
Abstract
The snub-nosed monkey genus Rhinopithecus includes five closely related species distributed across altitudinal gradients from 800 to 4,500 m. Rhinopithecus bieti, Rhinopithecus roxellana, and Rhinopithecus strykeri inhabit high-altitude habitats, whereas Rhinopithecus brelichi and Rhinopithecus avunculus inhabit lowland regions. We report the de novo whole-genome sequence of R. bieti and genomic sequences for the four other species. Eight shared substitutions were found in six genes related to lung function, DNA repair, and angiogenesis in the high-altitude snub-nosed monkeys. Functional assays showed that the high-altitude variant of CDT1 (Ala537Val) renders cells more resistant to UV irradiation, and the high-altitude variants of RNASE4 (Asn89Lys and Thr128Ile) confer enhanced ability to induce endothelial tube formation in vitro. Genomic scans in the R. bieti and R. roxellana populations identified signatures of selection between and within populations at genes involved in functions relevant to high-altitude adaptation. These results provide valuable insights into the adaptation to high altitude in the snub-nosed monkeys.
Similar content being viewed by others

Main
The snub-nosed monkey genus Rhinopithecus (Colobinae) comprises five closely related species (R. bieti, R. roxellana, R. brelichi, R. avunculus, and R. strykeri) that diverged in the Early Pleistocene1,2,3. All five species are among the world's rarest and most endangered primates (2000 International Union for Conservation of Nature (IUCN) Red List of Threatened Species; see URLs) and are confined to very limited areas in isolated regions of China and Vietnam1. The reference genome of R. roxellana and genomic resequencing data for all species included here, with the exception of R. avunculus, have been previously reported4.
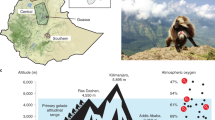
In addition to their conservation value, Rhinopithecus species are unique in the context of adaptive evolution. Besides having a folivorous diet with forestomach fermentation4, which is functionally analogous to that of ruminants, with molecular convergence of lysozyme and RNase functions demonstrated for leaf-eating monkeys and cows4,5,6, these species show a distribution across altitudinal gradients from 800 to 4,500 m. R. brelichi and R. avunculus inhabit lowland regions (<2,000 m) of Guizhou province in China and northern Vietnam, respectively, whereas R. bieti, R. roxellana, and R. strykeri live in separated highland zones that each have elevations above 3,000 m, including the Tibetan Plateau and the mountains of central China (Fig. 1). The most notable among these species is R. bieti, which is found exclusively on the Tibetan Plateau in a narrow area between the Yangtze and Mekong rivers and has been thought to be the non-human primate dwelling at the highest altitude, occurring at elevations of 3,500–4,500 m (refs. 7,8, 9, 10, 11). In addition, previous phylogenetic analyses of nuclear genes have shown that the high-altitude R. bieti and R. strykeri species are grouped with the low-altitude R. avunculus species and that the high-altitude R. roxellana species is grouped with the low-altitude R. brelichi species (Fig. 1)12. Accordingly, the Rhinopithecus genus offers an interesting study model not only to investigate the adaptive mechanisms of non-human primates to high altitude but also to examine the differences and similarities between the adaptive mechanisms in different high-altitude regions.

(a) Geographical distribution and sampling locations of the Rhinopithecus species. Sampling locations are represented by pink dots. The photographs of snub-nosed monkeys were taken by Y.C. Long and Z.F. Xiang. The map is from Google Maps, map data © 2016 Google, SK Planet, ZENRIN. (b) Phylogenetic relationships of the Rhinopithecus species inferred from a previous nuclear gene analysis12. The present whole-genome phylogeny supports the same relationships, regardless of the reference genomes and tree-building methods used. All nodes received support levels of 100%. The estimated divergence times are included above the nodes. MYA, million years ago.
Here we carried out de novo sequencing and genome assembly of R. bieti using the Illumina HiSeq 2000 platform, generating ∼270 Gb of genomic sequence with 76.5-fold coverage (Supplementary Table 1); R. bieti has an estimated genome size of 3.52 Gb on the basis of flow cytometry analysis (Supplementary Note). The assembly produced contig and scaffold N50 sizes of 20.5 kb and 2.2 Mb, respectively (Supplementary Table 2). The pattern of GC content for R. bieti is similar to those for macaque and human (Supplementary Fig. 1). We found genomic synteny between R. bieti and rhesus macaque (Macaca mulatta13) (Fig. 2a) and identified 21,812 recent segmental duplications with a total length of 72.1 Mb (2.43%) in the R. bieti genome (Fig. 2b).

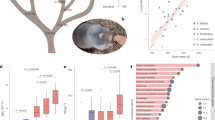
(a) Whole-genome synteny analysis for R. bieti and M. mulatta. Most R. bieti scaffolds (scaf1–scaf21) show high collinearity with the M. mulatta genome (chr1–chrX), except for the non-collinear regions indicated by red lines. (b) Distribution of the percentage identity of segmental duplications in the R. bieti genome. The horizontal axis represents the percentage identity, and the vertical axis represents the total length of the segmental duplications in each window of percentage identity. “Interscaffold” and “intrascaffold” correspond to segmental duplications between scaffolds and within scaffolds, respectively. (c) Gene family expansions and contractions in the R. bieti genome. The numbers on each branch correspond to the numbers of gene families that have expanded (orange) and contracted (blue). MRCA, most recent common ancestor.
The R. bieti genome is predicted to contain 22,834 protein-coding genes (Supplementary Fig. 2). We identified 1,187 gene families that were expanded in R. bieti in comparison to M. mulatta (Fig. 2c). InterPro classification of the genes from 231 significantly expanded gene families showed significant enrichment (P < 0.01) of these genes in categories involved in DNA repair and damage response (94 genes) and oxidative phosphorylation processes (107 genes) (Supplementary Table 3). This finding may be related to the increased exposure of snub-nosed monkeys to UV radiation and the potentially increased rate of energy metabolism required for high-altitude survival14,15,16,17,18. However, caution is needed in interpreting gene family expansions and contractions that were identified by comparing genomes assembled using different methods.
In addition to comparative genomic analyses, we performed a comparative transcriptomic analysis of R. bieti and M. mulatta based on ∼240 Gb of RNA-seq data from the same 11 tissues in both species (Supplementary Table 4 and Supplementary Note). We observed that the expression profiles of highly expressed genes from the digestive system (small intestine, large intestine, and stomach) and energy-consuming tissues (heart and skeletal muscle) were more similar in the same species than the expression patterns for the same tissue in the different species (Fig. 3), reflecting the genetic discrepancy between R. bieti and other primates with respect to dietary and high-altitude adaptation, respectively. Furthermore, analyses of highly expressed genes showed that those involved in the oxidative phosphorylation pathway (KEGG database, ko00190; 28 genes; P = 8.03 × 10−16) and the cardiac muscle contraction pathway (KEGG database, ko04260; 10 genes; P = 1.88 × 10−5) were significantly enriched for expression in energy-consuming tissues from R. bieti, indicating the possible involvement of energy metabolism and heart function in high-altitude adaptations of the snub-nosed monkeys.

The heat map was generated using hierarchical clustering and complete linkage of the top 500 most highly expressed one-to-one orthologous genes from R. bieti and M. mulatta. Distances, representing the relative similarity among genes and tissues, were calculated using Pearson's correlation coefficients. Color represents FPKM value of gene expression after scaling and centering.
To investigate the adaptive mechanisms of snub-nosed monkeys from different high-altitude regions, we performed a comparative genomic analysis of all snub-nosed monkeys by including resequencing genomes for R. brelichi, R. strykeri, and R. avunculus (30-fold coverage on average per species, with one individual per species; Supplementary Table 5) as well as the recently published R. roxellana genome4. Genomic sequence data for R. avunculus, with only 250 individuals in the wild, have to our knowledge not previously been reported. The genetic diversity among these snub-nosed monkeys is between 0.014% and 0.068%. R. brelichi has the highest diversity (0.068%), whereas R. strykeri has the lowest diversity (0.014%). R. roxellana (0.054%) and R. avunculus (0.044%) show similar levels of diversity, with the diversity in both species greater than that in R. bieti (0.030%) (Supplementary Fig. 3). On the basis of 69,586,645 SNPs in the five snub-nosed monkeys and macaque, we constructed a phylogenetic tree. Regardless of the reference genomes and tree-building methods used, the genome phylogeny strongly supported the interspecific relationships shown in a previous tree based on the analyses of several nuclear genes (Fig. 1) and significantly rejected the relationships represented in a tree based on the mitochondrial genome12 (P = 1 × 10−40 in an approximately uniform test). Estimation of divergence times suggests that the two high-altitude 'Himalaya' species R. bieti and R. strykeri diverged from the lowland R. avunculus species 0.80 million years ago and that the high-altitude northern species R. roxellana separated from the lowland R. brelichi species 1.07 million years ago (Fig. 1 and Supplementary Note). We then identified shared amino acid substitutions that occurred along the lineage containing R. bieti and R. strykeri and the lineage leading to R. roxellana, but not along the lineages leading to the low-altitude R. avunculus and R. brelichi species. Overall, 20 common amino acid substitutions for 18 genes were observed in all high-altitude snub-nosed monkeys (Supplementary Table 6), with this number of substitutions not resulting from chance (P = 3.3 × 10−16). Of the common substitutions identified, 18 occurred in 16 genes that were also identified as evolving under positive selection on the basis of likelihood-ratio tests for these two lineages (Supplementary Table 7). Positively selected genes had a significantly higher ratio of nonsynonymous substitutions to synonymous substitutions than other genes in the high-altitude snub-nosed monkeys (P = 3.84 × 10−3 for R. strykeri, 3.45 × 10−6 for R. bieti, and 5.10 × 10−8 for R. roxellana) (Supplementary Table 8). Further examination of these 16 genes found that 6 genes, including ARMC2, NT5DC1, RNASE4, CDT1, RTEL1, and DNAH11, have functional associations with lung function, angiogenesis, DNA repair, or respiratory cilia movement, suggesting a role in the high-altitude adaptation of the snub-nosed monkeys19,20,21,22,23,24 (Table 1). Moreover, when the common substitutions for these six genes were examined in more mammalian species with publicly available genomes, no identical substitutions were found in those species (Supplementary Table 9 and Supplementary Note).
To test the potential functional effects of these substitutions, we performed experimental validation of the substitutions identified in CDT1 (p.Ala537Val) and RNASE4 (p.Asn89Lys and p.Thr128Ile) (Fig. 4). CDT1 is a licensing factor for DNA replication that is tightly controlled to maintain genome integrity, and the encoded protein is rapidly degraded by the SCFSkp2 complex upon UV-induced DNA damage25. Using plasmids encoding human CDT1 proteins in a HeLa cell culture model, our UV irradiation assays showed that CDT1 Ala537Val was more stable than the reference CDT1 protein after irradiation with 100 J/m2 UV light (Fig. 4c,d), suggesting that the p.Ala537Val substitution might contribute to the increased resistance to UV radiation of the high-altitude snub-nosed monkeys. RNASE4 is a member of the RNase superfamily and has been reported to induce angiogenesis19,26. The coding regions of RNASE4 and its high-altitude variants were synthesized and cloned into an Escherichia coli expression vector. The purity of active proteins was demonstrated by Coomassie blue staining (Fig. 4e). We then examined the ability of cells to induce endothelial tube formation after addition of recombinant reference or variant (Asn89Lys and Thr128Ile) RNASE4 protein. Variant RNASE4 (Asn89Lys and Thr128Ile; 1 μg/ml concentration) showed higher activity in inducing tube formation in human umbilical vein endothelial cell (HUVEC) cultures in Matrigel than reference RNASE4 (Fig. 4f,g), indicating that the Asn89Lys and Thr128Ile variants may enhance angiogenesis for high-altitude adaptation in snub-nosed monkeys. The fact that heat inactivation or proteolysis abolishes RNASE4 protein activities suggests that intact peptide sequence and structure are also important (data not shown).

(a) Alignment of mammalian RNASE4 amino acid sequences. Amino acids unique to the three high-altitude snub-nosed monkey species (corresponding to residues 89 and 128 in human RNASE4) are shown in red; other amino acids at these positions are shown in yellow. (b) Alignment of mammalian CDT1 amino acid sequences. The amino acid unique to the three high-altitude snub-nosed monkey species (corresponding to residue 537 in human CDT1) is shown in red; other amino acids at this position are shown in yellow. (c) Immunoblot of C-terminally tagged human CDT1 and CDT1 Ala537Val in a time course of HeLa cells irradiated with 100 J/m2 UV. GAPDH is shown as a loading control. (d) In HeLa cells stably expressing C-terminally tagged human CDT1 and CDT1 Ala537Val, protein synthesis was inhibited with 100 μg/ml cycloheximide (CHX), and the fate of CDT1 protein was examined by SDS–PAGE and immunoblotting with antibody to the C-terminal FLAG tag. (e) Coomassie blue staining of E. coli recombinant RNASE4 and variant RNASE4 (Asn89Lys and Thr128Ile) proteins. (f) Microscopy images of endothelial cell tubes in HUVECs formed with various concentrations of recombinant RNASE4 protein. Representative images are shown from three independent experiments. Scale bar, 500 μm. (g) Number of circular tube structures per mm2 in HUVECs with various concentrations of recombinant RNASE4. Values are shown as means ± s.e.m. from three independent experiments, and statistical analysis was performed by Student's t test: *P < 0.05, **P < 0.01.
In addition to species-level investigation, we performed population-level genomic scans for selection in R. bieti (n = 20; ∼10-fold coverage per individual) and R. roxellana (n = 26; ∼12.29-fold coverage per individual) populations (Supplementary Tables 10–13). Three R. roxellana populations from Sichuan and Gansu (SG; n = 12; 2,500–3,500 m), Qinling (QL; n = 9; 2,000–3,000 m), and Shennongjia (SNJ; n = 5; 2,000–2,500 m), which showed obvious isolation and different demographic scenarios, were compared to one R. bieti population from Weixi (3,300–4,100 m) (Supplementary Fig. 4 and Supplementary Note). These populations occupy different elevations at high altitude and may face different levels of hypoxia stress, similar to those experienced by other high-altitude mammals at comparable altitudes27,28. The high-altitude R. strykeri population was not included because of its small size, with fewer than 300 individuals in the wild, which led to sample collection difficulties. By identifying loci and pathways that were outliers using three methods to scan for selection (Supplementary Note), we detected 60 genes in the R. bieti population (3.12 million SNPs), 247 genes in the SG population (5.85 million SNPs), 313 genes in the QL population (4.37 million SNPs), and 269 genes in the SNJ population (4.16 million SNPs) evolving under selection. Inspection of the genes overlapping between populations (Supplementary Fig. 5) identified two genes—BRCC3 (R. bieti, SG, and QL) and RYR2 (R. bieti and SG)—related to DNA repair and heart function, respectively29,30 (Table 2). Interestingly, RYR2 has also been reported as a key candidate gene related to high-altitude adaptation in Tibetan wolves31. In addition to the overlapping loci, we observed nine candidate high-altitude-related loci that were specific to particular populations, which may contribute to the development of unique and local adaptations32,33,34,35,36,37,38,39,40,41,42,43,44 (Table 2). Interestingly, ATR (R. bieti specific), which functions in DNA repair, regulates the expression of hypoxia-inducible factor (HIF)-1α, and confers hypoxia tolerance in vitro33,34,35, was identified here for the first time to our knowledge in R. bieti as a candidate target of selection in high-altitude adaptation. ARNT (SG specific), whose protein product binds to HIF-α to regulate adaptation to hypoxia37, has been mentioned as a target of positive selection in the human Tibetan population45. ACE (SG specific), which functions in blood pressure regulation and carries variants responsible for high-altitude pulmonary hypertension (HAPH)38, has been reported to have a role in high-altitude adaptation of human populations46,47,48. In comparison, no significant signatures of selection were observed in hemoglobin genes, which have been identified as critical genes for oxygen binding and resistance to hypoxia in other high-altitude mammals49,50,51,52,53. The occurrence of population-specific selection signatures mostly in genes in the R. bieti and SG populations, along with most of the overlapping genes being shared by these two populations, highlights the greater extent of signatures for genomic adaptation to high altitude in R. bieti and SG than in the QL and SNJ populations. This observation is consistent with the fact that the R. bieti and SG populations inhabit the highest altitudes.
Our study extends the genomic analysis of high-altitude adaptation to a unique branch of the non-human primate evolutionary tree. On the basis of a multilevel survey, the genomic sequence analyses from both the species level and population level, as well as the transcriptomic analyses and functional assays, find adaptive signatures that may reflect molecular adaptations consistent with the high-altitude environments of three snub-nosed monkey species, contributing to a new and more complete understanding of the complex biological features of high-altitude adaptation.
Methods
De novo genome sequencing and assembly.
Blood from an adult live male R. bieti (Rb0) that was caught in the wild and kept in the Centre of Experimental Primates at the Kunming Institute of Zoology (Chinese Academy of Sciences) was obtained for de novo sequencing. Genomic DNA was extracted using the QIAamp DNA Blood Mini kit (Qiagen). Thirteen libraries consisting of short paired-end inserts (200 bp, 300 bp, 400 bp, and 600 bp) and long mate-paired inserts (5 kb, 10 kb, and 15 kb) were constructed for genome sequencing according to the Illumina protocol. All libraries were sequenced on the HiSeq 2000 platform (Supplementary Table 1).
Illumina paired-end reads were assembled into contigs with the programs Anytag54 and Newbler55. To evaluate and improve the quality of the contigs, short reads were mapped to contigs using BWA56. Small indels and single-nucleotide variants (SNVs) were called from the short-read alignments by SAMtools57 and BCFtools57. In the first round, we used these indels and SNVs to correct the contigs. In the final round, we ran mapping/calling again and estimated the base accuracy of the assemblies to be 0.99954. Scaffolding was performed by our in-house scaffolder GOBOND. GapCloser 1.12 (ref. 58) was used to close gaps in the scaffolds.
Transcriptome sequencing.
Blood and 11 tissues (stomach, spleen, kidney, testicle, liver, brain, heart, lung, muscle, large intestine, and small intestine) were collected from the same R. bieti individual (Rb0) for genome sequencing after it died of old age. The study has been reviewed and approved by the internal review board of the Kunming Institute of Zoology, Chinese Academy of Sciences. Total RNA was extracted from each tissue using the TRIzol kit (Life Technologies). Libraries were constructed and sequenced according to the Illumina protocol (Supplementary Table 4).
Repeat identification, gene prediction, and annotation.
The R. bieti genome was searched for repeats using the program RepeatMasker and the Repbase library59. Repeat proteins were identified using RepeatProteinMask, and de novo repeat annotation was performed using RepeatModeler and RepeatMasker.
Using the repeat-masked genome, three approaches were combined to predict genes. AUGUSTUS60, Glimmer-HMM61, and SNAP62 were used for ab initio prediction. We also assembled R. bieti RNA-seq reads into transcripts using Trinity63 for gene structure annotation. Protein sequences from Homo sapiens, Mus musculus, and M. mulatta were aligned to the R. bieti genome using Exonerate64 for homology-based gene prediction. The gene sets predicted by the three approaches were integrated with EVM65 to produce consensus gene models, which were then updated by PASA66.
Gene functions were assigned according to the best match for each gene in alignment to the NCBI non-redundant (nr) protein database using BLAST (E value ≤ 1 × 10−5). Gene ontology analysis was performed with Blast2GO67 using default settings. We also compared the predicted proteins using BLAST (E value ≤ 1 × 10−5) with the KEGG database for pathway annotation.
Whole-genome synteny analysis.
The repeat-masked scaffolds of the R. bieti genome with sequence lengths >100,000 bp, which accounted for 92.45% of the assembled genome, were aligned to the M. mulatta genome13, which contains 21 chromosomes, using MUMmer v3.0 (ref. 68). The whole-genome syntenic relationships between the species were visualized using Circos69.
Segmental duplication identification.
Segmental duplications were detected from whole-genome assembly comparison. Self-alignment was generated using LASTZ70 with default parameters on the repeat-masked genome sequence, and the resulting alignment was filtered for maximum simultaneous gap ≤100 bp to identify fragments corresponding to recent segmental duplication (>90% identity and >1 kb in length).
Gene family construction.
The predicted genes in the R. bieti genome were used along with those from the genomes of H. sapiens (human), M. mulatta (macaque), and Canis lupus familiaris (dog), which were downloaded from the Ensembl public database (version 78)71. The longest translation form was chosen to represent each gene. We assigned the R. bieti genes to TreeFam9 proteins using BLAST. HMMER72 software was used to assign each gene to the corresponding profile HMM databases with default settings. The R. bieti genes were assigned to the gene family with the highest scoring HMM result.
Gene family expansions and contractions.
Expansion and contraction of gene clusters were determined using CAFÉ 2.2 (ref. 73) with the lambda option of 0.00596954. A phylogenetic tree from a previous study74 was used in CAFÉ to infer changes in gene family size using a probabilistic model73. Significantly expanded gene families (P < 0.05) were then classified by InterPro75.
Comparative transcriptomic analyses of R. bieti and M. mulatta.
RNA-seq data from 11 tissues of M. mulatta, the same as those used in the transcriptome sequencing of R. bieti (Supplementary Table 4 and Supplementary Note), were obtained. RNA extraction and sequencing library construction for M. mulatta used the same approaches as were applied for R. bieti. We aligned the high-quality RNA-seq reads from M. mulatta and those from R. bieti to their respective genomes (rhemac Ensembl release 72 and R. bieti de novo assembly) using TopHat76 with default parameters. The expression level of each gene in each RNA-seq library from R. bieti or M. mulatta was determined using Cufflinks77. The top 500 most highly expressed one-to-one orthologous genes in each tissue were selected and then merged. Clustering and generation of a heat map of FPKM values for the selected genes was performed using the pheatmap package in R.
Genome resequencing of three snub-nosed monkey species.
The genomic data for R. brelichi, R. strykeri, and R. avunculus were newly generated using the Illumina HiSeq 2000 platform. For each of the three snub-nosed monkey species, one paired-end library with an average insert size of 491 nt (range of 365–651 nt) was constructed. A total of 330 Gb of raw data were obtained. Library preparation and sequencing were performed according to the manufacturer's protocols.
Phylogenetic reconstruction for the five snub-nosed monkey species.
Raw paired-end reads for R. brelichi, R. strykeri, and R. avunculus as well as those published for R. roxellana4 and M. mulatta13 were aligned to the R. bieti assembled genome (Rb0) using BWA with default parameters56 (Supplementary Note). We performed SNP calling using SAMtools57 (Supplementary Note). To avoid potential tree estimation bias introduced by the reference genomes used, we also applied R. roxellana and M. mulatta, respectively, for comparison. Phylogenetic analysis was performed using RAxML78 for partitioned maximum-likelihood analyses. The GTR-GAMMA model was used, and 1,000 bootstrap replicates were conducted. Besides the concatenation methods, two coalescence-based species tree estimation methods—STAR79 (Species Tree Estimation using Average Ranks of Coalescences) and ASTRAL80 (Accurate Species Tree Algorithm)—were also used. Trees were rooted with M. mulatta. The incongruence between the topologies of the present nuclear genome tree and the previous mitochondrial genome tree was evaluated using the approximately uniform test81 as implemented in the CONSELV0.1i program82 with default scaling and replicate values. We implemented a Monte Carlo Markov chain (MCMC) algorithm for estimation of divergence times using the program MCMCtree from the PAML package83 (Supplementary Note).
Common amino acid substitutions.
Single–amino acid polymorphisms for R. bieti and R. roxellana genes were compared with known genes from human, dog, and macaque (Ensembl release 78). Protein sequences for R. strykeri, R. brelichi, and R. avunculus genes were predicted by aligning and substituting the raw reads to R. bieti scaffolds. Artifacts from the multiple-sequence alignment (PRANK84,85) were trimmed by the Gblocks program86. After trimming, any alignment that was shorter than 100 bp was discarded87. To exclude variation in individual species, only amino acid changes shared by the three high-altitude snub-nosed monkey species were used. Statistical analyses were conducted using the method of Zou and Zhang88.
dN/dS ratio and positive selection test.
The Codeml program from the PAML package69 was implemented to estimate the dN/dS (nonsynonymous to synonymous) ratio for genes with the branch-specific model and the branch-site model. For the branch-site model, the branch of interest on the phylogenetic tree was designated as the foreground branch. Genes with 2Δln L >3.84 in likelihood-ratio tests were designated as positively selected genes.
Plasmid construction, transfection, cell culture, UV irradiation assays, and immunoblot analysis.
A sequence encoding C-terminally 3×FLAG-tagged human CDT1 was synthesized and cloned into the pCDH-MSCV-E2F-eGFP lentiviral vector. The high-altitude variant form of CDT1 (Ala537Val) was generated by mutagenesis PCR. All constructs were verified via sequencing. Lentiviruses were generated according to the manufacturer's protocol. HeLa cells and HEK293T cells were provided by the Kunming Cell Bank, Kunming Institute of Zoology, Chinese Academy of Sciences. The cell lines were verified to be free of mycoplasma contamination by fluorescent staining. HeLa cells were infected by the different lentiviruses and cultured in high-glucose DMEM supplemented with 10% FBS, 1% penicillin-streptomycin and 1 μg/l puromycin (Sigma). All cells were incubated in a humidified atmosphere with 5% CO2 at 37 °C. For UV irradiation assays, HeLa cells stably expressing the different forms of CDT1-3×FLAG were synchronized by incubation with 50 ng/ml nocodazole (Sigma) for 16 h and then subjected to UV irradiation at 100 J/m2. Cells were collected at the indicated time points. Immunoblot analysis was performed according to the standard protocol. The antibodies used included rabbit antibody to CDT1 (Cell Signaling Technology, 8064; 1:500 dilution), mouse M2 antibody to FLAG (Sigma, F3165; 1:1,000 dilution), mouse antibody to GAPDH (ABclonal, AC002; 1:1,000 dilution), horseradish peroxidase (HRP)-conjugated goat anti-mouse antibody (Santa Cruz Biotechnology, sc-2055; 1:2,000 dilution), and HRP-conjugated goat anti-rabbit antibody (Santa Cruz Biotechnology, sc-2054; 1:2,000 dilution). Some cells were incubated with 100 μg/ml cycloheximide for the indicated periods of time.
In vitro angiogenesis assays.
The coding sequences for RNASE4 and variant RNASE4 (Asn89Lys and Thr128Ile) from human were synthesized and cloned into the pET21b E. coli expression vector. Recombinant proteins were expressed in BL21 (DE3) cells and purified following the protocol used for the generation of Ang89. The ability of RNASE4 and RNASE4 (Asn89Lys and Thr128Ile) to induce endothelial tube formation was examined. Commercially available Ang (Abcam, 114413) and PBS were used as positive and negative controls, respectively. A 48-well plate was precoated with 100 μl/well of growth-factor-reduced Matrigel. HUVEC cells were purchased from the American Type Culture Collection (CRL-1730). Primary culture passage 3 HUVECs were seeded onto the Matrigel (2 × 104 cells/well) and cultured in the presence of recombinant reference or variant (Asn89Lys and Thr128Ile) RNASE4 for 4 h. Cells were then fixed with 3.7% paraformaldehyde and photographed. Images were quantified using ImageJ software for capillary-like structures. Experiments were performed in triplicate, and significance was determined by Student's t test (two-tailed).
Genome resequencing of R. bieti and R. roxellana populations.
Libraries with an insert size of 500 bp were constructed to resequence R. bieti and R. roxellana individuals according to the Illumina protocol. All libraries were sequenced on the HiSeq 2000 platform (Supplementary Tables 10–13). In addition, a published resequenced R. bieti individual was also included in the present population genomic study4.
Scans of R. bieti and R. roxellana populations for selection.
Using BWA with default parameters56, raw paired-end reads from each of 20 R. bieti individuals were aligned to the R. bieti assembled genome (Rb0) and the reads from each of 26 R. roxellana individuals were aligned to the published R. roxellana genome4 (Supplementary Tables 10–13). We performed SNP calling using SAMtools57 (Supplementary Note). The θπ method was used to identify genomic regions as population outliers with VCFtools v0.1.11 (ref. 90). ZH and LSBL scans were processed as described in Ai et al.91 with the slipped windows strategy. Genes detected by at least two of the three scanning methods were identified as candidate genes under selection (Supplementary Note).
Population structure analyses and demographic history reconstruction.
SNPs in scaffolds longer than 50 kb were extracted. To avoid the effect of linkage disequilibrium, we selected one SNP for each interval of 50 kb and ran Admixture92 to perform genetic clustering. Demographic history was inferred using the pairwise sequentially Markovian coalescence (PSMC) model93 (Supplementary Note). The generation time (g = 5 years)94,95 and mutation rate (5 × 10−9 mutations per generation)96,97 were derived from previous studies.
URLs.
International Union for Conservation of Nature (IUCN) Red List of Threatened Species, http://www.iucnredlist.org/; GOBOND, http://software.big.ac.cn/gobond.html; WinMDI, http://facs.scripps.edu/software.html; RepeatMasker, http://www.repeatmasker.org/; Repbase, http://www.girinst.org/repbase/index.html; TreeFam, http://www.treefam.org/download/; InterPro, http://www.ebi.ac.uk/interpro/; macaque genome sequence, ftp://hgdownload.cse.ucsc.edu/goldenPath/rheMac3/bigZips/rheMac3.fa.gz; macaque transcriptomic sequence, http://www.ncbi.nlm.nih.gov/bioproject?LinkName=sra_bioproject&from_uid=260130; R. roxellana genome reference, ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF_000769185.1_Rrox_v1/.
Accession codes.
All raw data from de novo sequencing and resequencing are available through the NCBI Sequence Read Archive (SRA) under projects PRJNA247935, PRJNA283338, and PRJNA261768. RNA-seq reads are available under project PRJNA248058. The de novo genome assembly for R. bieti has been deposited in the DNA Databank of Japan/European Molecular Biology Laboratory/GenBank under project PRJNA315476.
Accession codes
References
Li, M.S., Wei, F.W., Huang, C.M., Pan, R.L. & deRuiter, J. Phylogeny of snub-nosed monkeys inferred from mitochondrial DNA, cytochrome B and 12S rRNA sequences. Int. J. Primatol. 25, 861–873 (2004).
Roos, C., Thanh, V.N., Walter, L. & Nadler, T. Molecular systematics of Indochinese primates. Vietn. J. Primatol 1, 41–53 (2007).
Geissmann, T. et al. A new species of snub-nosed monkey, genus Rhinopithecus Milne–Edwards, 1872 (Primates, Colobinae), from northern Kachin state, northeastern Myanmar. Am. J. Primatol. 73, 96–107 (2011).
Zhou, X. et al. Whole-genome sequencing of the snub-nosed monkey provides insights into folivory and evolutionary history. Nat. Genet. 46, 1303–1310 (2014).
Stewart, C.B., Schilling, J.W. & Wilson, A.C. Adaptive evolution in the stomach lysozymes of foregut fermenters. Nature 330, 401–404 (1987).
Zhang, J. & Kumar, S. Detection of convergent and parallel evolution at the amino acid sequence level. Mol. Biol. Evol. 14, 527–536 (1997).
Long, Y.C., Kirkpatrick, R.C., Zhong, T. & Xiao, L. Repport on the distribution, population, and ecology of the Yunnan snub-nosed monkey (Rhinopithecus bieti). Primates 35, 241–250 (1994).
Long, Y.C., Kirkpatrick, R.C., Zhong, T. & Xiao, L. Status and conservation strategy of the Yunnan snub-nosed monkey. Chin. Biodiv. 4, 145–152 (1996).
Li, B.G., Pan, R.L. & Oxnard, C.E. Extinction of snub-nosed monkeys in China during the past 400 years. Int. J. Primatol. 23, 1227–1244 (2002).
Liu, Z. et al. Phylogeography and population structure of the Yunnan snub-nosed monkey (Rhinopithecus bieti) inferred from mitochondrial control region DNA sequence analysis. Mol. Ecol. 16, 3334–3349 (2007).
Liu, Z. et al. The effect of landscape features on population genetic structure in Yunnan snub-nosed monkeys (Rhinopithecus bieti) implies an anthropogenic genetic discontinuity. Mol. Ecol. 18, 3831–3846 (2009).
Liedigk, R. et al. Evolutionary history of the odd-nosed monkeys and the phylogenetic position of the newly described Myanmar snub-nosed monkey Rhinopithecus strykeri. PLoS One 7, e37418 (2012).
Zimin, A.V. et al. A new rhesus macaque assembly and annotation for next-generation sequencing analyses. Biol. Direct 9, 20 (2014).
Fukuda, R. et al. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 129, 111–122 (2007).
Palomera-Sanchez, Z. & Zurita, M. Open, repair and close again: chromatin dynamics and the response to UV-induced DNA damage. DNA Repair (Amst.) 10, 119–125 (2011).
Alkorta-Aranburu, G. et al. The genetic architecture of adaptations to high altitude in Ethiopia. PLoS Genet. 8, e1003110 (2012).
Ge, R.L. et al. Draft genome sequence of the Tibetan antelope. Nat. Commun. 4, 1858 (2013).
Zhang, Z.Y., Chen, B., Zhao, D.J. & Kang, L. Functional modulation of mitochondrial cytochrome c oxidase underlies adaptation to high-altitude hypoxia in a Tibetan migratory locust. Proc. Biol. Sci. 280, 20122758 (2013).
Li, S. et al. Ribonuclease 4 protects neuron degeneration by promoting angiogenesis, neurogenesis, and neuronal survival under stress. Angiogenesis 16, 387–404 (2013).
Bartoloni, L. et al. Mutations in the DNAH11 (axonemal heavy chain dynein type 11) gene cause one form of situs inversus totalis and most likely primary ciliary dyskinesia. Proc. Natl. Acad. Sci. USA 99, 10282–10286 (2002).
Sakaguchi, H. et al. Checkpoint kinase ATR phosphorylates Cdt2, a substrate receptor of CRL4 ubiquitin ligase, and promotes the degradation of Cdt1 following UV irradiation. PLoS One 7, e46480 (2012).
Barber, L.J. et al. RTEL1 maintains genomic stability by suppressing homologous recombination. Cell 135, 261–271 (2008).
Soler Artigas, M. et al. Genome-wide association and large-scale follow up identifies 16 new loci influencing lung function. Nat. Genet. 43, 1082–1090 (2011).
Guo, Y. et al. Single-nucleotide polymorphisms in the TSPYL-4 and NT5DC1 genes are associated with susceptibility to chronic obstructive pulmonary disease. Mol. Med. Rep. 6, 631–638 (2012).
Kondo, T. et al. Rapid degradation of Cdt1 upon UV-induced DNA damage is mediated by SCFSkp2 complex. J. Biol. Chem. 279, 27315–27319 (2004).
Di Liddo, R. et al. In vitro biological activity of bovine milk ribonuclease-4. Mol. Med. Rep. 3, 127–132 (2010).
Beall, C.M. Two routes to functional adaptation: Tibetan and Andean high-altitude natives. Proc. Natl. Acad. Sci. USA 104 (suppl. 1), 8655–8660 (2007).
Ge, R.L., Kubo, K., Kobayashi, T., Sekiquchi, M. & Honda, T. Blunted hypoxic pulmonary vasoconstrictive response in the rodent Ochotona curzoniae (pika) at high altitude. Am. J. Physiol-Heart. C 274, H1792–H1799 (1998).
Dong, Y. et al. Regulation of BRCC, a holoenzyme complex containing BRCA1 and BRCA2, by a signalosome-like subunit and its role in DNA repair. Mol. Cell 12, 1087–1099 (2003).
Tiso, N. et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum. Mol. Genet. 10, 189–194 (2001).
Zhang, W. et al. Hypoxia adaptations in the grey wolf (Canis lupus chanco) from Qinghai–Tibet Plateau. PLoS Genet. 10, e1004466 (2014).
Arbiser, J.L. et al. Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proc. Natl. Acad. Sci. USA 99, 715–720 (2002).
Fallone, F., Britton, S., Nieto, L., Salles, B. & Muller, C. ATR controls cellular adaptation to hypoxia through positive regulation of hypoxia-inducible factor 1 (HIF-1) expression. Oncogene 32, 4387–4396 (2013).
Cliby, W.A. et al. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 17, 159–169 (1998).
Hammond, E.M., Dorie, M.J. & Giaccia, A.J. Inhibition of ATR leads to increased sensitivity to hypoxia/reoxygenation. Cancer Res. 64, 6556–6562 (2004).
International Stroke Genetics Consortium (ISGC) & Wellcome Trust Case Control Consortium 2 (WTCCC2). Genome-wide association study identifies a variant in HDAC9 associated with large vessel ischemic stroke. Nat. Genet. 44, 328–333 (2012).
Isaacs, J.S., Jung, Y.J. & Neckers, L. Aryl hydrocarbon nuclear translocator (ARNT) promotes oxygen-independent stabilization of hypoxia-inducible factor-1α by modulating an Hsp90-dependent regulatory pathway. J. Biol. Chem. 279, 16128–16135 (2004).
Ravn, L.S. et al. Angiotensinogen and ACE gene polymorphisms and risk of atrial fibrillation in the general population. Pharmacogenet. Genomics 18, 525–533 (2008).
Xie, P. et al. Histone methyltransferase protein SETD2 interacts with p53 and selectively regulates its downstream genes. Cell. Signal. 20, 1671–1678 (2008).
Prigione, A. et al. HIF1α modulates cell fate reprogramming through early glycolytic shift and upregulation of PDK1–3 and PKM2. Stem Cells 32, 364–376 (2014).
Lahat, H. et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am. J. Hum. Genet. 69, 1378–1384 (2001).
Wong, C.H., Koo, S.H., She, G.Q., Chui, P. & Lee, E.J. Genetic variability of RyR2 and CASQ2 genes in an Asian population. Forensic Sci. Int. 192, 53–55 (2009).
Watkins, P.A., Maiguel, D., Jia, Z. & Pevsner, J. Evidence for 26 distinct acyl–coenzyme A synthetase genes in the human genome. J. Lipid Res. 48, 2736–2750 (2007).
Valour, D. et al. Energy and lipid metabolism gene expression of D18 embryos in dairy cows is related to dam physiological status. Physiol. Genomics 46, 39–56 (2014).
Bigham, A. et al. Identifying signatures of natural selection in Tibetan and Andean populations using dense genome scan data. PLoS Genet. 6, e1001116 (2010).
Droma, Y. et al. Adaptation to high altitude in Sherpas: association with the insertion/deletion polymorphism in the angiotensin-converting enzyme gene. Wilderness Environ. Med. 19, 22–29 (2008).
Aldashev, A.A. et al. Characterization of high-altitude pulmonary hypertension in the Kyrgyz: association with angiotensin-converting enzyme genotype. Am. J. Respir. Crit. Care Med. 166, 1396–1402 (2002).
Qadar Pasha, M.A. et al. Angiotensin converting enzyme insertion allele in relation to high altitude adaptation. Ann. Hum. Genet. 65, 531–536 (2001).
Gou, X. et al. Whole-genome sequencing of six dog breeds from continuous altitudes reveals adaptation to high-altitude hypoxia. Genome Res. 24, 1308–1315 (2014).
Wang, G.D. et al. Genetic convergence in the adaptation of dogs and humans to the high-altitude environment of the Tibetan plateau. Genome Biol. Evol. 6, 2122–2128 (2014).
Yi, X. et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science 329, 75–78 (2010).
Galen, S.C. et al. Contribution of a mutational hot spot to hemoglobin adaptation in high-altitude Andean house wrens. Proc. Natl. Acad. Sci. USA 112, 13958–13963 (2015).
Storz, J.F. et al. The molecular basis of high-altitude adaptation in deer mice. PLoS Genet. 3, e45 (2007).
Ruan, J. et al. Pseudo-Sanger sequencing: massively parallel production of long and near error-free reads using NGS technology. BMC Genomics 14, 711 (2013).
Margulies, M. et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380 (2005).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Li, R. et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272 (2010).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32, W309–W312 (2004).
Majoros, W.H., Pertea, M. & Salzberg, S.L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Lukashin, A.V. & Borodovsky, M. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 26, 1107–1115 (1998).
Grabherr, M.G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Slater, G.S. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31 (2005).
Haas, B.J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Haas, B.J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Soderlund, C., Bomhoff, M. & Nelson, W.M. SyMAP v3.4: a turnkey synteny system with application to plant genomes. Nucleic Acids Res. 39, e68 (2011).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Zhang, Z., Berman, P. & Miller, W. Alignments without low-scoring regions. J. Comput. Biol. 5, 197–210 (1998).
Vilella, A.J. et al. EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 19, 327–335 (2009).
Finn, R.D., Clements, J. & Eddy, S.R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 (2011).
De Bie, T., Cristianini, N., Demuth, J.P. & Hahn, M.W. CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271 (2006).
dos Reis, M. et al. Phylogenomic datasets provide both precision and accuracy in estimating the timescale of placental mammal phylogeny. Proc. Biol. Sci. 279, 3491–3500 (2012).
Zdobnov, E.M. & Apweiler, R. InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Liu, L., Yu, L., Pearl, D.K. & Edwards, S.V. Estimating species phylogenies using coalescence times among sequences. Syst. Biol. 58, 468–477 (2009).
Mirarab, S. et al. ASTRAL: genome-scale coalescent-based species tree estimation. Bioinformatics 30, i541–i548 (2014).
Shimodaira, H. An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 51, 492–508 (2002).
Shimodaira, H. & Hasegawa, M. CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics 17, 1246–1247 (2001).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Löytynoja, A. & Goldman, N. An algorithm for progressive multiple alignment of sequences with insertions. Proc. Natl. Acad. Sci. USA 102, 10557–10562 (2005).
Löytynoja, A. & Goldman, N. Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science 320, 1632–1635 (2008).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000).
Shen, Y.Y. et al. Adaptive evolution of energy metabolism genes and the origin of flight in bats. Proc. Natl. Acad. Sci. USA 107, 8666–8671 (2010).
Zou, Z. & Zhang, J. Are convergent and parallel amino acid substitutions in protein evolution more prevalent than neutral expectations? Mol. Biol. Evol. 32, 2085–2096 (2015).
Holloway, D.E., Hares, M.C., Shapiro, R., Subramanian, V. & Acharya, K.R. High-level expression of three members of the murine angiogenin family in Escherichia coli and purification of the recombinant proteins. Protein Expr. Purif. 22, 307–317 (2001).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Ai, H. et al. Adaptation and possible ancient interspecies introgression in pigs identified by whole-genome sequencing. Nat. Genet. 47, 217–225 (2015).
Alexander, D.H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Li, H. & Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 475, 493–496 (2011).
Quan, G.Q. & Xie, J.Y. Research on the Golden Monkey (Science and Education Publishing House Press, 2002).
Li, H., Meng, S.J., Men, Z.M., Fu, Y.X. & Zhang, Y.P. Genetic diversity and population history of golden monkeys (Rhinopithecus roxellana). Genetics 164, 269–275 (2003).
Prado-Martinez, J. et al. Great ape genetic diversity and population history. Nature 499, 471–475 (2013).
Scally, A. & Durbin, R. Revising the human mutation rate: implications for understanding human evolution. Nat. Rev. Genet. 13, 745–753 (2012).
Acknowledgements
We thank Z.G. Wu for collection of the samples used for resequencing. We thank Z.H. Yang and J.Z. Zhang for assisting in data analyses. We thank Y.K. Wu and Y.H. Wang for help with the purification of RNASE4 proteins. We thank Y.C. Long and Z.F. Xiang for providing the photographs of snub-nosed monkeys. This study was supported jointly and equally by the Strategic Priority Research Program of the Chinese Academy of Sciences (to Y.-P.Z., G.-D.W., and Y.-B.C.; XDB13000000) and the National Nature Science Foundation of China (to L.Y.; 91131904). L.Y. was supported by the National Program for Support of Top-Notch Young Professionals, State Key Laboratory of Genetic Resources and Evolution (GREKF14-04) and by the program Innovative Research Team in University of Yunnan Province (IRTSTYN). Y.-B.C. was supported by the National Natural Science Foundation of China (81322030, U1502224), Yunnan Province High-Level Talents Introduced Program 2013HA021, and Yunnan Applied Basic Research Projects (2014FA038). This work was supported by the Animal Branch of the Germplasm Bank of Wild Species, Chinese Academy of Sciences (Large Research Infrastructure Funding).
Author information
Authors and Affiliations
Contributions
Y.-P.Z., L.Y., and C.-I.W. designed and managed the project. L.Z., L.W., Y.-H.L., and H.W. performed the genomic resequencing and RNA-seq, and carried out data submission. J.R., K.-L.W., L.F., and Z.-L.D. performed the genomic sequencing, assembly, and synteny and gene family analyses. N.Y. and B.B. performed the genome annotation. X.C., J.Y., and X.-P.W. performed the gene family and evolutionary analyses. X.W. and G.-D.W. performed comparative transcriptomic analyses. G.-D.W., Y.-H.L., and H.W. performed the population genomic analyses. Y.-B.C., C.-P.Y., and S.-C.C. designed and performed the functional experiments. S.-F.W., B.-G.L., J.-G.Z., Y.-Q.Y., C.-L.Z., Y.-C.L., H.-S.L., J.-Y.Y., and J.-Y.H. helped with sample collection. L.Y., C.-I.W., Y.-P.Z., G.-D.W., D.M.I., O.A.R., and Y.L. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 GC content of the R. bieti genome.
Distribution of GC content in the R. bieti (green), macaque (blue), human (brown), and dog (red) genomes. The proportion of 500-bp non-overlapping sliding windows with a given GC content is shown.
Supplementary Figure 2 Summary of the evidence for gene models merged from three types of gene sources.
P, ab initio prediction; H, homology based; R, RNA-seq.
Supplementary Figure 4 Population genomic analysis of R. roxellana.
(a) Population structure analysis of 26 R. roxellana individuals (K = 3). (b) Demographic history of R. roxellana reconstructed using the PSMC model. Blue lines represent the QL population, red lines represent the SNJ population, yellow lines represent the SG population, and orange line represents negative O18. O18 is inversely proportional to temperature.
Supplementary Figure 5 Genes under selection identified by the genome scans in one R. bieti population (DJ) and three R. roxellana populations (SG, QL, and SNJ).
Genes identified by at least two of the three scan methods (ZH, LSBL, and θπ scans) are shown as candidate genes under selection.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–5, Supplementary Tables 1–13 and Supplementary Note. (PDF 1420 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International licence. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, L., Wang, GD., Ruan, J. et al. Genomic analysis of snub-nosed monkeys (Rhinopithecus) identifies genes and processes related to high-altitude adaptation. Nat Genet 48, 947–952 (2016). https://doi.org/10.1038/ng.3615
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3615
This article is cited by
-
Gene expressions between obligate bamboo-eating pandas and non-herbivorous mammals reveal converged specialized bamboo diet adaptation
BMC Genomics (2023)
-
Metagenome and metabolome insights into the energy compensation and exogenous toxin degradation of gut microbiota in high-altitude rhesus macaques (Macaca mulatta)
npj Biofilms and Microbiomes (2023)
-
Enlarged fins of Tibetan catfish provide new evidence of adaptation to high plateau
Science China Life Sciences (2023)
-
Sex differences in habitat use, positional behavior, and gaits of Golden Snub-Nosed Monkeys (Rhinopithecus roxellana) in the Qinling Mountains, Shaanxi, China
Primates (2021)
-
Identification and expression analysis of lncRNA in seven organs of Rhinopithecus roxellana
Functional & Integrative Genomics (2021)
