
Abstract
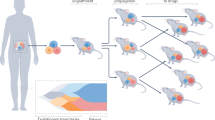
Patient-derived xenografts (PDXs) have become a prominent cancer model system, as they are presumed to faithfully represent the genomic features of primary tumors. Here we monitored the dynamics of copy number alterations (CNAs) in 1,110 PDX samples across 24 cancer types. We observed rapid accumulation of CNAs during PDX passaging, often due to selection of preexisting minor clones. CNA acquisition in PDXs was correlated with the tissue-specific levels of aneuploidy and genetic heterogeneity observed in primary tumors. However, the particular CNAs acquired during PDX passaging differed from those acquired during tumor evolution in patients. Several CNAs recurrently observed in primary tumors gradually disappeared in PDXs, indicating that events undergoing positive selection in humans can become dispensable during propagation in mice. Notably, the genomic stability of PDXs was associated with their response to chemotherapy and targeted drugs. These findings have major implications for PDX-based modeling of human cancer.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others


Accession codes
References
Tentler, J.J. et al. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 9, 338–350 (2012).
Siolas, D. & Hannon, G.J. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res. 73, 5315–5319 (2013).
Gao, H. et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 21, 1318–1325 (2015).
Hidalgo, M. et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 4, 998–1013 (2014).
Zhang, X. et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 73, 4885–4897 (2013).
Hodgson, J.G. et al. Comparative analyses of gene copy number and mRNA expression in glioblastoma multiforme tumors and xenografts. Neuro-oncol. 11, 477–487 (2009).
Eirew, P. et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 518, 422–426 (2015).
Bruna, A. et al. A biobank of breast cancer explants with preserved intra-tumor heterogeneity to screen anticancer compounds. Cell 167, 260–274.e22 (2016).
Kresse, S.H., Meza-Zepeda, L.A., Machado, I., Llombart-Bosch, A. & Myklebost, O. Preclinical xenograft models of human sarcoma show nonrandom loss of aberrations. Cancer 118, 558–570 (2012).
DeRose, Y.S. et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 17, 1514–1520 (2011).
Lin, D. et al. High fidelity patient-derived xenografts for accelerating prostate cancer discovery and drug development. Cancer Res. 74, 1272–1283 (2014).
Huynh, H. et al. Bevacizumab and rapamycin induce growth suppression in mouse models of hepatocellular carcinoma. J. Hepatol. 49, 52–60 (2008).
Zhang, X. et al. SRRM4 expression and the loss of REST activity may promote the emergence of the neuroendocrine phenotype in castration-resistant prostate cancer. Clin. Cancer Res. 21, 4698–4708 (2015).
Oakes, S.R. et al. Sensitization of BCL-2-expressing breast tumors to chemotherapy by the BH3 mimetic ABT-737. Proc. Natl. Acad. Sci. USA 109, 2766–2771 (2012).
Jäger, W. et al. Patient-derived bladder cancer xenografts in the preclinical development of novel targeted therapies. Oncotarget 6, 21522–21532 (2015).
Monsma, D.J. et al. Using a rhabdomyosarcoma patient-derived xenograft to examine precision medicine approaches and model acquired resistance. Pediatr. Blood Cancer 61, 1570–1577 (2014).
Wong, N.C. et al. Stability of gene expression and epigenetic profiles highlights the utility of patient-derived paediatric acute lymphoblastic leukaemia xenografts for investigating molecular mechanisms of drug resistance. BMC Genomics 15, 416 (2014).
Gu, Q. et al. Genomic characterization of a large panel of patient-derived hepatocellular carcinoma xenograft tumor models for preclinical development. Oncotarget 6, 20160–20176 (2015).
Smith, K.B. et al. Novel dedifferentiated liposarcoma xenograft models reveal PTEN down-regulation as a malignant signature and response to PI3K pathway inhibition. Am. J. Pathol. 182, 1400–1411 (2013).
Ma, C.X. et al. Targeting Chk1 in p53-deficient triple-negative breast cancer is therapeutically beneficial in human-in-mouse tumor models. J. Clin. Invest. 122, 1541–1552 (2012).
Morisot, S. et al. High frequencies of leukemia stem cells in poor-outcome childhood precursor-B acute lymphoblastic leukemias. Leukemia 24, 1859–1866 (2010).
Peng, S. et al. Tumor grafts derived from patients with head and neck squamous carcinoma authentically maintain the molecular and histologic characteristics of human cancers. J. Transl. Med. 11, 198 (2013).
Chou, J. et al. Phenotypic and transcriptional fidelity of patient-derived colon cancer xenografts in immune-deficient mice. PLoS One 8, e79874 (2013).
Ben-David, U., Mayshar, Y. & Benvenisty, N. Virtual karyotyping of pluripotent stem cells on the basis of their global gene expression profiles. Nat. Protoc. 8, 989–997 (2013).
Ben-David, U. et al. The landscape of chromosomal aberrations in breast cancer mouse models reveals driver-specific routes to tumorigenesis. Nat. Commun. 7, 12160 (2016).
Fehrmann, R.S. et al. Gene expression analysis identifies global gene dosage sensitivity in cancer. Nat. Genet. 47, 115–125 (2015).
Zack, T.I. et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 45, 1134–1140 (2013).
Carter, S.L., Eklund, A.C., Kohane, I.S., Harris, L.N. & Szallasi, Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 38, 1043–1048 (2006).
Clappier, E. et al. Clonal selection in xenografted human T cell acute lymphoblastic leukemia recapitulates gain of malignancy at relapse. J. Exp. Med. 208, 653–661 (2011).
Andor, N. et al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat. Med. 22, 105–113 (2016).
Bakker, B. et al. Single-cell sequencing reveals karyotype heterogeneity in murine and human malignancies. Genome Biol. 17, 115 (2016).
Mroz, E.A., Tward, A.D., Hammon, R.J., Ren, Y. & Rocco, J.W. Intra-tumor genetic heterogeneity and mortality in head and neck cancer: analysis of data from the Cancer Genome Atlas. PLoS Med. 12, e1001786 (2015).
Gibson, W.J. et al. The genomic landscape and evolution of endometrial carcinoma progression and abdominopelvic metastasis. Nat. Genet. 48, 848–855 (2016).
Kim, T.M. et al. Subclonal genomic architectures of primary and metastatic colorectal cancer based on intratumoral genetic heterogeneity. Clin. Cancer Res. 21, 4461–4472 (2015).
Bai, H. et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat. Genet. 48, 59–66 (2016).
Hedberg, M.L. et al. Genetic landscape of metastatic and recurrent head and neck squamous cell carcinoma. J. Clin. Invest. 126, 1606 (2016).
Um, S.W. et al. Molecular evolution patterns in metastatic lymph nodes reflect the differential treatment response of advanced primary lung cancer. Cancer Res. 76, 6568–6576 (2016).
Sveen, A. et al. Intra-patient inter-metastatic genetic heterogeneity in colorectal cancer as a key determinant of survival after curative liver resection. PLoS Genet. 12, e1006225 (2016).
Lee, S.Y. et al. Comparative genomic analysis of primary and synchronous metastatic colorectal cancers. PLoS One 9, e90459 (2014).
Lim, B. et al. Genome-wide mutation profiles of colorectal tumors and associated liver metastases at the exome and transcriptome levels. Oncotarget 6, 22179–22190 (2015).
Daniel, V.C. et al. A primary xenograft model of small-cell lung cancer reveals irreversible changes in gene expression imposed by culture in vitro. Cancer Res. 69, 3364–3373 (2009).
Günther, H.S. et al. Glioblastoma-derived stem cell–enriched cultures form distinct subgroups according to molecular and phenotypic criteria. Oncogene 27, 2897–2909 (2008).
Schulte, A. et al. A distinct subset of glioma cell lines with stem cell–like properties reflects the transcriptional phenotype of glioblastomas and overexpresses CXCR4 as therapeutic target. Glia 59, 590–602 (2011).
Cifola, I. et al. Renal cell carcinoma primary cultures maintain genomic and phenotypic profile of parental tumor tissues. BMC Cancer 11, 244 (2011).
Hollingshead, M.G. et al. Gene expression profiling of 49 human tumor xenografts from in vitro culture through multiple in vivo passages—strategies for data mining in support of therapeutic studies. BMC Genomics 15, 393 (2014).
Gillet, J.P., Varma, S. & Gottesman, M.M. The clinical relevance of cancer cell lines. J. Natl. Cancer Inst. 105, 452–458 (2013).
Birkbak, N.J. et al. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res. 71, 3447–3452 (2011).
Zhang, W. et al. Centromere and kinetochore gene misexpression predicts cancer patient survival and response to radiotherapy and chemotherapy. Nat. Commun. 7, 12619 (2016).
Silk, A.D. et al. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc. Natl. Acad. Sci. USA 110, E4134–E4141 (2013).
Zasadil, L.M. et al. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci. Transl. Med. 6, 229ra43 (2014).
Janssen, A., Kops, G.J. & Medema, R.H. Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc. Natl. Acad. Sci. USA 106, 19108–19113 (2009).
Burrell, R.A. et al. Targeting chromosomal instability and tumour heterogeneity in HER2-positive breast cancer. J. Cell. Biochem. 111, 782–790 (2010).
Tempest, H.G. et al. Sperm aneuploidy frequencies analysed before and after chemotherapy in testicular cancer and Hodgkin's lymphoma patients. Hum. Reprod. 23, 251–258 (2008).
Khan, F., Sherwani, A.F. & Afzal, M. Analysis of genotoxic damage induced by dacarbazine: an in vitro study. Toxin Rev. 29, 130–136 (2010).
Ben-David, U. et al. Aneuploidy induces profound changes in gene expression, proliferation and tumorigenicity of human pluripotent stem cells. Nat. Commun. 5, 4825 (2014).
Cai, Y. et al. Loss of chromosome 8p governs tumor progression and drug response by altering lipid metabolism. Cancer Cell 29, 751–766 (2016).
Chen, G. et al. Targeting the adaptability of heterogeneous aneuploids. Cell 160, 771–784 (2015).
Barretina, J. et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012).
Rees, M.G. et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 12, 109–116 (2016).
Tsherniak, A. et al. Defining a cancer dependency map. Cell 170, 564–576.e16 (2017).
Brastianos, P.K. et al. Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov. 5, 1164–1177 (2015).
Liu, W. et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat. Med. 15, 559–565 (2009).
Carter, S.L. et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 30, 413–421 (2012).
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Ben-Porath, I. et al. An embryonic stem cell–like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 40, 499–507 (2008).
Roschke, A.V. et al. Karyotypic complexity of the NCI-60 drug-screening panel. Cancer Res. 63, 8634–8647 (2003).
Ritchie, M.E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Larson, J.L. & Owen, A.B. Moment based gene set tests. BMC Bioinformatics 16, 132 (2015).
Bi, W.L. et al. Genomic landscape of high-grade meningiomas. NPJ Genom. Med. 2, 15 (2017).
Acknowledgements
We thank L. Franke for assistance with functional genomic mRNA profiling; A. Bass, K. Ligon, A.J. Aguirre and J. Lorch for providing the clinical samples for cell line derivation; A. Tubelli for assistance with figure preparation; M. Meyerson, A.J. Cherniack, A. Taylor, A. Pearson and Z. Tothova for helpful discussions; and W.J. Gibson for copy number data. U.B.-D. is supported by a Human Frontiers Science Program postdoctoral fellowship, R.B. received support from the US National Institutes of Health (R01 CA188228) and the Gray Matters Brain Cancer Foundation, and T.R.G. received support from the Howard Hughes Medical Institute.
Author information
Authors and Affiliations
Contributions
U.B.-D. conceived the project, collected the data and carried out the analyses. G.H., N.F.G. and J.M.M. assisted with computational analyses. Y.-Y.T. and J.S.B. provided cell line data. C.O. assisted with the copy number analysis of cell lines. B.W. assisted with figure design and preparation. J.S. assisted with the copy number analysis of TCGA samples. R.B. and T.R.G. directed the project. U.B.-D., R.B. and T.R.G. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–14, Supplementary Tables 1–6 and Supplementary Note (PDF 4534 kb)
Supplementary Data 1
CNA profiles of PDX samples. (XLSX 26968 kb)
Supplementary Data 2
Model-acquired CNAs in PDX samples. (XLSX 1425 kb)
Supplementary Data 3
CNA profiles of CLDX samples. (XLSX 15919 kb)
Supplementary Data 4
Model-acquired CNAs in CLDX samples. (XLSX 791 kb)
Rights and permissions
About this article

Cite this article
Ben-David, U., Ha, G., Tseng, YY. et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat Genet 49, 1567–1575 (2017). https://doi.org/10.1038/ng.3967
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3967
This article is cited by
-
Patient-derived organoids in human cancer: a platform for fundamental research and precision medicine
Molecular Biomedicine (2024)
-
Machine-learning analysis reveals an important role for negative selection in shaping cancer aneuploidy landscapes
Genome Biology (2024)
-
Patient derived tumoroids of high grade neuroendocrine neoplasms for more personalized therapies
npj Precision Oncology (2024)
-
Personalizing non-small cell lung cancer treatment through patient-derived xenograft models: preclinical and clinical factors for consideration
Clinical and Translational Oncology (2024)
-
Establishment and characterization of novel high mucus-producing lung tumoroids derived from a patient with pulmonary solid adenocarcinoma
Human Cell (2024)
