
Abstract
Silver birch (Betula pendula) is a pioneer boreal tree that can be induced to flower within 1 year. Its rapid life cycle, small (440-Mb) genome, and advanced germplasm resources make birch an attractive model for forest biotechnology. We assembled and chromosomally anchored the nuclear genome of an inbred B. pendula individual. Gene duplicates from the paleohexaploid event were enriched for transcriptional regulation, whereas tandem duplicates were overrepresented by environmental responses. Population resequencing of 80 individuals showed effective population size crashes at major points of climatic upheaval. Selective sweeps were enriched among polyploid duplicates encoding key developmental and physiological triggering functions, suggesting that local adaptation has tuned the timing of and cross-talk between fundamental plant processes. Variation around the tightly-linked light response genes PHYC and FRS10 correlated with latitude and longitude and temperature, and with precipitation for PHYC. Similar associations characterized the growth-promoting cytokinin response regulator ARR1, and the wood development genes KAK and MED5A.
Similar content being viewed by others

Main
Forest ecosystems maintain a large share of Northern Hemisphere biodiversity. Boreal forests comprise roughly 30% of global forest area1 and contain the highest tree density among climate zones2. Moreover, boreal regions are undergoing extensive climate change. Annual temperature increases exceeding 1.5 °C are projected to result in a warming of 4–11 °C by the end of this century, with little concomitant increase in precipitation1. At this pace, climate zones will shift northward at a greater speed than trees can migrate3. To understand how future populations of forest trees may respond to climate change, it is essential to uncover past and present signatures of molecular adaptation in their genomes. Silver birch, B. pendula, is a pioneer species in boreal forests of Eurasia. Flowering of the species can be artificially accelerated4, giving it an advantage over other tree species with published genome sequences (such as poplar5, spruce6, and pine7) for the optimization of fiber and biomass production.
Here we sequenced 150 birch individuals and assembled a B. pendula reference genome from a fourth-generation inbred line, resulting in a high-quality assembly of 435 Mb that was linked to chromosomes using a dense genetic map. We analyzed SNPs in the genomes of 80 birch individuals spanning most of the geographic range of B. pendula, as well as seven other members of Betulaceae. Population genomic analyses of these data provide insights into the deep-time evolution of the birch family and on recent natural selection acting on silver birch.
Results
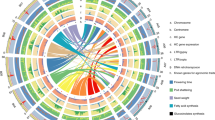
Genome assembly and gene duplication history
We constructed a hybrid nuclear genome assembly starting from 9× sequence coverage with Roche 454 technology. Assembled contigs were polished with Illumina paired-end data and connected and ordered using mate-pair libraries sequenced on both Illumina and SOLiD platforms, followed by further scaffolding and gap filling with 30× coverage of PacBio reads longer than 6,000 bp. This resulted in a first assembly consisting of 5,642 scaffolds with an N50 of 240 kb (Supplementary Figs. 1 and 2 and Supplementary Tables 1 and 2). Further scaffolding with additional PacBio reads resulted in 3,474 (super)scaffolds with N50 value of 527.7 kb. A total of 391 Mb of scaffolds (89% of the estimated 440-Mb genome) was assembled into 14 chromosomal linkage groups via an ultra-high-density genomic linkage map consisting of 3.4 million markers (Fig. 1 and Supplementary Note). In addition to the nuclear genome, organellar genomes were assembled and annotated (Supplementary Note and Supplementary Figs. 3–6). Evidence for birch gene models was obtained by sequencing EST libraries from 12 different birch tissues or growth conditions, providing 18,951 transcripts with an average length of 1,683 bp, and by carrying out de novo assembly of RNA-seq reads, yielding 16,906 transcripts (Supplementary Figs. 7 and 8, Supplementary Tables 3 and 4, and Supplementary Note). We annotated the nuclear genome in two stages. After initial automated gene prediction, 3,192 genes were manually annotated and used to train gene predictors for a second round, yielding 28,153 high-quality gene models (Supplementary Figs. 9–11, Supplementary Tables 5–7, and Supplementary Note), of which 17,746 were supported by nearly full-length transcriptomic evidence.

Center, syntenic links between gene duplicates and triplicates dating from the hexaploid event. Ancient triplicate links still preserved in the modern genome are shown in red. Bpe_Chr, B. pendula chromosome.
Transposable elements (TEs) constituted 49.23%, and retrotransposons 30.60%, of the genome (Supplementary Note and Supplementary Table 8). Superfamily Gypsy and Copia retrotransposons were less common (8.5% and 2.3%, respectively), and contained fewer young (<50,000 years) elements than other plant genomes of similar size, whereas the nonautonomous TRIM group of retrotransposons was significantly more abundant8, at 6.4% versus 1.26% (at most, in Pyrus, pear; P < 2.2 ×10−16, Grubbs test for one outlier). This suggests that the parasitic life cycle of TRIMs may attenuate replication of autonomous retrotransposons in B. pendula, thus limiting their contribution to genome size.
Syntenic alignment of the B. pendula genome with other eudicots, including grapevine (Supplementary Figs. 12 and 13 and Supplementary Table 9), demonstrated that the birch genome has not undergone whole-genome duplications (WGDs) subsequent to divergence from these species (Supplementary Note). As such, the only internally duplicated blocks in the B. pendula genome date from the ancient gamma hexaploidy event at the base of core eudicots9,10.
Gene duplication and divergence is a major source of functional novelty in eukaryotic genomes, and in plants both polyploidy and tandem duplications have been implicated in the evolution of phenotypic novelty11,12. Using self–self syntenic analysis, we separated the duplicated portion of the B. pendula genome into two pools: duplicate genes deriving from the ancient hexaploidy event, and those stemming from ongoing tandem (segmental) duplications (Supplementary Note). Gene ontology (GO) functional enrichment analysis (Supplementary Tables 10,11,12) revealed that transcription factors (TFs) were strongly overrepresented among polyploid duplicates (Supplementary Table 12), which corresponds with theoretical and empirical results indicating biased retention of highly interconnected genes following the duplication of entire functional modules13,14. This result appeared to hold for adaptive genome evolution in eudicots in general and after independent WGDs, as we obtained highly similar functional enrichments in corresponding analyses of the Arabidopsis thaliana and poplar genomes15 (Supplementary Table 12), which have experienced their own lineage-specific WGDs5,16 since diverging from a common ancestor with birch. In contrast to biased retention of modular TF function after WGDs, tandemly duplicated genes in these three species15 were enriched for environmental responses and secondary metabolism, which, although distinctive by species, were also significantly overlapping (P < 2.2 × 10−16, Fisher's test; Supplementary Note, Supplementary Fig. 14, and Supplementary Tables 11 and 12). Tandemly expanded gene families shared by all three species were enriched for secondary metabolism, bacterial defense, hormonal response, and hormone and nutrient transport. Adaptations possibly related to the arborescent habit were visible in convergent tandem expansions shared by B. pendula and Populus trichocarpa, including genes associated with fungal pathogen defense, cell wall biogenesis, and cellulose synthase activity (Supplementary Tables 11 and 12). Through evolutionary information stored within single genomes, these results suggest that whereas polyploid duplicates tend to diversify core processes in developmental and physiological regulation, tandem duplicates enhance the diversity of a plant's environmental response capacity, which is in concurrence with previous studies15,17,18.
Population-level signatures of interspecies admixture
To examine recent B. pendula adaptation and to place the species into perspective within its parent clade, we sequenced the genomes of five other diploid birch species (Betula nana, Betula platyphylla, Betula populifolia, Betula occidentalis, and Betula lenta), the tetraploid birch Betula pubescens, two alder species from the related genus Alnus (Alnus incana and Alnus glutinosa), and B. pendula individuals originating from 12 populations native to Ireland, Norway, Finland, and Russia (Fig. 2a and Supplementary Tables 13 and 14). Additionally, eight ornamental varieties of B. pendula were included to scan for candidate gene mutations. SNPs were called using GATK19 for this collection of 89 individuals (Supplementary Table 15). Principal component analysis (PCA) of fourfold degenerate neutrally evolving SNPs demonstrated that whereas most B. pendula individuals formed a single linear cline along PCA axis 2, another set consisting of eight B. pendula individuals was separated by PCA axis 1, following a trajectory suggestive of admixture from other birch species (Fig. 2b and Supplementary Fig. 15). Flow cytometric analysis of the latter set showed ploidy levels of four for two individuals where cambium material was available, and all atypical individuals showed high levels of heterozygosity (Supplementary Figs. 16 and 17), suggesting the possibility of novel polyploidies or admixture events with parents of polyploid origin.

(a) Dispersion of 80 silver birches sampled from 12 sites across most of the geographic range of B. pendula. Populations are plotted with dots color-coded based on dispersion by latitude and longitude. ADMIXTURE analysis of SNP variation (superimposed with bar plots; center line, median; box, interquartile range; whiskers, 1.5× interquartile range; points, outliers) shows that Finland is a mixing zone between European (blue) and Asian (green) source populations. Samples from Ireland were highly admixed with other birch species and/or polyploid and were removed from the analysis. Source: OpenStreetMap contributors. (b) PCA shows clear separation between B. pendula populations and other sampled birch species (open circles). Eight B. pendula individuals (gray shading) were putative polyploids or interspecies admixed individuals. These included all Irish individuals and two individuals from Punkaharju, Finland. The main B. pendula population formed a cline along PC2 (purple shading). Fin: Finland, Sco: Scotland.
Hybridization among birch species is well studied20,21. Explicit allele-sharing analyses with three-population F3 tests22 demonstrated traces of interspecific admixture within the main B. pendula population, suggesting that gene flow is ongoing and possibly bidirectional (Fig. 3 and Supplementary Table 16). Some B. pendula individuals appeared to be highly admixed, including a few individuals from Punkaharju, Finland, where the main population was not admixed (Fig. 3c). The diploid species B. occidentalis and B. populifolia displayed strong signatures of introgression from other species, including B. pendula (Fig. 3c and Supplementary Table 16). A phylogeny of diploid birches and alders estimated from SNP data (Fig. 3a) placed B. lenta at the first split within the Betula genus, as suggested previously23, and supported by limited ribosomal DNA (rDNA) internal transcribed spacer (ITS) data24. Notably, when SNP called as a diploid, the tetraploid B. pubescens was placed in the same clade as B. lenta. Further analysis with three-population tests showed high levels of allele sharing between these two species (Supplementary Table 16), suggesting that a species closely related to B. lenta may be one of the diploid ancestors of B. pubescens. In an ITS-based phylogeny24, B. pubescens was placed together with B. pendula, which suggests that it could be the second ancestor of the apparently allotetraploid B. pubescens.

(a) A phylogeny for Betula species, estimated from SNPs in neutrally evolving sites and noncoding sites 2 kb upstream and downstream of genes. Fin, Finland; Sco, Scotland. (b) PCA plot of 60 B. pendula individuals with low levels of admixture and known location information. Individuals diverging along PC2 are all from Punkaharju, Finland, indicating possible admixture from unknown source populations. (c) Heat maps for three-population F3 test statistics. Introgression is significant if the false discovery rate–adjusted Z-score (Z.adj; see Supplementary Note) is significantly negative (adjusted Z-score < − 1.96, purple).
For further analysis within B. pendula, we excluded the outlying individuals to reduce the confounding effects of interspecies admixture and polyploidy; this necessitated the removal of all Irish samples. Analysis using ADMIXTURE showed very weak population structure with a split into two ancestral populations, roughly divided into eastern and western groups (Fig. 2a and Supplementary Fig. 15), with some gene flow occurring between them, in Finland (Fig. 3c and Supplementary Table 16). This probably reflects allopatric division during the last Ice Age, followed by subsequent admixture when the two populations rejoined after ice-sheet retreat, as has been suggested on the basis of chloroplast DNA evidence25. The small number of ancestral populations is probably due to the high degree of interbreeding within birch populations; as a wind-pollinated species, birch pollen can spread more than 1,000 km26.
With the inclusion of ornamental cultivars, we sought to discover mutations in candidate genes that may account for their horticulturally interesting phenotypes. Among these were B. pendula 'Youngii', a weeping birch with a pendulous growth habit for which an in-frame stop codon was found in the birch AtLAZY1 ortholog (Supplementary Note and Supplementary Fig. 18). LAZY1 is a member of the IGT protein family27 and regulates tiller orientation in rice and maize as well as inflorescence branch angle in Arabidopsis28,29,30. It is thought to influence gravitropism through regulation of auxin transport and signaling28,29,30. Lateral organs in maize lazy1 mutants fail to grow vertically, giving rise to a phenotype similar to that observed in 'Youngii'. The stop codon in the birch LAZY1 ortholog could thus explain its weeping phenotype.
Population history shows ancient bottlenecks
For analyses of B. pendula effective population size (Ne) over time, we removed ornamental varieties, which do not have clear origin records, narrowing the analysis to 60 individuals. Within this set, linkage disequilibrium decay was slower (Supplementary Note and Supplementary Fig. 19), and nucleotide diversity (estimated to be 0.0088, Supplementary Table 17) was roughly 30% lower than in Populus tremula and Populus tremuloides31. The ancestral alleles for Betulaceae were reconstructed using the B. pendula reference genome and eight diploid Betula and Alnus species by estimating a phylogenetic tree and resolving ancestral states at nodes (Supplementary Note). The reconstruction was used to estimate the site frequency spectrum for the 60 B. pendula genomes using ANGSD32 and to generate a stairway plot33 elucidating Ne history over time (Fig. 4, Supplementary Fig. 20, and Supplementary Note). With a mutation rate estimate of 1 × 10−9 mutations per generation34 and a generation time of 20 years (Supplementary Note), the stairway plot revealed population bottlenecks over deep time that correspond roughly with known events of environmental upheaval (Fig. 4). An early Ne drop coincident with the great extinction event at the Cretaceous–Paleogene (K–Pg) boundary was clearly visible, followed by a rapid population expansion. Later Ne bottlenecks appeared during Eocene–Oligocene, mid-Miocene, and Pleistocene periods that correspond with other well-known episodes of environmental change35,36. The inferred history is largely supported by fossil evidence for Betulaceae23. Notably, the Ne dips we observed could be associated with cladogenetic events during Betulaceae history, as alder–birch speciation occurred soon after the K–Pg event, 60 million years ago (Mya), and the mid-Miocene event ∼14 Mya may have included the white-barked birches, for which there is fossil evidence from the late Miocene, 10 Mya23. In contrast, we observed no Ne bottlenecks during Holocene population history, for which a highly negative Tajima's D of −1.82 would imply ongoing population expansion. Taken together, the B. pendula reference genome, resequenced individuals, and additional resequenced species provide an overview of the population genomic history for the entire Betulaceae clade over the past 65 million years.

Stairway plot showing that the B. pendula population has undergone bottlenecks during four known periods of major climate upheaval: the K–Pg (green) the Eocene-Oligocene (E–Og; blue), the mid-Miocene (M; red), and the Pleistocene (Ple; purple). Data are median estimate from 200 bootstrap replicates (black line) and 95% confidence intervals (shading). Tick marks along the x axis show estimates for the Matuyama–Brunhes (0.77 Mya), Calabrian (1.8 Mya), and Gelasian (2.58 Mya) borders; the mid-Miocene disruption (14.5–14.8 Mya), and the E–Og (34 Mya) and K–Pg (66 Mya) events.
Selective sweeps reveal coordinated local adaptation
We analyzed the same population of 60 resequenced silver birch individuals for selective sweeps (Supplementary Note), where natural selection acting on a locus sweeps away variation across a genomic region surrounding the locus. Annotation of genes overlapping the sweep region or 2-kb flanking regions on either side indicated that some positive hits were probably artifacts resulting from recent insertions of chloroplast, mitochondrial, or TE DNA. Owing to their haploid nature at insertion, these horizontal transfers probably simulated homozygosity patterns reflective of selective sweeps. In total, 108 genes near organellar or TE inserts were excluded, resulting in a final collection of 913 genes at or around which selection may have swept variation (Supplementary Table 18). This set was enriched for nontandem duplicates and single-copy genes. Tandemly duplicated genes did not show significant enrichment, suggesting that selection among tandemly expanded genes acts through a different process. Regarding the age of the genes under selective sweeps, old syntenic orthologs were enriched for sweeps (P = 0.0018, Fisher's test) whereas young birch-specific genes were significantly depleted of them (P < 2.2 × 10−16). Additionally, birch-specific nontandem genes were depleted of sweeps (P = 3.352 × 10−15), excluding a possible confounding influence from tandem expansions. These results suggest that recent selective sweeps acted mostly on anciently diverged regulatory components.
Although GO categorization has known pitfalls, it provides one of the best means to objectively characterize gene sets37. Exploratory functional enrichment analysis of genes in the sweep regions revealed three significantly enriched GO categories: transmembrane receptor protein tyrosine kinase signaling pathway (P = 1.24 × 10−5, Fisher's test, Bonferroni adjusted); peptidyl-histidine phosphorylation P = 3.91 × 10−5; and longitudinal axis specification (P = 0.00212). These highlight known functions from model systems related to wood and fiber development, light sensing, embryogenesis, and reproductive isolation. The first GO category includes 23 genes influenced by selective sweeps (Supplementary Table 19), most of which are phylogenetically verified homologs of Arabidopsis genes encoding functionally characterized CLAVATA1-like receptor-like kinases (RLKs), including BAM3, PXC2, PXC3, MOL1, MIK1, and MDIS1. In Arabidopsis, BAM3 controls leaf shape, size, and symmetry, as well as protophloem development38. The PXC genes are known to be involved in secondary cell wall formation in developing wood39. MORE LATERAL GROWTH (MOL1) is involved in cambium homeostasis, normally repressing secondary growth40. MDIS1-INTERACTING RECEPTOR LIKE KINASE1 (MIK1) is related to the PXC genes and also has a role in stem vascular development. MDIS1 forms a receptor complex with MIK1 and MIK2 that mediates the male perception of female chemoattractant LURE1 during fertilization in Arabidopsis41 and contributes to reproductive isolation between species. Transformation of AtMDIS1 to Capsella rubella partially broke down the interspecific reproductive isolation barrier41. Natural selection affecting birch MDIS1 therefore could suggest possible involvement in determination of reproductive barriers between different birch species.
The second GO category highlighted by our exploratory analysis, peptidyl-histidine phosphorylation, includes nine phylogenetically verified orthologs of the phytochrome genes PHYA, PHYB, and PHYC, and genes encoding histidine kinases such as cytokinin receptors AHK2, AHK3, and AHK4 (CRE1), osmosensor AHK1, and ethylene receptors ERS1 and ETR2. The phytochromes are major mediators of red and far-red light responses that have vital roles in plant growth and reproduction42. The cytokinin and ethylene receptors control many key aspects of plant physiology and development, including acclimation to abiotic stress, shoot and root vascular development, flowering time, and longevity43,44.
The third GO category with putatively important functional enrichment, longitudinal axis specification, includes six phylogenetically verified genes including orthologs of Arabidopsis MONOPTEROS and GNL1, two homologs of TOADSTOOL 2, and two homologs of WRKY2. MONOPTEROS and GNL1 operate in embryo and vascular development45,46; TOADSTOOL 2 operates in meristem maintenance47, and the Arabidopsis WRKY2 protein acts in zygote polarization in embryo development48. Additionally, WRKY2 has a role in pollen development49 and growth arrest induced by ABA during seed germination50.
Candidate gene adaptation correlates with environment
To assess whether selective sweeps were confounded by population structure, we performed redundancy analysis (RDA)51 on SNPs in the putative sweep regions by comparing the PCA eigenvectors from their SNP variation to overall population structure from PCA of whole-genome SNPs (Fig. 5 and Supplementary Note). In total, 423 of the 913 genes had a statistically significant (Benjamini-Hochberg adjusted P < 0.05 from permutation test) proportion between— 9% and 100%— of their allelic variation explainable by overall B. pendula population structure (Supplementary Table 18), suggesting that these particular sweeps are at least partially confounded with drift processes. After controlling for population structure, we identified genes showing clinal variation associated with general environmental variables such as temperature and precipitation. These restricting criteria resulted in a small subset of six genes with significant associations and intriguing molecular functions. These genes were verified by phylogenetic analysis as orthologs of the A. thaliana genes SWEETIE, KAKTUS (KAK), ARABIDOPSIS RESPONSE REGULATOR 1 (ARR1), MED5A (encoding Mediator complex protein MED5A, also known as MED33A), PHYTOCHROME C (PHYC), and FAR1-RELATED SEQUENCE 10 (FRS10).

(a) Putative sweep regions (yellow) around PHYC and FRS10 were obtained by accumulating and filtering composite likelihood ratio (CLR) statistics (red). Gene models are shown for each gene (purple), with the corresponding mRNA region highlighted in gray (or blue for genes under selection). (b) PCA plot based on SNPs 2 kb upstream and downstream of the sweep related to PHYC. (Sampling locations are color-coded as in Fig. 2). (c) RDA plot of the PHYC SNP region. Axis RDA1 is the direction that best correlates with principal components of maximum temperature (Tmax). In this case, Tmax explains 26.3% of the variation in the SNP data; the remaining variation is explained by PCA, where PC1 explains 50.5% (y axis). Inset, differences in yearly Tmax between Yakutsk and Krasnoyarsk, the locations most separated along RDA1.
SWEETIE encodes a protein that may have a central role in sugar homeostasis52. In Arabidopsis, the sweetie mutant shows stunted growth, early senescence, flower sterility, and increased sugar levels. In particular, the mutant has high levels of trehalose, a metabolite associated with signaling in plant interactions with microbes and herbivorous insects, and in responses to cold and salinity. Additionally, sweetie shows altered expression for late embryogenesis-abundant (LEA) genes and many DREB2-type TFs. LEAs are anti-aggregation proteins that together with trehalose protect plant cells during the dehydration typical of abiotic stresses such as cold and drought53, whereas DREB2A and DREB2B are key transcription factors regulating responses to dehydration and high-salinity stresses54. In birch, DREB TFs have been associated with cold acclimation and winter hardiness55. These connections may relate to the strong correlation silver birch SWEETIE shows with environmental variables.
KAK was identified originally as an endoreduplication repressor in Arabidopsis trichomes. However, the kak1 mutant shows increased C-values in etiolated hypocotyls completely lacking trichomes, suggesting a broader role in the control of endoreduplication56. KAK is also expressed in cambium, a secondary meristem that gives rise to both phloem and secondary xylem, where the gene has been suggested to have a role in defining the balance between xylem and phloem formation during vascular development57. In leaves, endoreduplication is associated with an increase in cell size and rapid growth, and also higher stress tolerance58,59. If KAK is indeed a general regulator of endoreduplication, its correlation with temperature may be of adaptive significance to silver birch.
Together with cellulose and hemicellulose, lignin is an essential component of the secondary cell walls in structural fibers and water-conducting cells, determining their strength and rigidity. Lignin also interferes with the separation and breakdown of cellulose, hindering pulp and paper production and limiting the use of biomass crops for biofuel production. Attempts to reduce lignin production through genetic manipulation have so far resulted in plants with stunted growth and reduced yields60. MED5A was recently associated with lignin formation; the med5a mutation rescued the stunted growth, collapsed xylem vessels, and lignin deficiency phenotypes in the Arabidopsis phenylpropanoid pathway mutant ref8-1 (ref. 61). The double mutant med5a ref8-1 showed alleviated phenotypes, but cell wall properties were not restored to wild-type composition. Birch MED5A appears to be under positive selection, as several amino acid positions were significant by Bayes empirical Bayes positive selection analysis (Supplementary Note and Supplementary Table 20), its Tajima's D value was in the lower 10% quantile for the B. pendula genome, and its polymorphism correlated with latitude–longitude as well as temperature (Supplementary Table 18). A second component of the same mediator complex that appears among the putatively swept genes is MED12, which is involved in flowering time regulation in Arabidopsis62.
Cytokinin signaling is of pivotal importance for plant vascular development63,64; it is a major positive regulator of cambium activity and controls wood formation in the tree trunk65. We identified in our sweep list the birch ortholog of Arabidopsis transcription factor ARR1, a key regulator of Arabidopsis root meristem size66 that mediates the balance between cell division and differentiation by integrating auxin and cytokinin responses. ARR1 is involved in cold-induced inhibition of root growth and reduced auxin accumulation67, and it also controls Arabidopsis drought susceptibility68. This may explain the link to the geographic and temperature variables detected here. Variation in cytokinin signaling appears to have a large role in local adaption in silver birch, as the set of putatively swept genes also includes AHK2, AHK3, and AHK4, and their GO category (peptidyl-histidine phosphorylation) was enriched, as described above.
Finally, the orthologs of PHYC and FRS10 are closely linked in the silver birch genome (Fig. 5), which is also the case in the grapevine and tomato genomes (Supplementary Fig. 21). PHYC and FRS10 act in red and far-red light sensing, shade avoidance, canopy density, temperature-dependent adaptation, and flowering time regulation69. PHYC was recently connected to temperature-specific regulation of the circadian clock70, and in Arabidopsis it is strongly linked to altitudinal71 and latitudinal–longitudinal70,72,73 clines in flowering time. In addition to PHYC, FRIGIDA and FLC explain a large proportion of flowering time variation in Arabidopsis71. Birch homologs of their Arabidopsis regulators FES, TXR7, and VEL1 were included in the selective sweep gene set. Because bud burst and initiation of flowering depend mainly on (night) temperature74, the function of PHYC in birch may be related to the photoperiodic control of inflorescence initiation in the autumn, growth cessation, development of cold tolerance, and induction of senescence. PHYA and PHYB, encoding the other two main phytochromes, were also identified among the putatively swept genes, emphasizing the importance of light sensing for tree adaptation to varying environments. Phenotypic correlates of clinal variation in these and other genes remain obscure but are certainly worthy of more targeted analyses.
Discussion
Using the B. pendula reference genome and resequenced individuals spanning the geographic range of silver birch, we were able to characterize genomic adaptations at several levels. First, we detected enrichments of TF functions that date to the core-eudicot crown radiation. Second, we uncovered a suite of gene duplicates involved in environmental responses that were not polyploidy derived but instead stem from ongoing tandem duplication processes. Such duplicates are generated by the same mechanisms as copy number variants (CNVs), which have come under intense recent study (particularly in animal genomes) as adaptive “tuning knobs” at the inter-population level75. In the case of silver birch, the tandem duplicates we observed might be taken as a sort of 'species average' that reflects former CNVs fixed by selection and neo- or subfunctionalization.
After several bottlenecks at well-known times of global environmental change, the effective population size of silver birch has increased over the past 1 million years. As expected for a wind-pollinated species with high pollen dispersal, population structure across the species range was particularly weak, which should greatly facilitate future GWAS efforts. Although we found evidence of ongoing gene flow between birch species, they were still clearly separated, and even if hybridization and introgression occurred, it did not blur their genetic distinctiveness. This contrasts with birch cytoplasmic markers, where evidence for allele sharing is common and species limits weak. Similar discordance between nuclear and cytoplasmic markers has been observed in other plant species76.
To identify recent selection in silver birch, we analyzed selective sweeps at the intraspecific level. These acted mostly on genes dating from the ancient gamma hexaploidy event. Many of the genes located in the sweep regions were regulators and receptors that hold key positions in triggering developmental or physiological chains of events, suggesting that selection has acted during birch speciation by tuning the timing and cross-talk between different processes. However, tandemly duplicated genes were not enriched for sweeps, and recently duplicated birch-specific genes were significantly depleted of sweeps. Altogether, these findings suggest an 'exploration–exploitation' model for tandem duplicates in species evolution. Exploration would occur through generation of novel tandem CNVs within populations; with lineage splitting and lowering of Ne, polymorphic tandems may become fixed by drift. Another alternative is fixation by selection through a process analogous to soft sweeps. As often reported in mammalian genome analyses77, multiple alleles at a locus can be swept to fixation in a 'soft' event that evades detection by ordinary criteria. In our example, tandem CNVs among individuals would comprise the alternative (exploratory) 'allelic' states, perhaps maintained in populations by balancing selection75. Multiple CNV states could then be simultaneously selected for when opportunistic and rapid (i.e., exploitative) responses to environmental change are required. Strong ('hard') selective sweep patterns, in contrast, involve selection for a single allelic state and may be more likely among core regulatory components that coordinate developmental timing and physiological cross-talk. Here, particularly when intertwined with population bottlenecks engendered by environmental upheaval, perhaps only genotypes that are unique and have proper timing of responses can be exploited.
We further uncovered candidate genes that show selection associated with environmental responses and that are enriched for functions of practical relevance for forest biotechnology. Notably, several key components of cytokinin signaling, a major positive regulator of vascular cambium activity and radial tree-trunk growth65,78, show evidence of recent natural selection. Other examples are KAK and MED5A, which can elicit pleiotropic growth and cell wall phenotypes in Arabidopsis. If orthologs of these genes function similarly in birch, this information could used for selective engineering of forest trees for rapid generation of biomass. Similarly, natural variation in photoperiod regulation might be used to understand and alter cambial activity–dormancy cycling and wood production.
Methods
Sequencing and genome assembly.
For more detailed descriptions of the methods, see the Supplementary Note. The individual for the reference genome was a fourth-generation inbred line from the inbred collection of the Natural Resources Institute Finland. It originated from a seed collected in 1967 from an open-pollinated natural stand in Jäppilä, central Finland. Eleven microsatellite markers were used to assess the success of inbreeding. Several 454 libraries were constructed from leaf or cambium tissue for the initial single-end sequencing. Genomic DNA (5 μg) was sonicated on a Covaris S2. End repair, A-tailing, and adaptor ligation were done according to the manufacturer's protocol. Libraries were amplified using KAPA HiFi DNA Polymerase or Phusion HiFi DNA Polymerase. The amplified libraries were purified using AMPure XP beads and size-selected using carboxy-beads on a Magnatrix 1200 robot. Libraries were sequenced on a Genome Sequencer FLX and FLX+ platforms. Libraries for Illumina paired-end sequencing were constructed using 1 μg of starting material sonicated on a Covaris S2. After end repair, A-tailing, and ligation of Y-adaptors according to the manufacturer's instructions, PCR was performed using Phusion HiFi DNA Polymerase. PCR products were purified with AMPure XP beads and size selected. Insert libraries of 350 bp and 500 bp were generated and sequenced on HiScan SQ (100 + 100 bp) and MiSeq (250 + 250 bp) sequencers. For pooled sequencing, barcoded libraries (61 + 2) were prepared and pooled, size selected to inserts of 500 bp, and sequenced with a NextSeq 500 (150 + 150 bp). A SOLiD Mate-Pair kit was used to construct four different sized mate-pair libraries. For the 2-kb and 3-kb mate-pair libraries, Illumina Y-adaptors were ligated to the captured fragments and sequenced on a HiScan SQ Sequencer. For 4-kb and 5-kb mate-pair libraries, SOLiD adaptors were ligated and sequenced on a SOLiD 5500xl Sequencer. Library construction for PacBio sequencing was carried out using the manufacturer's protocols. Genomic DNA was sheared using a Megaruptor, followed by damage repair, end repair, hairpin ligation, and size selection using BluePippin. After primer annealing and polymerase binding, the DNA templates were sequenced on a PacBio RSII sequencer using P4/C2 chemistry and 120-min video time and later using P6/C4 chemistry and 360-min movie time. Contigs were assembled from 454 sequencing data using Newbler with large genome parameter settings. The accuracy of the contigs was improved using the Pilon software with Illumina paired-end sequences. Scaffolding was done using the Illumina and SOLiD mate-pair libraries and SSPACE 2 in a hierarchical manner; first the 2 kb library was mapped to the contigs, then contigs were scaffolded accordingly. Then the procedure was repeated with 3 kb, 4 kb, and 5 kb libraries. Before mapping and scaffolding, adaptors were removed using Cutadapt, and the reads were trimmed using Sickle with a Phred cutoff of 20 for paired-end data and 28 for mate-pair data. The SOLiD mate-pair libraries were converted to FASTQ sequences using XSQ-Tools. The scaffolds were further improved with PBJelly and ∼30× of PacBio sequence data with reads longer than 6 kb. The scaffolds were ordered into super-scaffolds using SSPACE-LongRead with a second set of PacBio sequencing data with approximately 20× coverage and read lengths of 10–75 kb (Supplementary Fig. 1 and Supplementary Table 1). Organellar genomes were assembled as part of the genomic assembly. Chloroplast contigs were connected using Gap4 and verified with PacBio data (Supplementary Figs. 3 and 4). A separate PacBio assembly was carried out for the mitochondrial genome using a modified Newbler assembler (Supplementary Note and Supplementary Figs. 5 and 6).
Linkage mapping.
For linkage mapping, low-coverage whole-genome sequencing data from 63 individuals were mapped to the genome using BWA-mem, and genotype posterior probabilities were calculated by the Lep-MAP3 pipeline using the output from samtools mpileup. The relatedness of individuals was checked using the IBD module of Lep-MAP3, and then the parental genotypes for each potential marker were estimated, providing the final data set for linkage mapping. Markers segregating identically on each contig were joined and collapsed into linkage groups separately for markers informative only paternally, maternally, or both. For each combination of parental markers, 14 linkage groups were found. The number of linkage groups matched the karyotype of the species (2n = 28), and thus can be considered pseudochromosomes.
Scaffolds were assigned to chromosomes by requiring a region of 4 kb including 8 markers assigned to the same linkage group; markers not fulfilling this criterion were removed. Contigs were assigned to intervals that were then mapped to a chromosome. Finally, two maps were constructed and ordered for each chromosome on the basis of paternally informative or maternally informative markers. The orderings were inspected and corrected manually, including orientation of the two maps in the same direction. Finally, the linkage groups and scaffolds assigned to them were arranged to form 14 pseudochromosomes. During the scaffold placement, super-scaffolds showing signs of possible chimeric assembly were split from the regions between markers and placed in their respective linkage groups.
Assembly validation.
The quality of the assembly was monitored during all stages and using several independent validation methods. Contig assembly was first tested using Scarpa software and the 2-kb Illumina mate-pair library and then validated by mapping a set of high-quality ESTs to the contigs using Exonerate. The completeness of the scaffold-level assembly was tested by mapping the unassembled Illumina reads against the scaffolds and by mapping Trinity de novo assembled RNA-sequencing transcripts to the assembly with PASA, as well as through CEGMA. An independent validation of the scaffold-level assembly quality was gap-filling with long Pacbio reads. The assembly was additionally evaluated with REAPR using paired-end libraries for error calling and 3 kb mate-pair Illumina libraries for estimating the library breakpoints. To assess the quality of the scaffolding, the assembly was also aligned to long, nonoverlapping PacBio-reads using BLASR by inspecting 12 reads of length 70–75 kb that had at least 20,000 bp 'best hit' mapping (Supplementary Fig. 2), with total sequence length of 863 kb. Final tests for assembly quality were carried out using QUAST (Supplementary Table 2).
Repeats in the contigs and pseudochromosomes were analyzed using RepeatModeler and RepeatMasker. For LTR retrotransposons, a library of elements was built by combining the output from Repet, LTRharvest/LTRdigest from genometools, our own custom pipeline, and a library of LTR retrotransposons. For MITEs, MITE-Hunter and RSPB were used. CACTA elements were mined on the basis of structural considerations. RepeatMasker was used to mask TEs using the classified repeat libraries. See Supplementary Table 8 for a summary of the TEs.
Sample preparation.
The tissues for EST libraries (Supplementary Table 3) were frozen in liquid nitrogen and stored at −80 °C until RNA extraction. Total RNA was isolated, and cDNA libraries were constructed with ZAP-cDNA Synthesis Kit and ZAP-cDNA Gigapack III Gold Cloning Kits according to the manufacturer's instructions. For sequencing, individual phages were excised to pBluescript SK plasmids in vivo. The EST clones were grown in colonies in 96-deep-well plates, and aliquots were taken for PCR amplification using universal primers from the excised pBluescript SK plasmids. Following purification of the PCR products, sequencing was performed using T3 primer and BigDye chemistry and analyzed on an ABI 3700 sequencer. Base calling was performed using Phred quality threshold value ≥20. EST libraries were also sequenced using the 454 and MiSeq sequencing platforms (Supplementary Table 4). PCR products from the libraries were pooled, and a 454 library was constructed and run on a FLX+ platform. From the glycerol stocks, PCR was performed with universal primers in 384-well plates in 20-ml volumes. After amplification, 5 μl from each plate was pooled and purified. A MiSeq library was constructed from the pool and sequenced as paired-end (300 bp + 300 bp). In the assembly, MiSeq paired-end reads were first overlapped and combined using FLASH, and then combined with existing Sanger reads from the sequenced EST library, followed by assembly using Newbler (Supplementary Fig. 7).
For the RNA sequencing, 12 genotypes were selected from southern and central parts of Finland. Birches were grown for 1 year and exposed to 150 nL L−1 of O3 for 8 h, and leaves were sampled at 0, 2, 8, 24, 48, and 96 h. Total RNA from leaves was isolated and pooled on the basis of clone tolerance to ozone (sensitive, tolerant, and moderate), and mRNA was isolated using the Dynabeads mRNA Purification Kit. RNA-seq libraries were constructed using a TruSeq stranded mRNA kit. mRNA (5 μl) was converted into cDNA using random hexamers, and the second strand was synthesized using DNA polymerase I and dUTP nucleotides. After purification, ends were repaired, and after A-tailing, Y-adaptors containing indexes from the kit were ligated to the fragments. After PCR amplification, the fragments were purified using AMPure XP, pooled in equal concentrations, and paired-end (100 bp + 100 bp) sequenced on a HiScan SQ sequencer. See Supplementary Table 5 for a summary of the reads. Preprocessing was done with Cutadapt to remove adapter sequences and to trim reads for low-quality sequence, ambiguous sequences, or poly(A) tails, using Phred threshold 33 and minimum length 60 bp. After preprocessing, paired-end information for each sequence was retrieved and orphaned reads were kept as single reads. A de novo transcriptome assembly was carried out using Trinity with 12 different k-mers. Sequences from all k-mer assemblies were pooled and merged using the Amos pipeline. Contigs shorter than 200 bp were discarded. To minimize redundancy, the final assembly was mapped to the ESTs, and fragments identical to particular ESTs were removed. See Supplementary Figure 8 for the assembly pipeline. The preprocessed reads were also aligned to the genome using TopHat, and the resulting BAM files were processed using HTSeq to obtain the final gene set.
Gene annotation.
A modular gene annotation framework was established to estimate gene models by combining evidence from ab initio gene predictors based on Hidden Markov Models, spliced transcript evidence from RNA-seq and EST data, protein evidence from orthologous proteins obtained from closely related and model plant species, and manually curated gene sequences. Gene prediction was carried out in two phases. The first phase (Supplementary Fig. 9) involved generation of an initial set of automated predictions, followed by manual curation of genes from select gene families to provide verified gene models for the second phase of predictions. RNA-seq transcripts generated using Trinity were splice aligned against the unmasked birch genome using PASA in a strand-specific manner to generate long ORFs. For the first phase of the training, these ORFs were directly used for training the ab initio gene predictors and obtaining an initial set of gene models. The EST libraries were splice-aligned against the unmasked birch genome in a similar manner and used as additional evidence. Four Hidden Markov Model–based ab initio gene predictors were used: Augustus, SNAP, GlimmerHMM, and GeneMark-ES. Gene prediction parameters were trained for the Augustus, SNAP, and GlimmerHMM using the nonredundant final training set from PASA-aligned RNA-seq transcripts. For Augustus, optimization of the parameters was carried out by eightfold cross-validation. For SNAP and GlimmerHMM, the parameters were optimized by carrying out one round of training with the training set. GeneMark-ES did not require an explicit training data set. Augustus, SNAP and GlimmerHMM were run on the masked birch genome using the optimized parameters, whereas GeneMark-ES was executed on the unmasked genome. Orthologous protein sequences from A. thaliana, Populus trichocarpa, and Prunus persica were splice-aligned against the unmasked birch genome using exonerate to obtain spliced protein evidence. EVidence Modeler was used to generate a single high-confidence gene model set, using ab initio gene predictions, spliced protein alignments, and spliced transcript alignments as inputs. Finally, UTR addition was carried out by running PASA with the RNA-seq and EST data.
Manual annotation was carried out using the WebApollo software (see Supplementary Table 6) for the list of manually annotated genes. For the second phase of the training, ORF generation using PASA was followed by filtering for complete ORFs and combining with core eukaryotic genes from CEGMA plus manually annotated genes to generate a nonredundant training set for the gene predictors (Supplementary Fig. 10). Gene predictors were trained as in the first phase and by passing the new parameters. Finally, UTR addition using PASA was re-executed. The manually curated genes and automated annotations were compared for overlaps using the overlap software. Annotations not having any overlap with the manual annotations were extracted and merged with the manually curated genes, thereby creating a single nonredundant set of merged annotations (Supplementary Table 6).
Protein functional analysis.
Protein functional analysis was carried out using Interproscan. Descriptions for the gene models were generated using AHRD with a custom database of A. thaliana and SWISS-PROT proteins. Finally, gene model predictions of 200 conserved single-copy genes were verified manually in CoGe by comparing orthologous birch gene models against A. thaliana and Vitis vinifera homologs.
Comparative genomic analyses.
Comparative genomic analyses for B. pendula were carried out against Arabidopsis thaliana, Coffea canephora, Populus trichocarpa, Prunus persica, Solanum lycopersicum, Theobroma cacao, and Vitis vinifera using OrthoMCL for ortholog clustering (Supplementary Tables 7 and 10 and Supplementary Data Set 1). Arabidopsis transcription factor family assignments were used to select the proper settings for the inflation parameter in Markov clustering to balance the precision versus recall rate in orthogroups (F-score, see Supplementary Fig. 11).
Syntenic path alignment (Supplementary Fig. 12) and syntenic depth analysis (Supplementary Table 9 and Supplementary Fig. 13) were carried out in CoGe by aligning the B. pendula pseudochromosomes to the chromosome-level Vitis vinifera genome using default parameters. Self-to-self SynMaps were generated within CoGe using the Quota Align algorithm with default parameters. DAGchainer provided the list of syntenic duplicates (Supplementary Data Set 2). Blast2raw, incorporated into CoGe's SynMap pipeline, was used to calculate tandem duplicates (Supplementary Data Set 2). Overlaps between orthogroups and syntenic and tandem gene sets were tested using Fisher's exact test (Supplementary Fig. 14 and Supplementary Table 11). For GO enrichment analyses, syntenic gene pairs and tandem duplicates for B. pendula, P. trichocarpa and A. thaliana were downloaded from CoGe and used as the foreground subset in GO enrichment analysis with GOATOOLS, using all the genes in each respective genome as background and Bonferroni-adjusted P < 0.05 as threshold for significance (Supplementary Table 12). Gene models from B. pendula and P. trichocarpa were annotated with the highest alignment score matches using tblastx versus Arabidopsis coding sequences with an E-value cutoff of 1 × 10−5. Whole-genome backgrounds for B. pendula and P. trichocarpa were custom generated by selecting the sets of genes, for each respectively, that were annotatable against Arabidopsis genes, accepting the topmost hit as the match (Supplementary Data Set 2).
Population analyses.
For the population analyses, individuals from six natural Finnish populations were sampled from Punkaharju, Loppi, Vehmersalmi, Posio, Rovaniemi, and Kittilä. Additionally, leaf or DNA samples were obtained from various natural sites in Europe and Russia. Individuals of different birch species were obtained from Helsinki Botanical Garden, Alnus glutinosa from Valkeakoski and Alnus incana from Huittinen, Finland. Finally, eight trees representing special horticultural forms were selected (Supplementary Table 13). For sequencing, DNA from leaf, bud or cambium tissue was isolated using the E.Z.N.A. SP Plant DNA Kit (Omega Bio-tek) whereafter Illumina paired-end libraries were constructed. After PCR amplification, the libraries were pooled and size-selected to an average of 500 bp. Paired-end sequencing was performed on a NextSeq 500 sequencer (150 bp + 150 bp). In addition to the resequenced population, the raw reads of a second Betula nana individual from Scotland sequenced earlier was retrieved from the Sequence Read Archive (ERP001867).
After quality control using FastQC, adaptors and low-quality bases were removed from the read ends using Trimmomatic (leading and trailing Phred score <20), and the reads were filtered with a sliding window of size 3, with average Phred score threshold of 15 within the window. Reads < 35 bp were removed, followed by a second round of quality control using FastQC. The trimmed reads were mapped to the unmasked B. pendula genome using Bowtie2 with default parameters. The mapped reads were sorted, and duplicated reads were removed using SAMtools (Supplementary Table 14). HaplotypeCaller from the Genome Analysis Toolkit GATK was used to estimate the general variant calling file for each individual, and then combined by GenotypeGVCFs to a single variant calling file.
Hard filtering of the SNP calls was carried out with Fisher strand bias (FS > 60.0), mapping quality MQ < 40.0, and thresholding by sequencing coverage based on minimum coverage (DP < 100) and maximum coverage (DP > 1,500). The SNPs were annotated with SnpEff (Supplementary Table 15). Linkage disequilibrium analysis was conducted using PLINK (Supplementary Fig. 19). For population analyses this set was further filtered to include only fourfold degenerate neutrally evolving sites.
The neutrally evolving SNPs were used to estimate population structure by principal component analysis in EIGENSOFT. Ancestral population structure was estimated from the same SNP set with ADMIXTURE using ancestral population sizes K = 1...10 and choosing the population with smallest leave-one-out-validation error (Supplementary Fig. 15). For further analysis, sample-wise admixture proportions were averaged into site-wise averages. For analyses of introgression, three-population F3 tests were run for all population configurations using ADMIXTOOLS (Supplementary Table 16) and adjusted for multiple testing using Benjamini-Hochberg correction (Supplementary Note).
Flow cytometry.
Flow cytometry (Supplementary Fig. 16) was performed using a BD LSR II flow cytometer as per instrument manufacturer's instructions. Cambium was isolated from frozen tree branches by free-hand section and chopped with woody plant buffer. The samples were filtered through 40-μm filter and stained with propidium iodide (PI) 50 μg ml−1 simultaneously with RNase at 50 μg ml−1. Greenhouse-grown rice leaf samples were used as reference.
Population data analysis.
A phylogeny for nine representatives of different birch species was estimated using SNPs called with GATK and hard filtering criteria. Only SNPs within 2 kb from coding regions and at neutrally evolving sites within coding regions were used. Phylogeny was estimated using SNPhylo, with SNPs having minor allele frequency >0.1. For ancestral state estimation, Illumina resequencing reads mapped against the birch reference genome were used to call consensus sequences using a combination of samtools, bcftools, and vcfutils. The consensus FASTA files were read into R, and the ancestral states estimated using the Phangorn package with maximum likelihood estimation and the GTR model of molecular evolution, where edge lengths were estimated separately for each contig. Marginal reconstruction of the ancestral character states was carried out using a maximum a posteriori estimate.
ANGSD was run to obtain the site frequency spectrum, heterozygosity (Supplementary Fig. 17) and sitewise estimates of allele frequency statistics following the protocols recommended by the software authors. The analysis was restricted to the set of 60 non-admixed B. pendula individuals with accurate information on their sampling origin. A custom R script was developed to calculate population genetic descriptive statistics for each gene (Supplementary Table 17). To filter out loci with low coverage and low numbers of called SNPs, only regions with >50 called nucleotides were used for computing the gene-wise statistics.
The site frequency spectrum from ANGSD was used to estimate the population history with Stairway plots using 200 bootstrap iterations, and a range of 0.7, 1.0, and 2.0 × 10−9 as the mutation rate, and 10, 20 and 40 years as the generation time (Supplementary Note and Supplementary Fig. 20). Selective sweeps were estimated with SweepFinder2. To prepare the input files for Sweepfinder, ANGSD was used to call allele frequencies for each position, given the ancestral states estimated earlier. Sweepfinder2 was run for each contig having mapping data from all individuals, with a grid size of 200 bp. For processing the composite likelihood ratio (CLR) scores into putative sweep regions, a custom R script was developed to carry out several filtering steps. First, the first three CLR scores in contig ends were set to 0. Then, to smoothe the fluctuations of CLR scores in neighboring sites, the scores were filtered with median filtering using a window size of 5. Then, CLR scores were merged into sweep regions if the neighboring scores exceeded the top 1% of CLR scores (CLR > 26). Regions where only one 200-bp site exceeded this threshold were removed from the analysis. The final score for each sweep region was the sum of CLR scores for the sites in the sweep region. Thereafter, sweep regions were excluded that contained too many nonsequenced positions (more than 10% of Ns in the sweep region ±2-kb flanking region on each side). Genes overlapping the sweep region or 2-kb flanking regions on either side of the sweep region were selected as genes putatively under selection. This gene set was then filtered for regions that may show artifacts of sweeps, such as sequences originating from organellar DNA or transposable element insertions (Supplementary Tables 18 and 19).
For verifying orthologies of genes identified by sweep analysis, B. pendula genes were used as queries for a NCBI local tblastx against the V. vinifera, Arabidopsis Col-0, C. canephora, S. lycopersicum, P. trichocarpa, and B. pendula coding sequence databases downloaded from CoGe, using E-value cutoff 1 × 10−10. The 10 topmost hits from each database were translated and aligned together using MUSCLE. The alignments were reverse translated, and ambiguous regions were removed using Gblocks, with stringency parameters to allow smaller blocks, gap positions within the final blocks, and less strict flanking positions. Phylogenetic analyses based on the alignments were performed using RAxML-HPC with the GTR substitution model (Supplementary Data Set 3).
To analyze whether a sweep could be explained by drift and population structure, a PCA of each of the sweep regions and 2-kb flanking regions was carried out for SNPs called by ANGSD. The principal components of the sweep regions were computed in R using data from ANGSD.
Weather information for each location was extracted from the National Center for Environmental Prediction (NCEP) website. Daily minimum, maximum, and average temperatures and precipitation information were downloaded based on the GPS coordinates of the closest weather station and processed into monthly averages per location. Redundancy analysis (RDA) was used to decouple the population structure from associations with environmental variables by using the first two principal components from overall population structure as covariates. For the climate data, principal component analysis was carried out in a similar manner using the (months × locations) matrix, and the first two principal components were extracted for minimum, maximum, and average temperatures as well as precipitation. The PCs of the climate variables and PCs from the population structure were then used as covariates to model the variation of the dependent variable, in this case the genomic variation around the sweep region and 2-kb flanking windows. The P values for the fits were estimated using a permutation test with 30,000 permutations, followed by adjustment for multiple comparisons using Benjamini-Hochberg correction. The coefficient of determination was reported together with the adjusted P value. Microsynteny analysis was carried out in CoGe (Supplementary Note and Supplementary Fig. 21).
Samtools mpileup was used to call the variant sites in the cultivars. Bcftools and vcfutils from samtools htslib were used to obtain a variant calling file. The variant sites were annotated using snpEff, and loci having a stop codon and a SNP quality score >20 were selected. The mutation in B. pendula 'Youngii' (Supplementary Fig. 18) was verified by genotyping; the product was digested with MboI, which at the mutation site cuts the WT gene but not the mutated version.
For molecular evolutionary analyses, repredictions for dubious gene models were conducted using default settings of AUGUSTUS and the genomic sequences of the previously predicted B. pendula gene models, plus 500–1,000 bp of upstream and downstream genomic sequence. Tests were conducted on a restricted subset of each gene tree (produced as described above), which in most cases amounted to only orthologous genes from the above species, or in some cases, their paralogs as well. We estimated ω (dN/dS) values for each CDS alignment and RAxML (subset) phylogeny using the codeml part of the PAML package. Gaps in the alignment were excluded by PAML. Two types of models were implemented: branch-specific and branch-site. Comparisons of two nested models were performed using a likelihood ratio test to test for the following: asymmetric sequence evolution versus two-ratio model 2, divergent selection (model 3 versus clade model D (K = 3)), and positive selection (model A null (ω2 = 1) versus model A (0 < ω0 < 1). Additionally, Bayes–empirical–Bayes analyses were conducted to estimate individual amino acids that might have evolved under positive selection (Supplementary Table 20).
Data availability.
OpenStreetMap data are available under the Open Database License (http://www.openstreetmap.org/copyright). The genome assembly is available from Ensembl Plants (http://plants.ensembl.org/) and the CoGe comparative genomics platform (https://genomevolution.org/CoGe/GenomeInfo.pl?gid=35079 and https://genomevolution.org/CoGe/GenomeInfo.pl?gid=35080). All sequencing data have been deposited in the European Nucleotide Archive (ENA) under accession code PRJEB14544.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
13 June 2019
"In the version of this article initially published, there was a mistake in the calculation of the nucleotide mutation rate per site per generation: 1 × 10−9 mutations per site per generation was used, whereas 9.5 × 10−9 was correct. This error affects the interpretation of population-size changes over time and their possible correspondence with known geological events, as shown in the original Fig. 4 and supporting discussion in the text, as well as details in the Supplementary Note. Neither the data themselves nor any other results are affected. Figure 4 has been revised accordingly. Images of the original and corrected figure panels are shown in the correction notice."
13 June 2019
"In the version of this article initially published, there was a mistake in the calculation of the nucleotide mutation rate per site per generation: 1 × 10−9 mutations per site per generation was used, whereas 9.5 × 10− was correct. This error affects the interpretation of population-size changes over time and their possible correspondence with known geological events, as shown in the original Fig. 4 and supporting discussion in the text, as well as details in the Supplementary Note. Neither the data themselves nor any other results are affected. Figure 4 has been revised accordingly. Images of the original and corrected figure panels are shown in the correction notice."
References
Gauthier, S., Bernier, P., Kuuluvainen, T., Shvidenko, A.Z. & Schepaschenko, D.G. Boreal forest health and global change. Science 349, 819–822 (2015).
Crowther, T.W. et al. Mapping tree density at a global scale. Nature 525, 201–205 (2015).
McKenney, D.W., Pedlar, J.H., Lawrence, K., Campbell, K. & Hutchinson, M.F. Potential impacts of climate change on the distribution of North American trees. Bioscience 57, 939–948 (2007).
Longman, K.A. & Wareing, P.F. Early induction of flowering in birch seedlings. Nature 184, 2037–2038 (1959).
Tuskan, G.A. et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313, 1596–1604 (2006).
Nystedt, B. et al. The Norway spruce genome sequence and conifer genome evolution. Nature 497, 579–584 (2013).
Neale, D.B. et al. Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol. 15, R59 (2014).
Gao, D., Li, Y., Kim, K.D., Abernathy, B. & Jackson, S.A. Landscape and evolutionary dynamics of terminal repeat retrotransposons in miniature in plant genomes. Genome Biol. 17, 7 (2016).
Jaillon, O. et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467 (2007).
Jiao, Y. et al. A genome triplication associated with early diversification of the core eudicots. Genome Biol. 13, R3 (2012).
Jiao, Y. & Paterson, A.H. Polyploidy-associated genome modifications during land plant evolution. Phil. Trans. R. Soc. Lond. B 369, 20130355 (2014).
Flagel, L.E. & Wendel, J.F. Gene duplication and evolutionary novelty in plants. New Phytol. 183, 557–564 (2009).
Papp, B., Pál, C. & Hurst, L.D. Dosage sensitivity and the evolution of gene families in yeast. Nature 424, 194–197 (2003).
Freeling, M. & Thomas, B.C. Gene-balanced duplications, like tetraploidy, provide predictable drive to increase morphological complexity. Genome Res. 16, 805–814 (2006).
Rodgers-Melnick, E. et al. Contrasting patterns of evolution following whole genome versus tandem duplication events in Populus. Genome Res. 22, 95–105 (2012).
Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815 (2000).
Hanada, K., Zou, C., Lehti-Shiu, M.D., Shinozaki, K. & Shiu, S.-H. Importance of lineage-specific expansion of plant tandem duplicates in the adaptive response to environmental stimuli. Plant Physiol. 148, 993–1003 (2008).
Chae, L., Kim, T., Nilo-Poyanco, R. & Rhee, S.Y. Genomic signatures of specialized metabolism in plants. Science 344, 510–513 (2014).
DePristo, M.A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Anamthawat-Jónsson, K. & Thór Thórsson, A. Natural hybridisation in birch: triploid hybrids between Betula nana and B. pubescens. Plant Cell Tissue Organ Cult. 75, 99–107 (2003).
Eidesen, P.B., Alsos, I.G. & Brochmann, C. Comparative analyses of plastid and AFLP data suggest different colonization history and asymmetric hybridization between Betula pubescens and B. nana. Mol. Ecol. 24, 3993–4009 (2015).
Patterson, N. et al. Ancient admixture in human history. Genetics 192, 1065–1093 (2012).
Ashburner, K., McAllister, H.A. & Rix, M. The Genus Betula: A Taxonomic Revision of Birches (Royal Botanic Gardens, 2013).
Wang, N., McAllister, H.A., Bartlett, P.R. & Buggs, R.J.A. Molecular phylogeny and genome size evolution of the genus Betula (Betulaceae). Ann. Bot. 117, 1023–1035 (2016).
Lascoux, M., Palmé, A.E., Cheddadi, R. & Latta, R.G. Impact of Ice Ages on the genetic structure of trees and shrubs. Phil. Trans. R. Soc. Lond. B 359, 197–207 (2004).
Sofiev, M., Siljamo, P., Ranta, H. & Rantio-Lehtimäki, A. Towards numerical forecasting of long-range air transport of birch pollen: theoretical considerations and a feasibility study. Int. J. Biometeorol. 50, 392–402 (2006).
Hollender, C.A. & Dardick, C. Molecular basis of angiosperm tree architecture. New Phytol. 206, 541–556 (2015).
Li, P. et al. LAZY1 controls rice shoot gravitropism through regulating polar auxin transport. Cell Res. 17, 402–410 (2007).
Dong, Z. & Jin, W. Pleiotropic effects of ZmLAZY1 on the auxin-mediated responses to gravity and light in maize shoot and inflorescences. Plant Signal. Behav. 8, e27452 (2013).
Yoshihara, T., Spalding, E.P. & Iino, M. AtLAZY1 is a signaling component required for gravitropism of the Arabidopsis thaliana inflorescence. Plant J. 74, 267–279 (2013).
Wang, J., Street, N.R., Scofield, D.G. & Ingvarsson, P.K. Natural selection and recombination rate variation shape nucleotide polymorphism across the genomes of three related Populus species. Genetics 202, 1185–1200 (2016).
Korneliussen, T.S., Albrechtsen, A. & Nielsen, R. ANGSD: Analysis of Next Generation Sequencing Data. BMC Bioinformatics 15, 356 (2014).
Liu, X. & Fu, Y.-X. Exploring population size changes using SNP frequency spectra. Nat. Genet. 47, 555–559 (2015).
Lynch, M. Evolution of the mutation rate. Trends Genet. 26, 345–352 (2010).
Wolfe, J.A. A paleobotanical interpretation of Tertiary climates in the Northern Hemisphere: data from fossil plants make it possible to reconstruct Tertiary climatic changes, which may be correlated with changes in the inclination of the Earth's rotational axis. Am. Sci. 66, 694–703 (1978).
Head, M.J. & Gibbard, P.L. Formal subdivision of the Quaternary System/Period: past, present, and future. Quat. Int. 383, 4–35 (2015).
Gaudet, P. & Dessimoz, C. in The Gene Ontology Handbook (eds. Dessimoz, C. & Škunca, N.) 189–205 (Springer, 2017).
DeYoung, B.J. et al. The CLAVATA1-related BAM1, BAM2 and BAM3 receptor kinase–like proteins are required for meristem function in Arabidopsis. Plant J. 45, 1–16 (2006).
Wang, J. et al. The Arabidopsis LRR-RLK, PXC1, is a regulator of secondary wall formation correlated with the TDIF-PXY/TDR-WOX4 signaling pathway. BMC Plant Biol. 13, 94 (2013).
Gursanscky, N.R. et al. MOL1 is required for cambium homeostasis in Arabidopsis. Plant J. 86, 210–220 (2016).
Wang, T. et al. A receptor heteromer mediates the male perception of female attractants in plants. Nature 531, 241–244 (2016).
Wang, H. & Wang, H. Phytochrome signaling: time to tighten up the loose ends. Mol. Plant 8, 540–551 (2015).
Bartrina, I. et al. Gain-of-function mutants of the cytokinin receptors AHK2 and AHK3 regulate plant organ size, flowering time and plant longevity. Plant Physiol. 173, 1783–1797 (2017).
Merchante, C., Alonso, J.M. & Stepanova, A.N. Ethylene signaling: simple ligand, complex regulation. Curr. Opin. Plant Biol. 16, 554–560 (2013).
Schlereth, A. et al. MONOPTEROS controls embryonic root initiation by regulating a mobile transcription factor. Nature 464, 913–916 (2010).
Doyle, S.M. et al. An early secretory pathway mediated by GNOM-LIKE 1 and GNOM is essential for basal polarity establishment in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 112, E806–E815 (2015).
Kinoshita, A. et al. RPK2 is an essential receptor-like kinase that transmits the CLV3 signal in Arabidopsis. Development 137, 3911–3920 (2010).
Ueda, M., Zhang, Z. & Laux, T. Transcriptional activation of Arabidopsis axis patterning genes WOX8/9 links zygote polarity to embryo development. Dev. Cell 20, 264–270 (2011).
Guan, Y. et al. Phosphorylation of a WRKY transcription factor by MAPKs is required for pollen development and function in Arabidopsis. PLoS Genet. 10, e1004384 (2014).
Jiang, W. & Yu, D. Arabidopsis WRKY2 transcription factor mediates seed germination and postgermination arrest of development by abscisic acid. BMC Plant Biol. 9, 96 (2009).
Legendre, P. & Fortin, M.-J. Comparison of the Mantel test and alternative approaches for detecting complex multivariate relationships in the spatial analysis of genetic data. Mol. Ecol. Resour. 10, 831–844 (2010).
Veyres, N. et al. The Arabidopsis sweetie mutant is affected in carbohydrate metabolism and defective in the control of growth, development and senescence. Plant J. 55, 665–686 (2008).
Goyal, K., Walton, L.J. & Tunnacliffe, A. LEA proteins prevent protein aggregation due to water stress. Biochem. J. 388, 151–157 (2005).
Sakuma, Y. et al. Functional analysis of an Arabidopsis transcription factor, DREB2A, involved in drought-responsive gene expression. Plant Cell 18, 1292–1309 (2006).
Welling, A. & Palva, E.T. Involvement of CBF transcription factors in winter hardiness in birch. Plant Physiol. 147, 1199–1211 (2008).
El Refy, A. et al. The Arabidopsis KAKTUS gene encodes a HECT protein and controls the number of endoreduplication cycles. Mol. Genet. Genomics 270, 403–414 (2003).
Bensussan, M. et al. Suppression of dwarf and irregular xylem phenotypes generates low-acetylated biomass lines in Arabidopsis. Plant Physiol. 168, 452–463 (2015).
Sugimoto-Shirasu, K. & Roberts, K. “Big it up”: endoreduplication and cell-size control in plants. Curr. Opin. Plant Biol. 6, 544–553 (2003).
Gegas, V.C. et al. Endopolyploidy as a potential alternative adaptive strategy for Arabidopsis leaf size variation in response to UV-B. J. Exp. Bot. 65, 2757–2766 (2014).
Bonawitz, N.D. & Chapple, C. Can genetic engineering of lignin deposition be accomplished without an unacceptable yield penalty? Curr. Opin. Biotechnol. 24, 336–343 (2013).
Bonawitz, N.D. et al. Disruption of Mediator rescues the stunted growth of a lignin-deficient Arabidopsis mutant. Nature 509, 376–380 (2014).
Imura, Y. et al. CRYPTIC PRECOCIOUS/MED12 is a novel flowering regulator with multiple target steps in Arabidopsis. Plant Cell Physiol. 53, 287–303 (2012).
Mähönen, A.P. et al. Cytokinin signaling and its inhibitor AHP6 regulate cell fate during vascular development. Science 311, 94–98 (2006).
Bishopp, A. et al. A mutually inhibitory interaction between auxin and cytokinin specifies vascular pattern in roots. Curr. Biol. 21, 917–926 (2011).
Immanen, J. et al. Cytokinin and auxin display distinct but interconnected distribution and signaling profiles to stimulate cambial activity. Curr. Biol. 26, 1990–1997 (2016).
Dello Ioio, R. et al. A genetic framework for the control of cell division and differentiation in the root meristem. Science 322, 1380–1384 (2008).
Zhu, J. et al. Low temperature inhibits root growth by reducing auxin accumulation via ARR1/12. Plant Cell Physiol. 56, 727–736 (2015).
Nguyen, K.H. et al. Arabidopsis type B cytokinin response regulators ARR1, ARR10, and ARR12 negatively regulate plant responses to drought. Proc. Natl. Acad. Sci. USA 113, 3090–3095 (2016).
Lin, R. & Wang, H. Arabidopsis FHY3/FAR1 gene family and distinct roles of its members in light control of Arabidopsis development. Plant Physiol. 136, 4010–4022 (2004).
Edwards, K.D., Guerineau, F., Devlin, P.F. & Millar, A.J. Low-temperature-specific effects of PHYTOCHROME C on the circadian clock in Arabidopsis suggest that PHYC underlies natural variation in biological timing. Preprint at bioRxiv https://doi.org/10.1101/030577 (2015).
Méndez-Vigo, B., Picó, F.X., Ramiro, M., Martínez-Zapater, J.M. & Alonso-Blanco, C. Altitudinal and climatic adaptation is mediated by flowering traits and FRI, FLC, and PHYC genes in Arabidopsis. Plant Physiol. 157, 1942–1955 (2011).
Samis, K.E., Heath, K.D. & Stinchcombe, J.R. Discordant longitudinal clines in flowering time and phytochrome C in Arabidopsis thaliana. Evolution 62, 2971–2983 (2008).
Balasubramanian, S. et al. The PHYTOCHROME C photoreceptor gene mediates natural variation in flowering and growth responses of Arabidopsis thaliana. Nat. Genet. 38, 711–715 (2006).
Myking, T. & Heide, O.M. Dormancy release and chilling requirement of buds of latitudinal ecotypes of Betula pendula and B. pubescens. Tree Physiol. 15, 697–704 (1995).
Iskow, R.C., Gokcumen, O. & Lee, C. Exploring the role of copy number variants in human adaptation. Trends Genet. 28, 245–257 (2012).
Fogelqvist, J. et al. Genetic and morphological evidence for introgression between three species of willows. BMC Evol. Biol. 15, 193 (2015).
Messer, P.W. & Petrov, D.A. Population genomics of rapid adaptation by soft selective sweeps. Trends Ecol. Evol. 28, 659–669 (2013).
Matsumoto-Kitano, M. et al. Cytokinins are central regulators of cambial activity. Proc. Natl. Acad. Sci. USA 105, 20027–20031 (2008).
Acknowledgements
We thank A. Korpijaakko, A. Korpilahti, S. Koskela, K. Lipponen, P. Laamanen, H. Kangas, M. Rantanen, and E.-M. Turkki for excellent assistance, and L. Schulman and L. Junikka (University of Helsinki Botanical Gardens) for providing birch species samples. Birch sequencing was supported by a Finnish Technology Development Agency (TEKES) grant to J.K., Y.H., and P.A. J.K. and Y.H. were supported by the Finnish Centre of Excellence in Molecular Biology of Primary Producers (Academy of Finland CoE program 2014-2019, decision 271832). Y.H. was funded by the Gatsby Foundation and the European Research Council Advanced Investigator Grant SYMDEV. V.A.A. acknowledges support from the US National Science foundation (0922742 and 1442190). J.S. acknowledges a University of Helsinki 3-year grant. A.H.S. and J.T. acknowledge Academy of Finland decision (266430). EST libraries were created with TEKES funding to E.T.P.
Author information
Authors and Affiliations
Contributions
J.K., P.A., and Y.H. conceived the study, and led the work together with J.S. and V.A.A. J.S. managed and coordinated all bioinformatics activities. L.G.P., A.L., and J.I. performed library construction and sequencing, and R.H., J.I., K.N., J.A.S., C.T.K., C.v.d.S., L.V., M.R., E.O., J.M., S.K.-S., G.E., A.P.M., and B.J.H.M.P. participated in various aspects of biological sample collection, preparation, and quality control. O.-P.S. assembled the genome, with P.A. leading the work. O.-P.S. assembled and P.S. and A.A. annotated the organellar genomes. P.R. carried out linkage mapping and anchored the genome into pseudochromosomes with J.S., O.-P.S., S.R., P.S., J.I.K., O.S. and A.A. J.I., R.R., L.K., A.W., T.P., P.I.H., L.G.P., E.T.P., Y.H., and J.K. produced EST libraries, which were analyzed by O.-P.S., J.S., and S.R. J.I. and L.G.P. produced RNA-seq libraries, and O.-P.S., B.J., and S.R. analyzed the RNA sequencing data. J.S. and S.R. contributed to functional annotation, and P.J.G., H.F., J.V., T.H.T., S.E., A.V., A.K., M.W., S.O.K., A.G., A.S., V.P., M.M.R., M.B., M.P., S.K., O.S., T.B., K.M., K.H., J.P.K., S.S., F.O.A., P.H., F.C., K.V.F., K.-J.L., P.S., V.H.A., L.M., P.E., S.B., D.B., E.X., T.P.S., J.A.S., K.O., O.-P.S., O.B., A.K.K., J.I.K., M.S., A.I.T., J.L., J.d.D.B.-L., P.P., L.P., Ü.N., S.E.E., and K.N. participated in manual annotation, which was coordinated by P.S. and J.S. J.T. and A.H.S. annotated and analyzed the transposable elements. J.S., V.A.A., T.L., and S.R. performed comparative genomics analyses. J.S., V.A.A., O.S., and M.L. analyzed population genomics data. J.S., V.A.A., and T.L. prepared figures. J.S. and V.A.A. wrote the paper, with input from J.K., M.L., K.N., Y.H., P.A., A.H.S., O.-P.S., C.D., and J.I. J.S., O.-P.S., V.A.A., L.G.P., S.R., A.L., R.H., J.I., O.S., K.N., M.R., A.H.S., P.R., P.S., A.A., and G.E. provided text for the supplement. K.N. and S.R. had equivalent overall roles in the project. All authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–21, Supplementary Tables 1, 4, 5, 7, 15, and Supplementary Note (PDF 4752 kb)
Supplementary Tables
Supplementary Tables 2, 3, 6, 8, 9, 10, 11, 12, 13, 14 and 16–20. (ZIP 3930 kb)
Supplementary Data Sets
Supplementary Data Set 1–3. (ZIP 154503 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) License. The images or other third party material in this article is included in the article’s Creative Commons license, unless otherwise indicated in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Salojärvi, J., Smolander, OP., Nieminen, K. et al. Genome sequencing and population genomic analyses provide insights into the adaptive landscape of silver birch. Nat Genet 49, 904–912 (2017). https://doi.org/10.1038/ng.3862
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3862
This article is cited by
-
Allopolyploid origin and diversification of the Hawaiian endemic mints
Nature Communications (2024)
-
Dissection of figured wood trait in curly birch (Betula pendula Roth var. carelica (Mercklin) Hämet-Ahti) using high-throughput genotyping
Scientific Reports (2024)
-
The genome and population genomics of allopolyploid Coffea arabica reveal the diversification history of modern coffee cultivars
Nature Genetics (2024)
-
Genome structure-based Juglandaceae phylogenies contradict alignment-based phylogenies and substitution rates vary with DNA repair genes
Nature Communications (2023)
-
Divergent mechanisms of reduced growth performance in Betula ermanii saplings from high-altitude and low-latitude range edges
Heredity (2023)
