
Abstract
The molecular mechanisms by which different mutations in the same gene can result in distinct disease phenotypes remain largely unknown. Truncating mutations of SOX10 cause either a complex neurocristopathy designated PCWH or a more restricted phenotype known as Waardenburg-Shah syndrome (WS4; OMIM 277580). Here we report that although all nonsense and frameshift mutations that cause premature termination of translation generate truncated SOX10 proteins with potent dominant-negative activity, the more severe disease phenotype, PCWH, is realized only when the mutant mRNAs escape the nonsense-mediated decay (NMD) pathway. We observe similar results for truncating mutations of MPZ that convey distinct myelinopathies. Our experiments show that triggering NMD and escaping NMD may cause distinct neurological phenotypes.
Similar content being viewed by others

Main
Different mutations in the same gene often cause distinct disease phenotypes in humans. Generally, such variations in the clinical phenotypes have been considered to be a consequence of the function or dysfunction of mutant proteins. Thus, a primary emphasis in genotype-phenotype correlation studies has been placed on determining the unique functional properties of encoded mutant proteins. But in vitro functional assays of mutant proteins often show discordance between predicted protein function and clinical outcome. Little is known about the many factors that are potentially involved in this discrepancy, but loss-of-function versus gain-of-function effects are often invoked as a possible mechanism.
We previously identified two unrelated individuals with an unusual phenotype that combined four distinct syndromes—peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, Waardenburg syndrome and Hirschsprung disease—that are characterized by deficiencies of Schwann cells, oligodendrocytes, melanocytes and enteric ganglia neurons, respectively1,2. Here we describe four more individuals and propose that this complex disorder is a newly described neurocristopathy called PCWH.
We previously identified mutations in SOX10 in all affected individuals1,2. SOX10 is a transcription factor that contains a central high mobility group (HMG) DNA-binding domain and a transactivation domain at its C terminus3. SOX10 is essential for the development of cells in the neural crest lineage, including melanocytes and enteric ganglia neurons4,5; it also controls the proliferation and differentiation of Schwann cells and oligodendrocytes6,7,8. Notably, some mutations in SOX10 also cause a distinct and more restricted disease that does not involve either the peripheral (PNS) or the central (CNS) nervous systems9,10,11. This less complicated neurocristopathy, called WS4, combines Waardenburg and Hirschsprung diseases12. Most SOX10 disease-associated mutations, regardless of whether they cause PCWH or WS4, result in premature termination codons (PTCs).
As in SOX10, different mutations in MPZ are responsible for distinct neurological diseases, which each affect the myelin of the PNS. These neuropathies include early onset congenital hypomyelinating neuropathy (CHN; OMIM 605253), Dejerine-Sottas neuropathy (DSN; OMIM 145900) and the less severe, adult onset Charcot-Marie-Tooth disease type 1B (CMT1B; OMIM 118200; ref. 13). It has been suggested that the severity of alleles in CHN and DSN is due to dominant-negative effects, whereas the reduced severity of alleles in CMT1B is due to loss of function. But although some nonsense and frameshift alleles cause CMT1B, several truncating mutations have been reported that convey either a CHN or a DSN phenotype.
We investigated the molecular mechanisms underlying the neurological phenotypes of the PCWH and WS4 neurocristopathies resulting from allelic SOX10 truncating mutations, as well as those underlying the CHN, DSN and CMT1B myelinopathies caused by allelic MPZ truncating mutations. Unexpectedly, we found that the function of the truncated SOX10 proteins has little effect on clinical outcome. Essentially all truncated SOX10 proteins, irrespective of their associated phenotypes, had a similar dominant-negative effect in vitro.
We also examined SOX10 and MPZ mRNAs that contain truncating mutations resulting in distinct neurological phenotypes. Our findings show that mutant mRNAs that result in a less severe disease had decreased stability, suggesting that the underlying mechanism is haploinsufficiency. By contrast, the more severe neurological disease seemed to result from a stable mRNA that is translated into a mutant protein with potent dominant-negative activity. We show that the unstable mRNAs are degraded by the NMD pathway. Thus, NMD is responsible for distinct neurological phenotypes arising from allelic truncating mutations and, by extension, may contribute to allelic affinity where multiple diseases have been associated with different mutations in the same gene.
Results
Clinical characterization of individuals with PCWH
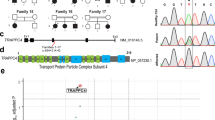
We identified four unrelated individuals with an unusual but uniform clinical phenotype consisting of four syndromes: peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, Waardenburg syndrome and Hirschsprung disease (PCWH). Here we define PCWH as a unique clinical presentation. Table 1 summarizes the clinical features of the four individuals with PCWH (see also case reports in Supplementary Note online), together with those of previously identified individuals1,2,14,15. The peripheral demyelinating neuropathy in these individuals was objectively shown by nerve biopsy2,15 and nerve conduction velocity (NCV) studies (Table 1). The CNS dysmyelinating leukodystrophy was identified by magnetic resonance imaging1 (MRI; Fig. 1a) and neuropathological studies2: both analyses showed that the individuals had a marked deficit in myelination.

(a) T2-weighted images of individual BAB1474 at 16 years of age (left) and individual BAB1916 at 2 years of age (right). Note the bilateral diffuse hyperintensity of cerebral white matter, especially in the periventricular white matter (arrowheads), representing dysmyelinating leukodystrophy. The volume of the white matter was preserved. Brain stem, cerebellum and spinal cord had normal myelination (not shown). (b) Pedigrees, restriction analysis and sequence chromatograms of four families with SOX10 mutations. Filled symbols indicate probands. Lanes in the restriction analyses represent the individual directly above in the pedigree. In each family, the proband showed the expected mutation-specific restriction pattern (arrowheads), which was not present in any other family members, indicating that all mutations occurred de novo. The mutations and predicted outcomes (in parentheses) are shown above the sequence chromatograms. (c) SOX10 mutations associated with PCWH and WS4. The three coding exons of SOX10 are indicated by shades of blue. The HMG and transactivation (TA) domains are indicated. Open symbols, WS4-associated mutations; filled symbols, PCWH-associated mutations; circles, nonsense mutations; squares, missense and in-frame insertion or deletion mutations; triangles, frameshift mutations. Arrows indicate putative terminations after frameshift mutations. Graph shows disease severity and the clinical diagnosis of individuals with each mutation. Green indicates a WS4 phenotype, red indicates a PCWH phenotype with a spectrum of severity decreasing from left to right.
5′ and 3′ SOX10 mutations convey distinct phenotypes
We identified heterozygous de novo mutations in exon 5 of SOX10 in each of the four individuals with sporadic PCWH (Fig. 1b). We found two frameshift and two nonsense mutations that result in premature termination at codon 313 (in two individuals), codon 297 or codon 364 (Fig. 1b). We also sequenced EDN3 and EDNRB, mutations of which also cause a WS4 phenotype, but found no alterations in these genes.
The SOX10 mutations in individuals with PCWH were located mainly in the last exon (exon 5). By contrast, mutations associated with the WS4 phenotype were clustered in the upstream coding exons (exons 3 and 4; Fig. 1c). To determine the generality of this correlation between the location of the mutation in SOX10 and phenotype, we examined a wider data set obtained from previous studies on people with SOX10 mutations1,2,9,10,11,14,16,17,18,19. We identified several individuals possessing clinical features that resembled PCWH but that were not well defined by objective electrophysiological (NCV) and imaging (MRI) studies.
We think that most or all of these published cases are probably PCWH because the neurological features largely overlap with those observed in the individuals with PCWH that we studied (e.g., areflexia, muscle wasting, dysautonomia, ataxia, nystagmus, spastic paraplegia and developmental delay; Supplementary Table 1 online). Similar to the four individuals with PCWH, most of these previously studied individuals had nonsense mutations or frameshift mutations that resulted in PTCs in the last exon of SOX10 (e.g., Y313X, Q234X, S251X, 778delG, S376X, Q377X and X467K; refs. 10,11,16,17,19; Fig. 1c).
In the studies described below, we investigated the molecular mechanism underlying why mutations in the 3′ region of SOX10 result in the PCWH phenotype, whereas mutations in the 5′ region result in the more restricted WS4 phenotype.
Truncated SOX10 mutants have dominant-negative activity
We considered that PCWH might not result from haploinsufficiency, but rather might be caused by dominant-negative effects1. To investigate the molecular basis of the PCWH-associated SOX10 mutations, we used a series of functional assays. First, to determine the ability of the SOX10 mutant proteins to interfere with wild-type SOX10 function, we carried out competition assays by co-transfecting vectors expressing the mutants and wild-type SOX10 into U138 human glioblastoma cells together with a vector containing SOX10-binding sites upstream of a minimal β-globin promoter3.
The SOX10 mutants resulting in PCWH (Q250X, 795delG, 847insT, Y313X, Q364X and Q377X) suppressed the transcriptional activity of cotransfected wild-type SOX10 in a dose-dependent manner (Fig. 2a,b), supporting the idea that PCWH is caused by dominant-negative mutations in the SOX10 gene. Unexpectedly, however, we observed similar suppression for mutants associated with the more restricted WS4 phenotype (E189X and Y207X). Below we provide evidence that, although these mutant proteins possess potent dominant-negative activity, their activity is not realized because their expression is downregulated by the NMD RNA surveillance pathway.

(a) SOX10 mutants used in the assays. Mutations shown in green lead to WS4, those in red to PCWH. Two clones, 939C→G and 938insA, result in the same amino acid change, Y313X. WT, wild-type. (b) Competition assays. Increasing amounts of mutant SOX10 expression plasmid were mixed with a fixed amount of wild-type SOX10 expression plasmid and cotransfected with the luciferase reporter plasmid 3xSXluc (ref. 3), which contains binding sites for SOX proteins. The total amount of DNA per well was kept constant by adding empty expression plasmid. The mean activity from transfections with the luciferase reporter and wild-type SOX10 plasmid was set as 1, and the other data are shown relative to this activity. (c) Transcriptional activities of SOX10 mutants determined by luciferase reporter assay. Plasmid 3xSXluc (ref. 3) was cotransfected into U138 human glioblastoma cells with a plasmid expressing wild-type SOX10 or mutant SOX10. The y axis corresponds to the clones in a, the x axis shows the luciferase activities as the relative induction above the mean activity from transfections with luciferase reporter and empty expression plasmid, which was set as 1. (d) Synergistic activities of SOX10 mutants. The luciferase reporter plasmid 3xFXOluc (ref. 3), which contains binding sites for both SOX and POU proteins, was transfected with the indicated combinations of plasmids expressing POU3F1, wild-type SOX10 and SOX10 mutants, and the luciferase activity was measured as in c. In b–d, bars represent the mean ± s.d.
Notably, two other WS4-associated mutants (482ins6 and 1076delGA) showed an additive transcriptional activation effect similar to that of wild-type SOX10. Their lack of dominant-negative activity may explain why they are associated with the weaker phenotype (WS4), but precisely how these mutations cause disease is not clear from this assay. In a separate test, we used a GJB1 promoter (a naturally occurring SOX10-responsive promoter) in a luciferase reporter assay20 and observed comparable competition effects for each of the mutants (Supplementary Fig. 1 online).
We evaluated the transcriptional activity of each SOX10 mutant by itself. All mutants that we tested showed diminished transcriptional activity relative to wild-type SOX10 (Fig. 2a,c). We also tested the ability of SOX10 mutants to increase synergistically the transcriptional activity of another transcription factor, which is a known property of wild-type SOX10 (ref. 3). All mutants retaining the HMG domain partially enhanced the transcriptional activity of POU3F1 (also known as SCIP, OCT6 and Tst-1; Fig. 2d). Notably, the transcriptional activation phenotype of a mutant associated with WS4 (E189X) did not significantly differ from that of mutants associated with PCWH (Q250X, 795delG, 847insT, Y313X and Q364X). Thus, it is unlikely that a difference in the POU3F1-activating ability of SOX10 mutants is responsible for the difference in these two phenotypes.
In summary, most truncated SOX10 mutants have reduced transcriptional activity and a dominant-negative effect regardless of whether they cause the PCWH or WS4 phenotype. Thus, whereas a dominant-negative mechanism is responsible for disease, an alternative mechanism must underlie the different phenotypic consequences of mutation (i.e., PCWH versus WS4).
SOX10 mutants have enhanced DNA-binding affinity
We used electrophoretic mobility shift assays (EMSAs) to investigate further the molecular mechanism of the dominant-negative activity of the truncated SOX10 mutant proteins. All mutants retaining a complete HMG domain had enhanced apparent DNA-binding affinity in comparison to wild-type SOX10 (Fig. 3a). As reported previously21, the 1076delGA mutant had a DNA-binding affinity similar to that of the wild type, and the 482ins6 mutant did not detectably bind DNA at all (data not shown).

(a) DNA-binding affinities of each SOX10 mutant. Top, western blotting of SOX10 proteins in nuclear extracts from HeLa cells transfected with wild-type (WT) or mutant SOX10 expression plasmids. Associated phenotypes are color coded as in Figure 2a. Molecular markers on the right show 86, 44 and 33 kDa from top to bottom. Bottom, equal amounts of SOX10 proteins (1:30 dilution) were used for EMSA. DNA binding is more prominent for the shorter SOX10 mutant proteins than for wild-type SOX10 (arrowhead). (b) Mutant SOX10 proteins interfere with the DNA binding of wild-type SOX10. Top, western blot of SOX10 proteins in nuclear extracts from HeLa cells cotransfected with the indicated combinations of wild-type and mutant SOX10 expression plasmids. The amount of wild-type SOX10 plasmid was constant, whereas that of the E189X, Y207X, Q250X, 847insT and Y313X mutant plasmids varied (1:25, 1:5 and 1:1 dilutions), as indicated by the triangle. Bottom, equal amounts of nuclear extracts (1:10 dilution) were used for EMSA. The wild-type SOX10-probe complex (WT) and the mutant SOX10-probe complex (Mut) are indicated. Increasing amounts of each SOX10 mutant seem to inhibit the binding of wild-type SOX10 protein.
As a better measure of relative DNA-binding affinity, we carried out competition experiments between the mutants and wild-type SOX10. All mutants tested inhibited the binding of wild-type SOX10 despite an excess amount of free target DNA (Fig. 3b). Thus, all mutants bind to DNA with high affinity and can interfere with the DNA binding of wild-type SOX10.
Downregulation of dominant-negative SOX10 transcripts
We sought to determine why some SOX10 mutant proteins are associated with the WS4 phenotype despite having potent dominant-negative activity. The dominant-negative mutants associated with the WS4 phenotype have nonsense mutations in internal exons (exon 3 and 4), whereas those associated with PCWH have nonsense mutations in the final exon (exon 5). We therefore considered that nonsense codons located in internal exons might trigger mRNA decay, thereby reducing the amount of dominant-negative protein that could be produced. Such a scheme would be consistent with the NMD RNA surveillance pathway, which typically degrades only transcripts containing nonsense codons that are followed by at least one intron22,23.
We addressed this hypothesis by using minigene constructs containing intron 4, which normally separates the last two exons of SOX10 (Fig. 4a). Northern-blot analysis showed that nonsense mutations upstream of the intron (Y83X, E189X or Y207X) led to reduced mRNA levels. By contrast, nonsense mutations downstream of the intron (Q250X or 795delG) led to no such reduction in mRNA (Fig. 4b). Comparable results were obtained in two different cell lines, HeLa adenocarcinoma (Fig. 4) and U138 glioblastoma (data not shown). These results support our hypothesis that WS4-associated mutations, but not PCWH-associated mutations, lead to a reduction in mRNA, thereby preventing dominant-negative effects.

(a) Minigene construct. Coding exons are indicated by shades of blue, with the last 55 bp of exon 4 in a darker shade. Intron 4 (unbroken line) is between exons 4 and 5. The CMV promoter was used to drive expression. Symbols and colors for each mutation are as Figure 1c. G218X is a synthetic mutation. Ter, normal termination codon. (b) Northern blot showing degradation of SOX10 transcripts carrying PTCs in upstream exons. The transcript level of each mutant is shown as a ratio of the level of wild-type SOX10 after normalization. Results shown are typical of five experiments. WS4-associated mutations (Y83X, E189X and Y207X), but not PCWH-associated mutations (Q250X and 795delG), result in reduced mRNA levels as compared with wild-type (WT) SOX10. The expression of G218X mutant mRNA, in which the PTC occurs in the last 55 bp of the penultimate exon, indicates that NMD probably mediates the mRNA degradation. (c) Removal of intron 4 (−intron) results in escape from NMD in WS4-associated mutations. The transcript level of each clone is shown as a ratio of the level of wild-type (WT) SOX10 after normalization. (d) NMD-mediated mRNA degradation results in reduced protein expression. Western blotting was done on nuclear extracts from HeLa cells transfected with the indicated SOX10 plasmids either with (NMD) or without (−intron) an intron. SOX10 protein was reduced by nonsense mutations only when an intron was present downstream of the mutation. Molecular markers on the right show 86, 44 and 33 kDa from top to bottom. WT, wild-type. (e) Cycloheximide treatment (CHX) rescues WS4-associated mutations from degradation. The transcript level of each mutant is shown as a ratio of the level of wild-type (WT) SOX10 without CHX treatment after normalization (n = 2). In each blot, either lacZ (3.1 kb) or neo (1.3 kb) was used for normalization. SOX10 transcripts are 2.8 kb.
To provide more evidence that NMD is responsible for the selective downregulation of WS4-associated nonsense mutations, we tested whether removal of intron 4 would abrogate NMD. Deleting the intron prevented downregulation of the mRNA of all of the WS4-specific nonsense mutants tested (Y83X, E189X and Y207X; Fig. 4c), indicating that NMD may be responsible for this downregulation. A strong intron-dependent reduction was also observed at the protein level: SOX10 protein was reduced by nonsense mutations only when an intron was present downstream of the mutation (Fig. 4d, compare E189X and Y207X with and without the intron).
As another test of whether NMD is responsible for downregulating SOX10 expression, we incubated the transfected cells with the protein synthesis inhibitor cycloheximide, which has been shown to reverse NMD24. Cycloheximide upregulated the expression of nonsense codon-bearing SOX10 transcripts (Y83X, Y207X and E189X) to a level similar to that of wild-type transcripts expressed in cycloheximide-treated cells (Fig. 4e).
Another characteristic of NMD is that it is not activated by nonsense codons in the penultimate exon that are located less than ∼55 bp from the final intron23. To test whether this rule applies to SOX10, we introduced a nonsense mutation 25 bp upstream of the intron in our construct (G218X). This construct showed no reduction in SOX10 mRNA expression (Fig. 4b). Thus, the mutants that are associated with the more restricted WS4 phenotype (Y83X, Y207X or E189X) are downregulated by the NMD mRNA surveillance pathway, whereas those that are associated with the more complex and severe PCWH phenotype (Q250X and 795delG) escape NMD and express large amounts of dominant-negative protein. Verification in human tissues in vivo would strengthen these in vitro findings; however, such samples of the nervous system were not available for this study.
Polarity of truncating mutations causing PCWH
Notably, we found that the severity of neurological symptoms in PCWH correlates with the location of the mutations in a polar manner, such that PTCs in the 5′ end of exon 5 result in a most severe and sometimes lethal phenotype, whereas the clinical outcome is milder for mutations located towards the 3′ end of exon 5. Specifically, the Q234X, Q250X and S251X mutations are associated with a lethal phenotype2,11,16, whereas the 847insT, Y313X and Q364X mutations are associated with infantile- or juvenile-onset myelin deficiencies that cause only modest neurological disabilities (Fig. 1c). Mutations nearest the 3′ end (S376X and Q377X) are associated with even milder neurological manifestations (refs. 10,19 and data not shown). Notably, the most 3′ mutation (1076delGA), which generates the longest truncated protein, causes no measurable dysmyelinating phenotype9.
Experimental evidence provides a potential explanation for this polarity. EMSAs showed that the enhanced DNA-binding affinity of the truncated SOX10 proteins was also polar; that is, DNA binding was more prominent for short truncation mutants (E189X, Y207X, Q234X, Q250X and S251X) than for long truncation mutants (Y313X, Q364X and Q377X; Fig. 3a). In addition, competition assays showed that individual mutants varied in their competitive binding: short truncation mutants (E189X, Y207X and Q250X) tended to compete more strongly than did long truncation mutants (847insT and Y313X).
Although we do not know how stronger DNA binding increases the ability of SOX10 to cause disease, the strong correlation between the DNA-binding affinity of SOX10 and its disease phenotype indicates that this variable may be important. Either stronger DNA binding may enhance the dominant-negative activity of SOX10 by a mechanism that is not detectable in our assays (for example, promoter-specific dominant-negative effects) or it may elicit effects that remain to be defined.
NMD downregulates PTC-bearing MPZ transcripts
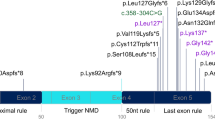
To examine whether NMD can explain why other distinct neurological diseases result from allelic truncating mutations, we tested PTCs in the gene MPZ by similar expression assays. Allelic point mutations of MPZ cause a spectrum of genotypically related, dominantly inherited, peripheral neuropathies with variable severity, including the more severe, early onset CHN and DSN, as well as the less severe, adult onset CMT1B. Most disease-associated mutations in MPZ mutations are missense alterations, but 18 truncating MPZ mutations have been reported to cause either mild or severe forms of peripheral neuropathy (ref. 25 and Inherited Peripheral Neuropathies Mutation Database; see URL in Methods). Molecular studies of mutant MPZ have not provided a plausible explanation for the association between mutation and disease severity (that is, genotype-phenotype correlations). As in SOX10, we identified an apparent correlation between phenotype and the location of PTCs in MPZ. The PTCs in the 5′ end result in mild neuropathy, whereas most of those in the 3′ end cause severe diseases, with the exception of a few cases for which detailed clinical information was not available (Fig. 5a).

(a) MPZ truncating mutations associated with inherited peripheral neuropathies26. The six coding exons of MPZ are indicated by shades of blue. The transmembrane domain (TM) is encoded by exon 4. Open symbols, mutations associated with CMT1B; filled symbols, mutations associated with DSN/CHN; circles, nonsense mutations; triangles, frameshift mutations. Arrows indicate putative terminations after frameshift mutations. Green indicates a CMT1B phenotype, red indicates a DSN/CHN phenotype. (b) MPZ genomic expression construct used for northern blotting. Six mutations associated with either CMT1B or DSN/CHN are shown (symbols as in a). Ter, normal termination codon. (c) Northern blots showing degradation of MPZ transcripts carrying PTCs in internal exons. The transcript level of each mutant is shown as a ratio of the level of wild-type (WT) MPZ after normalization. Results shown are typical of three experiments. The 5′ PTCs (223delG, Y154X, Y181X and Q206X), but not the 3′ PTCs (Q215X and 662insGC), result in reduced mRNA levels as compared with wild-type MPZ. The DSN/CHN-associated Q215X PTC located 2 bp from the end of exon 4 escaped mRNA downregulation, whereas the CMT1B-associated G206X PTC located 28 bp upstream of Q215X in the same exon underwent mRNA downregulation. These data suggest that MPZ does not obey the '−55 bp rule'23, as has been observed for some genes44. (d) Removal of intron 4 (−intron) recovers the expression of CMT1B-associated mutations. Experiments were done as in Figure 4c. WT, wild-type. (e) Cycloheximide treatment (CHX) rescues mRNAs carrying CMT1B-associated mutations from degradation. Experiments were done as in Figure 4e (n = 2). WT, wild-type. (f) RNAi-mediated suppression of UPF1 rescues mRNAs carrying CMT1B-associated PTCs from degradation (n = 3). Western blot (inset) shows decreased expression of UPF1 protein after siRNA treatment. In the northern blots, neo (1.3 kb) was used for normalization. MPZ transcripts are 1.8 kb. For all constructs that showed mRNA instability owing to NMD, quantitative RT-PCR experiments were done independently to validate the results of the northern blots (not shown). WT, wild-type.
We examined the mRNA stability of MPZ PTC mutations that convey distinct peripheral neuropathies. Of six mutations (Fig. 5b), two (Y181X and G206X) cause truncation at a position similar to truncation clones that have been shown in vitro to behave as dominant-negative alleles, despite causing mild CMT1B (ref. 26). Each PTC associated with a mild phenotype is located in an internal exon and resulted in a downregulation of transcripts (Fig. 5c). This downregulation was prevented by each of the following: first, elimination of all introns by using a cDNA construct; second, cycloheximide treatment; and third, suppression of UPF1 (ref. 27), an essential factor in the NMD pathway, by RNA-mediated interference (RNAi; Fig. 5d–f).
These findings suggest that the MPZ transcripts carrying these PTCs selectively undergo NMD and are likely to be degraded in vivo. By contrast, the PTCs associated with severe diseases are all located in the last exon (except for one that is located at the distal end of the penultimate exon) and resulted in an accumulation of mRNA at levels equivalent to the wild-type allele. These finding suggest that the MPZ transcripts containing these PTCs escape NMD and are probably translated into truncated proteins that may have a dominant-negative function26.
Discussion
We propose that the phenotypes of genetic diseases are commonly influenced by NMD. First, PTCs are common: nonsense and frameshift mutations are present in approximately one-third of mutations that cause human genetic disease28,29. Second, nonsense codons in all internal exons are capable of triggering NMD23, which means that mRNA levels will be reduced by most nonsense and frameshift mutations28,29,30; therefore, PTCs generally result in milder phenotypes as compared with missense mutations, as has been postulated in several human diseases including osteogenesis imperfecta31, Stickler syndrome32 and Marfan syndrome33,34. Third, on the basis that NMD has a potential role in most disease-causing PTCs, it has been proposed that haploinsufficiency is the most predictable pathogenetic mechanism underlying heterozygous nonsense alleles29. Last, PTCs located in the last coding exon specifically escape NMD23 and so are processed distinctly from those in internal exons, leading to the stable translation of truncated proteins.
β-thalassemia is a classic example in which 5′ PTCs in the gene encoding β-globin result in a recessive trait, whereas 3′ PTCs result in a specific dominant form of disease, because 5′ PTCs but not 3′ PTCs trigger β-globin NMD35,36. We think that this previously underappreciated mechanism has an important role not only in phenotypic variation, but also in the expression of distinct phenotypes from allelic truncating mutations. In support of this idea, we have shown experimentally that PTCs that trigger NMD versus those that escape NMD result in distinct neurological phenotypes in two different disease-causing genes, SOX10 and MPZ.
A corollary to our study is that many genetic diseases that were previously thought to be the result of dominant-negative mutations on the basis of functional assays may actually result from haploinsufficiency in vivo because of NMD. In fact, previous genotype-phenotype studies of MPZ mutations could not explain the mild phenotype associated with dominant-negative truncated proteins26,37,38. Invoking NMD not only delineates the genotype-phenotype correlation for truncating mutations, but it also facilitates a functional analysis of missense alterations by comparison with clinical phenotypes. Thus, NMD must be considered when formulating and testing explanations for phenotypic differences associated with disease-causing mutations. We have shown that triggering NMD versus escaping NMD may cause distinct neurological phenotypes in addition to contributing to the variability of expression in human genetic diseases.
Three case reports previously identified an association between SOX10 mutations and deficiencies in peripheral and/or central myelin1,2,14. Here, by combining those data with data from four additional individuals, we have provided evidence for the existence of a distinct genetic disease, PCWH, caused by SOX10 mutations. We consider that PCWH is a distinct neurocristopathy because it can be distinguished from other neurocristopathies, including WS4, on the basis of the following observations.
First, there are no other syndromes that simultaneously show the clinical findings of peripheral myelinopathy, central dysmyelination, Waardenburg syndrome and Hirschsprung disease. In particular, the involvement of both the PNS and the CNS is a distinguishing characteristic of PCWH. These neurological findings predominate in determining the degree of disability in individuals and also significantly influence the prognosis: severe hypomyelination in the PNS and CNS has even resulted in early death in some cases2,11,16. Second, PCWH is unique because so far it is known only to be caused by a single gene, whereas WS4 is associated with at least two other genes, EDN3 and EDNRB (refs 9,39,40). Notably, neither EDN3 nor EDNRB has been associated with some of the symptoms characteristic of PCWH, such as peripheral and central myelin deficiencies. Last, we have shown that the SOX10 mutations that cause PCWH represent a subset that are distinct from those that cause WS4.
Our study indicates that the two phenotypes associated with SOX10 mutations, PCWH and WS4, may be caused by two distinct molecular mechanisms. The more severe phenotype, PCWH, is caused by nonsense mutations that generate truncated SOX10 mutant proteins possessing enhanced DNA-binding affinity and potent dominant-negative activity. The more modest phenotype, WS4, is caused by nonsense mutations that activate the NMD RNA surveillance pathway, thereby reducing dominant-negative expression and resulting in SOX10 haploinsufficiency.
This interpretation of the data from SOX10-deficient humans is consistent with the finding that Sox10−/− homozygous mice have profound defects in both peripheral and central myelin lineages that result in embryonic lethality, whereas heterozygotes show a phenotype corresponding to the WS4 phenotype8. The dominant-negative activity of the SOX10 mutant proteins that cause PCWH probably does not block SOX10 function completely, because the PCWH phenotype is milder than that seen in homozygous Sox10−/− mice. It is also possible, however, that mice and humans differ subtly in their reliance on SOX10 for neurological development. Although our data are most consistent with dominant-negative interference, a gain-of-function effect cannot be absolutely ruled out.
We found that for mutations in SOX10 exon 5, the severity of the PCWH phenotype inversely correlates with the length of SOX10 protein (Fig. 1c). Our transfection studies identified a potential molecular mechanism for this correlation: the shorter SOX10 mutants had stronger DNA-binding affinity and dominant-negative activity than did the longer SOX10 mutants (Figs. 2b and 3a,b). This polarity may explain the differential severity observed in individuals with PCWH; that is, it may explain why Q250X, one of the early truncating mutations, completely diminishes myelin development in both the PNS and the CNS2, whereas a mutant that is 63 residues longer, Y313X, causes a much milder phenotype (patients 1 and 2). In addition, later truncations (i.e., longer SOX10 mutants) might also have a smaller interference effect and thus would not result in the PCWH phenotype9 (e.g., 1076delGA in Figs. 1c and 2a).
Much evidence indicates that the more restricted WS4 phenotype is caused by SOX10 nonsense mutations that activate the protective NMD RNA surveillance pathway. Two WS4-associated mutations in exon 4, E189X and Y207X, resulted in truncated proteins possessing strong dominant-negative activity when expressed from cDNAs lacking introns. However, cellular mRNAs bearing these mutations were diminished when transcribed from a construct with an intron-containing minigene, suggesting that NMD degrades mRNAs containing PTCs before translation, which seems to mitigate the dominant-negative action of these mutations. As a result, these mutant alleles become equivalent to null alleles and thus haploinsufficiency is probably the underlying mechanism causing the disease phenotype in vivo. By contrast, mRNAs with nonsense mutations in the final exon escape NMD because there is no intron downstream of the stop codon. Large amounts of dominant-negative protein are therefore produced, leading to the severe (PCWH) disease phenotype. Consistent with this interpretation, all truncating mutations associated with PCWH are located in exon 5 and are anticipated to escape NMD (Fig. 1c).
Both SOX10 and MPZ PTCs encode proteins with dominant-negative effects, but only those PTCs that escape NMD realize that potential. This finding, in conjunction with previous results31,32,33,34,35,36, suggests that one of the physiological roles of NMD may be to protect against severe disease phenotypes by converting dominant-negative effects to haploinsufficiency. The extent of the beneficial effect may vary depending on both the toxicity of truncated proteins encoded by different genes and the nature of traits. Rarely, as exemplified by the hemizygous X-linked DMD gene mutated in muscular dystrophy, NMD may result in more severe disease by abrogating the hypomorphic protein function41. Nevertheless, individuals potentially carry several silent PTCs in their genome and the suppression of these PTC-carrying alleles by NMD may be crucial for preventing disease expression, as implied by the embryonic lethality caused by congenital suppression of NMD in mice42.
Methods
SOX10 mutation analysis.
Peripheral blood was obtained from individuals and family members with informed consent approved by the Institutional Review Board at Baylor College of Medicine. Genomic DNA was extracted from white blood cells by a standard method. The genomic DNA from each individual was used as a template for PCR to amplify each coding exon of the SOX10 gene1. We sequenced PCR products directly by using DyePrimer chemistry and an ABI377 sequencer (Applied Biosystems). Gene mutations were examined in each family member by restriction enzyme digestion of PCR products to determine the de novo occurrence or segregation within the family. DdeI, MwoI, BssSI and StyI were used to determine each mutation. Using the same restriction analyses, we also examined at least 180 normal chromosomes to exclude the possibility of benign polymorphism. Paternity was confirmed in each family by microsatellite haplotyping analyses done by IdentiGene.
Recombinant constructs.
We incorporated each mutation into pCMV-huSOX10, which contains human wild-type SOX10 cDNA under a cytomegalovirus (CMV) promoter, either by PCR-directed mutagenesis or by subcloning PCR products generated from patient DNA. The POU3F1 expression plasmid pCMV/Tst-1 and the luciferase reporter plasmids 3xSXluc and 3xFXOluc have been described3. A 750-bp fragment from the GJB1 promoter was subcloned into the luciferase reporter plasmid pGL3 (Promega) as described20. To construct intron-containing SOX10 minigenes, we used PCR to fuse two overlapping fragments, one from cDNA spanning exons 3–4 and the other from genomic DNA spanning a portion of exon 4, intron 4 and exon 5, and subcloned the product into the mammalian expression vector pcDNA3.1 (Invitrogen) to generate pcDNASOX10NMD. We also subcloned the human SOX10 cDNA into pcDNA3.1 to generate pcDNASOX10-intron. Full-length human MPZ cDNA (IMAGE 3926008) was obtained (OpenBiosystems) and subcloned into pcDNA3.1 (Invitrogen) to generate pcDNAMPZ. The whole genomic coding region was also subcloned from genomic DNA by PCR to generate pcDNAMPZNMD. Mutations were generated in each construct with the QuikChange Site-Directed mutagenesis kit (Stratagene). A β-galactosidase expression plasmid, pCMVβ (Clontech), was used as a reference for transfections.
Functional assays.
U138 human glioblastoma cells and HeLa cells were grown in DMEM medium supplemented with 10% fetal bovine serum and were transfected by using PolyFect or Effectene transfection reagents (Qiagen). For the luciferase assay, we collected U138 cells from 24-well trays after 48 h of transfection and then assayed luciferase activity3. Nuclear extract was obtained from HeLa cells after 48 h of transfection and used for western blotting and EMSA studies. Rabbit antiserum to SOX10 (1:3,000 dilution; Chemicon) was used as a primary antibody for western blots. This antiserum recognizes an epitope at amino acids 165–181 of human SOX10; therefore, the signals were quantitative for both wild-type SOX10 and the truncated proteins used in this study. We used oligonucleotide probes containing a single SOX10-binding site (site B)43 for EMSAs.
For northern blotting, total RNA was extracted from HeLa cells after 24 h of transfection using 0.1 μg of SOX10 or MPZ expression plasmids along with 0.9 μg of pCMVβ reference plasmid per 60-mm dish. Two hours before the cells were collected, cycloheximide was added to a concentration of 1 mg/ml. Knockdown of UPF1 by RNAi was done as described27. Polyclonal antibody to UPF1 was a gift from J. Lykke-Andersen (University of Colorado, Boulder). Expression of the neomycin phosphotransferase gene (neo) gene from pcDNA3.1 or the cotransfected β-galactosidase gene (lacZ) was used as a reference control for normalization. After electrophoretic separation and transfer to a nylon membrane, mRNA was probed by 32P-labeled SOX10, neo and lacZ probes. The value for each signal was measured with a Personal Densitometer SI (Molecular Dynamics).
URL.
The Inherited Peripheral Neuropathies Mutation Database is available at http://molgen-www.uia.ac.be/CMTMutations/.
Note: Supplementary information is available on the Nature Genetics website.
References
Inoue, K., Tanabe, Y. & Lupski, J.R. Myelin deficiencies in both the central and the peripheral nervous systems associated with a SOX10 mutation. Ann. Neurol. 46, 313–318 (1999).
Inoue, K. et al. Congenital hypomyelinating neuropathy, central dysmyelination, and Waardenburg-Hirschsprung disease: phenotypes linked by SOX10 mutation. Ann. Neurol. 52, 836–842 (2002).
Kuhlbrodt, K., Herbarth, B., Sock, E., Hermans-Borgmeyer, I. & Wegner, M. Sox10, a novel transcriptional modulator in glial cells. J. Neurosci. 18, 237–250 (1998).
Herbarth, B. et al. Mutation of the Sry-related Sox10 gene in Dominant megacolon, a mouse model for human Hirschsprung disease. Proc. Natl. Acad. Sci. USA 95, 5161–5165 (1998).
Southard-Smith, E.M., Kos, L. & Pavan, W.J. Sox10 mutation disrupts neural crest development in Dom Hirschsprung mouse model. Nat. Genet. 18, 60–64 (1998).
Sonnenberg-Riethmacher, E. et al. Development and degeneration of dorsal root ganglia in the absence of the HMG-domain transcription factor Sox10. Mech. Dev. 109, 253–265 (2001).
Stolt, C.C. et al. Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor Sox10. Genes Dev. 16, 165–170 (2002).
Britsch, S. et al. The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev. 15, 66–78 (2001).
Pingault, V. et al. SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nat. Genet. 18, 171–173 (1998).
Southard-Smith, E.M. et al. The Sox10Dom mouse: modeling the genetic variation of Waardenburg-Shah (WS4) syndrome. Genome Res. 9, 215–225 (1999).
Pingault, V. et al. SOX10 mutations in chronic intestinal pseudo-obstruction suggest a complex physiopathological mechanism. Hum. Genet. 111, 198–206 (2002).
Omenn, G.S. & McKusick, V.A. The association of Waardenburg syndrome and Hirschsprung megacolon. Am. J. Med. Genet. 3, 217–223 (1979).
Warner, L.E. et al. Clinical phenotypes of different MPZ (P0) mutations may include Charcot-Marie-Tooth type 1B, Dejerine-Sottas, and congenital hypomyelination. Neuron 17, 451–460 (1996).
Pingault, V. et al. Peripheral neuropathy with hypomyelination, chronic intestinal pseudo-obstruction and deafness: a developmental 'neural crest syndrome' related to a SOX10 mutation. Ann. Neurol. 48, 671–676 (2000).
Jacobs, J.M. & Wilson, J. An unusual demyelinating neuropathy in a patient with Waardenburg's syndrome. Acta Neuropathol. 83, 670–674 (1992).
Touraine, R.L. et al. Neurological phenotype in Waardenburg syndrome type 4 correlates with novel SOX10 truncating mutations and expression in developing brain. Am. J. Hum. Genet. 66, 1496–1503 (2000).
Sham, M.H., Lui, V.C.H., Chen, B.L.S., Fu, M. & Tam, P.K.H. Novel mutations of SOX10 suggest a dominant negative role in Waardenburg-Shah syndrome. J. Med. Genet. 38, E30 (2001).
Bondurand, N. et al. A molecular analysis of the Yemenite deaf-blind hypopigmentation syndrome: SOX10 dysfunction causes different neurocristopathies. Hum. Mol. Genet. 8, 1785–1789 (1999).
Toki, F. et al. Intestinal agangliosis associated with the Waardenburg syndrome: report of two cases and review of the literature. Pediatr. Surg. Int. 19, 725–728 (2003).
Bondurand, N. et al. Human Connexin 32, a gap junction protein altered in the X-linked form of Charcot-Marie-Tooth disease, is directly regulated by the transcription factor SOX10. Hum. Mol. Genet. 10, 2783–2795 (2001).
Kuhlbrodt, K. et al. Functional analysis of Sox10 mutations found in human Waardenburg-Hirschsprung patients. J. Biol. Chem. 273, 23033–23038 (1998).
Carter, M.S., Li, S. & Wilkinson, M.F. A splicing-dependent regulatory mechanism that detects translation signals. EMBO J. 15, 5965–5975 (1996).
Nagy, E. & Maquat, L.E. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem. Sci. 23, 198–199 (1998).
Carter, M.S. et al. A regulatory mechanism that detects premature nonsense codons in T-cell receptor transcripts in vivo is reversed by protein synthesis inhibitors in vitro. J. Biol. Chem. 270, 28995–29003 (1995).
Saifi, G.M., Szigeti, K., Snipes, G.J., Garcia, C.A. & Lupski, J.R. Molecular mechanisms, diagnosis, and rational approaches to management and therapy for Charcot-Marie-Tooth disease and related peripheral neuropathies. J. Investig. Med. 51, 261–283 (2003).
Wong, M-H. & Filbin, M.T. Dominant-negative effect on adhesion by myelin Po protein truncated in its cytoplasmic domain. J. Cell Biol. 134, 1531–1541 (1996).
Mendell, J.T., ap Rhys, C.M.J. & Dietz, H.C. Separable roles for rent1/hUpf1 in altered splicing and decay of nonsense transcripts. Science 298, 419–422 (2002).
Frischmeyer, P.A. & Dietz, H.C. Nonsense-mediated mRNA decay in health and disease. Hum. Mol. Genet. 8, 1893–1900 (1999).
Mendell, J.T. & Dietz, H.C. When the message goes awry: disease-producing mutations that influence mRNA content and performance. Cell 107, 411–414 (2001).
Byers, P.H. Killing the messenger: new insights into nonsense-mediated mRNA decay. J. Clin. Invest. 109, 3–6 (2002).
Körkkö, J. et al. Analysis of the COL1A1 and COL1A2 genes by PCR amplification and scanning by conformation-sensitive gel electrophoresis identifies only COL1A1 mutations in 15 patients with osteogenesis imperfecta type I: identification of common sequences of null-allele mutations. Am. J. Hum. Genet. 62, 98–110 (1998).
Snead, M.P. & Yates, J.R.W. Clinical and Molecular genetics of Stickler syndrome. J. Med. Genet. 36, 353–359 (1999).
Dietz, H.C. et al. Four novel FBN1 mutations: significance for mutant transcript level and EGF-like domain calcium binding in the pathogenesis of Marfan syndrome. Genomics 17, 468–475 (1993).
Schrijver, I. et al. Premature termination mutations in FBN1: distinct effects on differential allelic expression and on protein and clinical phenotypes. Am. J. Hum. Genet. 71, 223–237 (2002).
Baserga, S.J. & Benz, E.J. Jr. Nonsense mutations in the human β-globin gene affect mRNA metabolism. Proc. Natl. Acad. Sci. USA 85, 2056–2060 (1988).
Forget, B.G., Benz, E.J., Jr., Skoultchi, A., Baglioni, C. & Housman, D. Absence of messenger RNA for β globin chain in β0-thalassaemia. Nature 247, 379–381 (1974).
Shames, I., Fraser, A., Colby, J., Orfali, W. & Snipes, G.J. Phenotypic differences between peripheral myelin protein-22 (PMP22) and myelin protein zero (P0) mutations associated with Charcot-Marie-Tooth-related diseases. J. Neuropathol. Exp. Neurol. 62, 751–764 (2003).
Yoshida, M. & Colman, D.R. Rapid functional analysis in Xenopus oocytes of Po protein adhesive interactions. Neurochem. Res. 26, 703–712 (2001).
Edery, P. et al. Mutation of the endothelin-3 gene in the Waardenburg-Hirschsprung disease (Shah-Waardenburg syndrome). Nat. Genet. 12, 442–444 (1996).
Puffenberger, E.G. et al. A missense mutation of the endothelin-B receptor gene in multigenic Hirschsprung's disease. Cell 79, 1257–1266 (1994).
Kerr, T.P., Sewry, C.A., Robb, S.A. & Roberts, R.G. Long mutant dystrophins and variable phenotypes: evasion of nonsense-mediated decay? Hum. Genet. 109, 402–407 (2001).
Medghalchi, S.M. et al. Rent1, a trans-effector of nonsense-mediated mRNA decay, is essential for mammalian embryonic viability. Hum. Mol. Genet. 10, 99–105 (2001).
Peirano, R.I., Goerich, D.E., Riethmacher, D. & Wegner, M. Protein zero gene expression is regulated by the glial transcription factor Sox10. Mol. Cell. Biol. 20, 3198–3209 (2000).
Wang, J., Gudikote, J.P., Olivas, O.R. & Wilkinson, M.F. Boundary-independent polar nonsense-mediated decay. EMBO Rep. 3, 274–279 (2002).
Acknowledgements
We thank the affected individuals and their families for their cooperation; M. Quanrud for collecting blood samples from family members; T. Shimotake and Y. Tsuchida for providing clinical information; R. Peirano and E. Sock for advice and assistance; and A. Beaudet, H. Bellen, K. Szigeti and H. Zoghbi for critical reviews. This study was supported in part by grants from the US National Institute for Neurological Disorders and Strokes, the US National Institutes of Health and the Muscular Dystrophy Association to J.R.L. K.I. was a fellow of the Charcot-Marie-Tooth Association when this study was initiated and is currently supported by a development grant from the Muscular Dystrophy Association.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
About this article
Cite this article
Inoue, K., Khajavi, M., Ohyama, T. et al. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat Genet 36, 361–369 (2004). https://doi.org/10.1038/ng1322
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng1322
This article is cited by
-
A novel PAX6 variant as the cause of aniridia in a Chinese patient with SRRRD
BMC Medical Genomics (2023)
-
Identification of molecular targets of Trigonelline for treating breast cancer through network pharmacology and bioinformatics-based prediction
Molecular Diversity (2023)
-
Whole exome analysis of patients in Japan with hearing loss reveals high heterogeneity among responsible and novel candidate genes
Orphanet Journal of Rare Diseases (2022)
-
Three novel variants in SOX10 gene: Waardenburg and PCWH syndromes
Egyptian Journal of Medical Human Genetics (2022)
-
Genetic insights, disease mechanisms, and biological therapeutics for Waardenburg syndrome
Gene Therapy (2022)
