for western blotting or RNA extraction (RNeasy, Qiagen), as previously described50. For western blotting, we probed membranes with rabbit antibody to human heparanase (1:1,000, Ins-26-2, ProSpec), goat antibody to mouse ICAM-1 (1:200, clone M-19, Santa Cruz), rabbit antibody to mouse VCAM-1 (1:200, clone H-276, Santa Cruz) or rabbit antibody to human β-actin (1:10,000, 4967, Cell Signaling). We performed quantitative PCR using ICAM-1 (ref. 51) and cyclophilin A (Applied Biosystems) primers, carrying out reverse transcription reactions using a SuperScript III First-Strand Synthesis System for RT-PCR kit (Invitrogen). PCR conditions included initial denaturation at 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 15 s, annealing at 60 °C for 1 min. We calculated relative abundance of mRNA expression in each sample as (ref. 52).
Immunofluorescence.
We performed immunofluorescence on 5-μm frozen or 4-μm paraffin-embedded sections from perfusion-fixed isolated lungs or agarose-inflated lungs from intact mice, as previously described50. Primary antibodies included mouse monoclonal antibody to heparan sulfate (1:50, HepSS-1, USBiological), mouse antibody to human CD31 (1:200, clone JC70A, Dako), rabbit polyclonal antibody to human heparanase (1:1,000, Ins-26-2, ProSpec), goat antibody to mouse thrombomodulin (1:1,000, AF3894, R&D Systems), rat antibody to mouse Ly-6B.2 (1:300, clone MCA771G, AbD Serotec) and biotinylated hyaluronic acid–binding protein (2 μg ml−1, Seikagaku). Isotype antibodies served as negative controls: mouse IgMκ, MM-30, 1:50, BioLegend (control for HepSS-1); mouse IgG1κ, X0931, 1:200, Dako (control for CD31); rabbit IgG, 27472, 1:1,000, Abcam (control for heparanase); goat IgG, AB-108-C, 1:1,000, R&D Systems (control for thrombomodulin); rat IgG2a, MCA1212, 1:300, AbD Serotec (control for Ly-6B.2). We performed image quantification using Metamorph as previously described53.
Flow cytometry.
Specimens were washed twice in PBS, incubated with Fc block (BD Biosciences) for 20 min on ice and fixed with 1% paraformaldehyde. We incubated neutrophils with preconjugated monoclonal antibodies (all from BD Biosciences) to CD11b (1:200, clone M1/70), Ly6G (1:100, clone 1A8), Gr1 (1:100, clone RB6-8C5), ICAM-1 (1:100, clone 3E2) and VCAM-1 (1:100, clone 429 MVCAM.A) or matched isotype controls (rat IgG2b, A95-1, 1:200 (control for CD11b) or 1:100 (control for Gr1); rat 1gG2a, R35-95, 1:100 (control for Ly6G and VCAM-1); hamster IgG1κ, A-19-3, 1:100 (control for ICAM-1) for 45 min and then washed three times. We identified neutrophils on the basis of forward and side scatter and staining for CD11b and Ly6G. We incubated lung digestates with antibodies to CD45 (1:50, clone 30-F11, eBioscience), CD31 (1:20, clone MEC7.46, Abcam), CD141 (1:50, AF3894, R&D Systems), ICAM-1 (1:50) and VCAM-1 (1:50) or matched isotype controls: rat IgG2a (1:200 (matching concentration used for CD31), 18540, Abcam); goat IgG (1:250 (matching concentration used for CD141), AB-108-C, R&D Systems); VCAM-1 and ICAM-1 controls as above (1:50). No isotype control was used for CD45, as staining was used for negative selection. We identified endothelial cells as cells negative for CD45 and positive for both CD31 and CD141. We performed cytometry on a LSRII (Becton Dickinson) and analysis using FlowJo software (Treestar).
Nuclear factor-κB promoter assay.
We grew 1 × 104 mouse lung epithelial cells (MLE-15 cells, kindly provided by J. Whitsett, Children's Hospital Cincinnati54) to 70–90% confluence and transfected them with a pHTS nuclear factor-κB reporter vector (kindly provided by S. Biswal, Johns Hopkins University50) encoding the firefly luciferase gene. We cotransfected cells with a pRL-TK vector (S. Biswal50) encoding Renilla luciferase to control for efficiency of transfection. We performed all transfections using Lipofectamine 2000 (Invitrogen). Forty-eight hours after transfection, we treated cells for 4 h with saline, LPS (1 μg ml−1) or LPS (1 μg ml−1) with heparin (0.4 U ml−1). We lysed cells in 200 μl lysis buffer (Promega) and performed a luciferase assay (Dual-Glo, Promega) as previously described50. We used the same plate to determine Renilla luciferase activity; data represent the ratio of firefly to Renilla luciferase activities.
Statistical analyses.
Data are represented as means ± s.e.m. We performed multiple comparisons by analysis of variance with Dunnet's post-hoc testing. We used two-way analysis of variance to compare group differences in ESL thickness over time. We used Student's two-tailed t-test when comparing two groups. We analyzed survival data by log-rank. Differences were statistically significant if P < 0.05. We performed all calculations using Prism (GraphPad).
References
Cohen, J. The immunopathogenesis of sepsis. Nature 420, 885–891 (2002).
Hudson, L.D., Milberg, J.A., Anardi, D. & Maunder, R.J. Clinical risks for development of the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 151, 293–301 (1995).
Iscimen, R. et al. Risk factors for the development of acute lung injury in patients with septic shock: an observational cohort study. Crit. Care Med. 36, 1518–1522 (2008).
Herridge, M.S. et al. Functional disability 5 years after acute respiratory distress syndrome. N. Engl. J. Med. 364, 1293–1304 (2011).
Weinbaum, S., Tarbell, J.M. & Damiano, E.R. The structure and function of the endothelial glycocalyx layer. Annu. Rev. Biomed. Eng. 9, 121–167 (2007).
Pries, A.R. & Kuebler, W.M. Normal endothelium. Handb. Exp. Pharmacol. 1–40 (2006).
Jacob, M. et al. Albumin augmentation improves condition of guinea pig hearts after 4 hr of cold ischemia. Transplantation 87, 956–965 (2009).
Chappell, D. et al. Glycocalyx protection reduces leukocyte adhesion after ischemia/reperfusion. Shock 34, 133–139 (2010).
Constantinescu, A.A., Vink, H. & Spaan, J.A. Endothelial cell glycocalyx modulates immobilization of leukocytes at the endothelial surface. Arterioscler. Thromb. Vasc. Biol. 23, 1541–1547 (2003).
Mulivor, A.W. & Lipowsky, H.H. Role of glycocalyx in leukocyte-endothelial cell adhesion. Am. J. Physiol. Heart Circ. Physiol. 283, H1282–H1291 (2002).
Steppan, J. et al. Sepsis and major abdominal surgery lead to flaking of the endothelial glycocalyx. J. Surg. Res. 165, 136–141 (2011).
Chappell, D. et al. The glycocalyx of the human umbilical vein endothelial cell: an impressive structure ex vivo but not in culture. Circ. Res. 104, 1313–1317 (2009).
Potter, D.R. & Damiano, E.R. The hydrodynamically relevant endothelial cell glycocalyx observed in vivo is absent in vitro. Circ. Res. 102, 770–776 (2008).
Evans, T.J. et al. Protective effect of 55- but not 75-kD soluble tumor necrosis factor receptor–immunoglobulin G fusion proteins in an animal model of Gram-negative sepsis. J. Exp. Med. 180, 2173–2179 (1994).
Chappell, D. et al. Heparinase selectively sheds heparan sulphate from the endothelial glycocalyx. Biol. Chem. 389, 79–82 (2008).
Vlodavsky, I., Ilan, N., Naggi, A. & Casu, B. Heparanase: structure, biological functions and inhibition by heparin-derived mimetics of heparan sulfate. Curr. Pharm. Des. 13, 2057–2073 (2007).
Doerschuk, C.M., Mizgerd, J.P., Kubo, H., Qin, L. & Kumasaka, T. Adhesion molecules and cellular biomechanical changes in acute lung injury*. Chest 116, 37S–43S (1999).
Phillipson, M. & Kubes, P. The neutrophil in vascular inflammation. Nat. Med. 17, 1381–1390 (2011).
Lever, R., Hoult, J.R. & Page, C.P. The effects of heparin and related molecules upon the adhesion of human polymorphonuclear leucocytes to vascular endothelium in vitro. Br. J. Pharmacol. 129, 533–540 (2000).
Matute-Bello, G., Frevert, C.W. & Martin, T.R. Animal models of acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 295, L379–L399 (2008).
Witzenbichler, B., Westermann, D., Knueppel, S., Schultheiss, H.P. & Tschope, C. Protective role of angiopoietin-1 in endotoxic shock. Circulation 111, 97–105 (2005).
Alvira, C.M., Abate, A., Yang, G., Dennery, P.A. & Rabinovitch, M. Nuclear factor-κB Activation in neonatal mouse lung protects against lipopolysaccharide-induced inflammation. Am. J. Respir. Crit. Care Med. 175, 805–815 (2007).
Berger, G. et al. Sepsis impairs alveolar epithelial function by downregulating Na-K-ATPase pump. Am. J. Physiol. Lung Cell. Mol. Physiol. 301, L23–L30 (2011).
Aggarwal, N.R. et al. Moderate oxygen augments lipopolysaccharide-induced lung injury in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 298, L371–L381 (2010).
Javaid, K. et al. Tumor necrosis factor-α induces early-onset endothelial adhesivity by protein kinase Cζ–dependent activation of intercellular adhesion molecule-1. Circ. Res. 92, 1089–1097 (2003).
Reitsma, S., Slaaf, W., Vink, H., van Zandvoort, M.A. & oude Egbrink, M.G. The endothelial glycocalyx: composition, functions and visualization. Pflugers Arch. 454, 345–359 (2007).
Smith, M.L., Long, D.S., Damiano, E.R. & Ley, K. Near-wall micro-PIV reveals a hydrodynamically relevant endothelial surface layer in venules in vivo. Biophys. J. 85, 637–645 (2003).
Stevens, A.P., Hlady, V. & Dull, R.O. Fluorescence correlation spectroscopy can probe albumin dynamics inside lung endothelial glycocalyx. Am. J. Physiol. Lung Cell. Mol. Physiol. 293, L328–L335 (2007).
King, J. et al. Structural and functional characteristics of lung macro- and microvascular endothelial cell phenotypes. Microvasc. Res. 67, 139–151 (2004).
Vlodavsky, I. et al. Heparanase, heparin and the coagulation system in cancer progression. Thromb. Res. 120 (suppl. 2), S112–S120 (2007).
Lerner, I. et al. Heparanase powers a chronic inflammatory circuit that promotes colitis-associated tumorigenesis in mice. J. Clin. Invest. 121, 1709–1721 (2011).
Rao, G. et al. Reactive oxygen species mediate high glucose–induced heparanase-1 production and heparan sulphate proteoglycan degradation in human and rat endothelial cells: a potential role in the pathogenesis of atherosclerosis. Diabetologia 54, 1527–1538 (2011).
Bashenko, Y., Ilan, N., Krausz, M.M., Vlodavsky, I. & Hirsh, M.I. Heparanase pretreatment attenuates endotoxin-induced acute lung injury in rats. Shock 28, 207–212 (2007).
Fux, L., Ilan, N., Sanderson, R.D. & Vlodavsky, I. Heparanase: busy at the cell surface. Trends Biochem. Sci. 34, 511–519 (2009).
Wang, L., Fuster, M., Sriramarao, P. & Esko, J.D. Endothelial heparan sulfate deficiency impairs L-selectin– and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat. Immunol. 6, 902–910 (2005).
Massena, S. et al. A chemotactic gradient sequestered on endothelial heparan sulfate induces directional intraluminal crawling of neutrophils. Blood 116, 1924–1931 (2010).
Potter, D.R., Jiang, J. & Damiano, E.R. The recovery time course of the endothelial cell glycocalyx in vivo and its implications in vitro. Circ. Res. 104, 1318–1325 (2009).
Zcharia, E. et al. Newly generated heparanase knock-out mice unravel co-regulation of heparanase and matrix metalloproteinases. Plos One 4, 1–13 (2009).
Tabuchi, A., Mertens, M., Kuppe, H., Pries, A.R. & Kuebler, W.M. Intravital microscopy of the murine pulmonary microcirculation. J. Appl. Physiol. 104, 338–346 (2008).
Vink, H. & Duling, B.R. Identification of distinct luminal domains for macromolecules, erythrocytes, and leukocytes within mammalian capillaries. Circ. Res. 79, 581–589 (1996).
Marechal, X. et al. Endothelial glycocalyx damage during endotoxemia coincides with microcirculatory dysfunction and vascular oxidative stress. Shock 29, 572–576 (2008).
Hostettler, N. et al. P-selectin– and heparanase-dependent antimetastatic activity of non-anticoagulant heparins. FASEB J. 21, 3562–3572 (2007).
Takahashi, H. et al. A comparison of the effects of unfractionated heparin, dalteparin and danaparoid on vascular endothelial growth factor-induced tumour angiogenesis and heparanase activity. Br. J. Pharmacol. 146, 333–343 (2005).
Nick, J.A. et al. Role of p38 mitogen-activated protein kinase in a murine model of pulmonary inflammation. J. Immunol. 164, 2151–2159 (2000).
Bagher, P. & Segal, S.S. The mouse cremaster muscle preparation for intravital imaging of the microcirculation. J. Vis. Exp. e2874 (2011).
Adamson, R.H. Permeability of frog mesenteric capillaries after partial pronase digestion of the endothelial glycocalyx. J. Physiol. (Lond.) 428, 1–13 (1990).
Tepper, R.I., Coffman, R.L. & Leder, P. An eosinophil-dependent mechanism for the antitumour effect of interleukin-4. Science. 257, 548–551 (1992).
Schmidt, E.P. et al. Soluble guanylyl cyclase contributes to ventilator-induced lung injury in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 295, L1056–L1065 (2008).
Stephens, R.S. et al. cGMP increases antioxidant function and attenuates oxidant cell death in mouse lung microvascular endothelial cells by a protein kinase G–dependent mechanism. Am. J. Physiol. Lung Cell. Mol. Physiol. 299, L323–L333 (2010).
Yoshida, T. et al. Rtp801, a suppressor of mTOR signaling, is an essential mediator of cigarette smoke–induced pulmonary injury and emphysema. Nat. Med. 16, 767–773 (2010).
Wu, X., Guo, R., Chen, P., Wang, Q. & Cunningham, P.N. TNF induces caspase-dependent inflammation in renal endothelial cells through a Rho- and myosin light chain kinase-dependent mechanism. Am. J. Physiol. Renal Physiol. 297, F316–F326 (2009).
Yang, Y., Cui, Y., Fan, Z., Cook, G.A. & Nishimura, H. Two distinct aquaporin-4 cDNAs isolated from medullary cone of quail kidney. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 147, 84–93 (2007).
Petrache, I. et al. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat. Med. 11, 491–498 (2005).
Wikenheiser, K.A. et al. Production of immortalized distal respiratory epithelial cell lines from surfactant protein C/simian virus 40 large tumor antigen transgenic mice. Proc. Natl. Acad. Sci. 90, 11029–11033 (1993).
Acknowledgements
We thank J.P. Li (Uppsula University) for providing Hpse−/− mice, C. Silliman (University of Colorado) for providing human donor plasma samples and R. Coffman (DNAX Research Institute) for donation of a Gr-1–specific monoclonal antibody (RB6-8C5) hybridoma. MLE-15 cells were kindly provided by J. Whitsett (Children's Hospital Cincinnati). pHTS nuclear factor-κB reporter vector and pRL-TK vector were kindly provided by S. Biswal (Johns Hopkins University). We additionally thank W. Kuebler and A. Tabuchi for instruction on mouse lung IVM techniques, K. Queensland for instruction regarding cecal ligation and puncture, P. Henson and G. Downey for manuscript review and the Tuder lab for support and discussions of experimental design. This work was supported by the US National Institutes of Health (NIH) /National Heart, Lung and Blood Institute (NHLBI) grants P30 HL101295 (to E.P.S. and R.M.T.); K08 HL105538 (to E.P.S.); Chronic Obstructive Pulmonary Disease Specialized Centers of Clinically Oriented Research P01–NHLBI 085609 (to R.M.T.); and R01 ES016285 (to R.M.T.). Additional funding was provided by the Flight Attendant Medical Research Institute (to R.M.T.) and the Colorado Clinical and Translational Sciences Institute UL1 RR025780 (to E.P.S.).
Author information
Authors and Affiliations
Contributions
E.P.S. designed all experiments, performed isolated lung experiments, analyzed the data and composed the manuscript. Y.Y. performed IVM, CLP, immunofluorescence and in vitro experiments. W.J.J. analyzed flow cytometry data and circulating neutrophil counts, supervised neutrophil isolation experiments and assisted in manuscript preparation. A.G. performed mouse and human tissue immunofluorescence. M.J.P. and L.P.S. assisted with transgenic mouse breeding and performed lung inflation and fixation. L.B. performed neutrophil isolation experiments, neutrophil flow cytometry and circulating neutrophil counts. R.L.Z. performed neutrophil depletion experiments. J.C.B. performed nuclear factor-κB reporter experiments. D.E.K. performed tissue processing and sectioning. Z.X.Y. performed lung digestion and endothelial flow cytometry experiments. S.S.C. assisted with CLP surgeries. K.H.O., K.R.T. and I.S.D. obtained plasma samples from individuals with respiratory failure. M.W.G. provided support for intravital microscopy. D.B.P. provided support for pilot studies and assisted in manuscript preparation. R.M.T. supervised the overall execution of the project, performed histologic and morphometric analyses and assisted in manuscript preparation.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–12 and Supplementary Tables 1 and 2 (PDF 3015 kb)
Rights and permissions
About this article
Cite this article
Schmidt, E., Yang, Y., Janssen, W. et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med 18, 1217–1223 (2012). https://doi.org/10.1038/nm.2843
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nm.2843
This article is cited by
-
Effect of 5% albumin on endothelial glycocalyx degradation during off-pump coronary artery bypass
Canadian Journal of Anesthesia/Journal canadien d'anesthésie (2024)
-
Bone mesenchymal stem cell extracellular vesicles delivered miR let-7-5p alleviate endothelial glycocalyx degradation and leakage via targeting ABL2
Cell Communication and Signaling (2023)
-
GPR109A controls neutrophil extracellular traps formation and improve early sepsis by regulating ROS/PAD4/Cit-H3 signal axis
Experimental Hematology & Oncology (2023)
-
Capillary leak and endothelial permeability in critically ill patients: a current overview
Intensive Care Medicine Experimental (2023)
-
Chemical mimetics of the N-degron pathway alleviate systemic inflammation by activating mitophagy and immunometabolic remodeling
Experimental & Molecular Medicine (2023)
