
Abstract
Aire is a transcription factor that controls T cell tolerance by inducing the expression of a large repertoire of genes specifically in thymic stromal cells. It interacts with scores of protein partners of diverse functional classes. We found that Aire and some of its partners, notably those implicated in the DNA-damage response, preferentially localized to and activated long chromatin stretches that were overloaded with transcriptional regulators, known as super-enhancers. We also identified topoisomerase 1 as a cardinal Aire partner that colocalized on super-enhancers and was required for the interaction of Aire with all of its other associates. We propose a model that entails looping of super-enhancers to efficiently deliver Aire-containing complexes to local and distal transcriptional start sites.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others

Accession codes
Primary accessions
Gene Expression Omnibus
Sequence Read Archive
References
Klein, L., Kyewski, B., Allen, P.M. & Hogquist, K.A. Positive and negative selection of the T cell repertoire: what thymocytes see (and don't see). Nat. Rev. Immunol. 14, 377–391 (2014).
Sansom, S.N. et al. Population and single-cell genomics reveal the Aire dependency, relief from Polycomb silencing, and distribution of self-antigen expression in thymic epithelia. Genome Res. 24, 1918–1931 (2014).
Meredith, M., Zemmour, D., Mathis, D. & Benoist, C. Aire controls gene expression in the thymic epithelium with ordered stochasticity. Nat. Immunol. 16, 942–949 (2015).
Brennecke, P. et al. Single-cell transcriptome analysis reveals coordinated ectopic gene-expression patterns in medullary thymic epithelial cells. Nat. Immunol. 16, 933–941 (2015).
Anderson, M.S. et al. Projection of an immunological self shadow within the thymus by the aire protein. Science 298, 1395–1401 (2002).
Peterson, P., Org, T. & Rebane, A. Transcriptional regulation by AIRE: molecular mechanisms of central tolerance. Nat. Rev. Immunol. 8, 948–957 (2008).
Mathis, D. & Benoist, C. Aire. Annu. Rev. Immunol. 27, 287–312 (2009).
Guerau-de-Arellano, M., Mathis, D. & Benoist, C. Transcriptional impact of Aire varies with cell type. Proc. Natl. Acad. Sci. USA 105, 14011–14016 (2008).
Derbinski, J., Pinto, S., Rösch, S., Hexel, K. & Kyewski, B. Promiscuous gene expression patterns in single medullary thymic epithelial cells argue for a stochastic mechanism. Proc. Natl. Acad. Sci. USA 105, 657–662 (2008).
Villaseñor, J., Besse, W., Benoist, C. & Mathis, D. Ectopic expression of peripheral-tissue antigens in the thymic epithelium: probabilistic, monoallelic, misinitiated. Proc. Natl. Acad. Sci. USA 105, 15854–15859 (2008).
Koh, A.S. et al. Aire employs a histone-binding module to mediate immunological tolerance, linking chromatin regulation with organ-specific autoimmunity. Proc. Natl. Acad. Sci. USA 105, 15878–15883 (2008).
Org, T. et al. The autoimmune regulator PHD finger binds to non-methylated histone H3K4 to activate gene expression. EMBO Rep. 9, 370–376 (2008).
Oven, I. et al. AIRE recruits P-TEFb for transcriptional elongation of target genes in medullary thymic epithelial cells. Mol. Cell. Biol. 27, 8815–8823 (2007).
Giraud, M. et al. Aire unleashes stalled RNA polymerase to induce ectopic gene expression in thymic epithelial cells. Proc. Natl. Acad. Sci. USA 109, 535–540 (2012).
Abramson, J., Giraud, M., Benoist, C. & Mathis, D. Aire's partners in the molecular control of immunological tolerance. Cell 140, 123–135 (2010).
Giraud, M. et al. An RNAi screen for Aire cofactors reveals a role for Hnrnpl in polymerase release and Aire-activated ectopic transcription. Proc. Natl. Acad. Sci. USA 111, 1491–1496 (2014).
Blecher-Gonen, R. et al. High-throughput chromatin immunoprecipitation for genome-wide mapping of in vivo protein-DNA interactions and epigenomic states. Nat. Protoc. 8, 539–554 (2013).
Org, T. et al. AIRE activated tissue specific genes have histone modifications associated with inactive chromatin. Hum. Mol. Genet. 18, 4699–4710 (2009).
Hnisz, D. et al. Super-enhancers in the control of cell identity and disease. Cell 155, 934–947 (2013).
Witte, S., O'Shea, J.J. & Vahedi, G. Super-enhancers: asset management in immune cell genomes. Trends Immunol. 36, 519–526 (2015).
Whyte, W.A. et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013).
LaFlam, T.N. et al. Identification of a novel cis-regulatory element essential for immune tolerance. J. Exp. Med. 212, 1993–2002 (2015).
Buenrostro, J.D., Giresi, P.G., Zaba, L.C., Chang, H.Y. & Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013).
Liu, W. et al. Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell 155, 1581–1595 (2013).
Halonen, M. et al. APECED-causing mutations in AIRE reveal the functional domains of the protein. Hum. Mutat. 23, 245–257 (2004).
Yoshida, H. et al. Brd4 bridges the transcriptional regulators, Aire and P-TEFb, to promote elongation of peripheral-tissue antigen transcripts in thymic stromal cells. Proc. Natl. Acad. Sci. USA 112, E4448–E4457 (2015).
Saunders, A., Core, L.J. & Lis, J.T. Breaking barriers to transcription elongation. Nat. Rev. Mol. Cell Biol. 7, 557–567 (2006).
King, I.F. et al. Topoisomerases facilitate transcription of long genes linked to autism. Nature 501, 58–62 (2013).
Polo, S.E. & Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 25, 409–433 (2011).
Lai, J.S. & Herr, W. Ethidium bromide provides a simple tool for identifying genuine DNA-independent protein associations. Proc. Natl. Acad. Sci. USA 89, 6958–6962 (1992).
Taniguchi, R.T. et al. Detection of an autoreactive T-cell population within the polyclonal repertoire that undergoes distinct autoimmune regulator (Aire)-mediated selection. Proc. Natl. Acad. Sci. USA 109, 7847–7852 (2012).
Jiang, W., Anderson, M.S., Bronson, R., Mathis, D. & Benoist, C. Modifier loci condition autoimmunity provoked by Aire deficiency. J. Exp. Med. 202, 805–815 (2005).
Dowen, J.M. et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 159, 374–387 (2014).
Björses, P. et al. Localization of the APECED protein in distinct nuclear structures. Hum. Mol. Genet. 8, 259–266 (1999).
Saare, M., Rebane, A., Rajashekar, B., Vilo, J. & Peterson, P. Autoimmune regulator is acetylated by transcription coactivator CBP/p300. Exp. Cell Res. 318, 1767–1778 (2012).
Chapuy, B. et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 24, 777–790 (2013).
Lovén, J. et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334 (2013).
Ashour, M.E., Atteya, R. & El-Khamisy, S.F. Topoisomerase-mediated chromosomal break repair: an emerging player in many games. Nat. Rev. Cancer 15, 137–151 (2015).
Baranello, L., Kouzine, F. & Levens, D. DNA topoisomerases beyond the standard role. Transcription 4, 232–237 (2013).
Strumberg, D. et al. Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5′-phosphorylated DNA double-strand breaks by replication runoff. Mol. Cell. Biol. 20, 3977–3987 (2000).
Sordet, O. et al. Ataxia telangiectasia mutated activation by transcription- and topoisomerase I-induced DNA double-strand breaks. EMBO Rep. 10, 887–893 (2009).
Trotter, K.W., King, H.A. & Archer, T.K. Glucocorticoid receptor transcriptional activation via the BRG1-dependent recruitment of TOP2beta and Ku70/86. Mol. Cell. Biol. 35, 2799–2817 (2015).
Husain, A. et al. Chromatin remodeller SMARCA4 recruits topoisomerase 1 and suppresses transcription-associated genomic instability. Nat. Commun. 7, 10549 (2016).
Puc, J. et al. Ligand-dependent enhancer activation regulated by topoisomerase-I activity. Cell 160, 367–380 (2015).
Baranello, L. et al. RNA polymerase II regulates topoisomerase 1 activity to favor efficient transcription. Cell 165, 357–371 (2016).
Madabhushi, R. et al. Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell 161, 1592–1605 (2015).
Teves, S.S. & Henikoff, S. Transcription-generated torsional stress destabilizes nucleosomes. Nat. Struct. Mol. Biol. 21, 88–94 (2014).
Bunch, H. et al. Transcriptional elongation requires DNA break-induced signalling. Nat. Commun. 6, 10191 (2015).
Heo, K. et al. FACT-mediated exchange of histone variant H2AX regulated by phosphorylation of H2AX and ADP-ribosylation of Spt16. Mol. Cell 30, 86–97 (2008).
Garber, M. et al. A high-throughput chromatin immunoprecipitation approach reveals principles of dynamic gene regulation in mammals. Mol. Cell 47, 810–822 (2012).
Brind'Amour, J. et al. An ultra-low-input native ChIP-seq protocol for genome-wide profiling of rare cell populations. Nat. Commun. 6, 6033 (2015).
Langmead, B. & Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Robinson, J.T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Ramírez, F., Dündar, F., Diehl, S., Grüning, B.A. & Manke, T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–91 (2014).
Shen, L., Shao, N., Liu, X. & Nestler, E. ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics 15, 284 (2014).
Lara-Astiaso, D. et al. Immunogenetics. Chromatin state dynamics during blood formation. Science 345, 943–949 (2014).
Yang, S., Bansal, K., Lopes, J., Benoist, C. & Mathis, D. Aire's plant homeodomain(PHD)-2 is critical for induction of immunological tolerance. Proc. Natl. Acad. Sci. USA 110, 1833–1838 (2013).
Acknowledgements
We thank G. Buruzula, K. Rothamel, A. Rhoads, K. Hattori, A. Lopez, G. Gopalan, K. Waraska, M. Thorsen for experimental assistance and C. Laplace for help with manuscript preparation. The NIH Tetramer Core Facility (contract HHSN272201300006C) kindly provided tetramers. This work was supported by NIH grants R01 DK060027 and R01 AI088204. K.B. was supported by American Diabetes Association Mentor-Based Postdoctoral Fellowship #7-12-MN-51 to D.M.
Author information
Authors and Affiliations
Contributions
K.B. and H.Y. performed the experiments. K.B., C.B. and D.M. designed the study, analyzed and interpreted the data, and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Consistency of Aire ChIP-seq in mTECs.
a) Venn diagram displaying number of overlapping Aire peaks identified in two independent experiments. b) Exemplar genome browser views of two independent Aire ChIP-seq experiments in B6.Aire+/+ mTEChi.
Supplementary Figure 2 The mTEC super-enhancer repertoire.
a) Histogram depicting size density of super-enhancers (SEs) across mTEC genome. Dotted yellow line depicts the median length for all super-enhancers. Dotted black line shows a smoothed curve for super-enhancer size density. b) Normalized ChIP-seq profiles at exemplar super-enhancers in B6.Aire+/+ mTEChi. Numbers to the right indicate the ranges of normalized tag densities. SE, super-enhancer. Data are representative of two independent experiments. Data for one of the H3K27ac ChIP-seq experiments in (a, b) came from (ref.22).
Supplementary Figure 3 Super-enhancer repertoire of HEK293T cells.
a) Hockey plot for delineation of super-enhancers in HEK293T cells with H3K27ac ChIP-seq experiment reported in (ref. 24) SE, super-enhancer; CE, conventional enhancer. b) Heatmaps of tag density for the indicated proteins 500kb up- or down-stream of H3K27ac-delimited super-enhancers in HEK293T cells, H3K27ac and H3K4me1 data came from experiments reported in (ref. 24) while Aire, RNA-PolII and IgG data were reported in (ref. 14).
Supplementary Figure 4 shRNA knockdown of Aire-partners to reveal the sequence of Aire-partner associations in HEK293T cells.
Primary data supporting Fig. 3e. IP, immunoprecipitation. Representative immunoblots from two independent experiments.
Supplementary Figure 6 Differential distribution of TOP1 and TOP2A at super-enhancers in mTECs.
a, b) Normalized ChIP-seq profiles of indicated proteins at exemplar super-enhancers (a) or non-super-enhancer regions (b) in B6.Aire+/+ mTEChi to emphasize the lack of enrichment of TOP2A at super-enhancers. Numbers to the right indicate the ranges of normalized tag densities. SE, super-enhancer. Data are representative of two independent experiments. Data for one of the H3K27ac ChIP-seq experiments in (a, b) came from (ref. 22).
Supplementary Figure 7 Lack of thymic stromal cell and thymocyte perturbations after inhibition of topoisomerases.
a, b) Representative cytofluorimetric plots and summary quantitative data for mTEChi (a) and various thymocyte compartments (b) of B6.Aire+/+ mice treated with the indicated topoisomerase inhibitor/s or just vehicle (DMSO) every day for 3 days. Data are representative of three independent experiments with similar results (error bars, mean ± s.e.m. from n=3 measurements pooled from three experiments).
Supplementary Figure 8 Effect of topoisomerase inhibitors on mTEC gene expression and autoimmunity.
a, b) Representative cytofluorimetric plots gated on CD45-Ly51loMHCIIhi mTECs (b) and summary quantitative data (a, b) for mTEC expression of Aire (a) or the topoisomerases (b) in B6.Aire+/+ mice treated with the indicated topoisomerase inhibitor/s or just vehicle (DMSO) every day for 3 days. MFI, mean fluorescence intensity. c) As per Fig. 7a, except total RNA was isolated from mTECs after three weeks of treatment of mice with topoisomerase inhibitors. Gene-expression analyses were performed by quantitative PCR using SYBR Green. d) As per Fig. 6a, except 4-week-old B6.Aire-/- mice were intra-peritoneally injected with topotecan (n=3), etoposide (n=2), both drugs (n=2) or just vehicle (DMSO) (n=3). Orange: transcripts increased >2-fold in vehicle-treated Aire+/+ vs Aire-/- mice; green: transcripts decreased >2-fold. Numbers refer to transcripts up- (right) or down- (left) regulated by the indicated inhibitors (etoposide, P = 1.6 x 10-45; topotecan, P = 4 x 10-58; etoposide + topotecan, P = 9 x 10-101, P values for Aire-induced genes from χ2 test). e) Organ histology scores at 8-12 weeks of age for NOD/LtJ pups (Aire-/-) intra-peritoneally injected with topotecan, etoposide or just vehicle on the 2nd, 4th and 8th day after birth (6-7 mice per group). Scores reflect the scale described in Methods. *P < 0.05 (unpaired Student’s t-test). Data are representative of at least two (d, e) or three independent experiments (a-c) (error bars, mean ± s.e.m. from n = 3 (a-c) or n ≥ 6 (e) measurements).
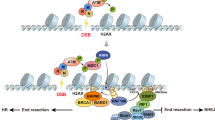
Supplementary Figure 9 A simplified model of Aire’s mechanism of action.
A detailed scenario is presented in the Discussion section of the text. Not all Aire partners are depicted here, just those highlighted in the text.
1) At the super-enhancer, Aire and TOP1 interact, resulting in stabilization of DNA DSBs, mobilization of the DNA-damage response (e.g. γH2AX, DNA-PKcs, Ku80, PARP-1) and recruitment of general transcription factors (e.g. CBP, BRD4). 2) At the TSS or just downstream of it, additional DSBs occur due to contortion induced by paused RNA-PolII. The super-enhancer delivers its abundant Aire-containing complexes to the TSS, which overcomes pausing and promotes elongation. 3) Effective elongation necessitates relief of torsional stress up- and down-stream of the advancing RNA-PolII as well as eviction of interfering nucleosomes, likely performed by the “histone eviction” complex (including TOP2, DNA-PKcs, Ku80, PARP-1. etc). 4) An as-yet poorly defined Aire-containing complex promotes pre-mRNA processing. Ac = an acetylated residue; me1 = addition of a single methyl group; me3 = a trimethylated site; P = a phosphorylation site; TSS = transcriptional start-site; pTEFb is depicted as having two subunits, CycT1 and CDK9; bold red stretch = super-enhancer; bold black stretch = TSS; bold green = emerging pre-mRNA transcript.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–9 (PDF 2368 kb)
Supplementary Table 1
Efficiency and robustness of various ChIP-seq experiments (PDF 344 kb)
Rights and permissions
About this article

Cite this article
Bansal, K., Yoshida, H., Benoist, C. et al. The transcriptional regulator Aire binds to and activates super-enhancers. Nat Immunol 18, 263–273 (2017). https://doi.org/10.1038/ni.3675
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ni.3675
This article is cited by
-
Tipping the balance in autoimmunity: are regulatory t cells the cause, the cure, or both?
Molecular and Cellular Pediatrics (2024)
-
Z-DNA marks the spot for AIRE
Cell Research (2024)
-
AIRE relies on Z-DNA to flag gene targets for thymic T cell tolerization
Nature (2024)
-
Insm1 regulates mTEC development and immune tolerance
Cellular & Molecular Immunology (2023)
-
BRD9 determines the cell fate of hematopoietic stem cells by regulating chromatin state
Nature Communications (2023)
