
Abstract
We report a high-quality chromosome-scale assembly and analysis of the carrot (Daucus carota) genome, the first sequenced genome to include a comparative evolutionary analysis among members of the euasterid II clade. We characterized two new polyploidization events, both occurring after the divergence of carrot from members of the Asterales order, clarifying the evolutionary scenario before and after radiation of the two main asterid clades. Large- and small-scale lineage-specific duplications have contributed to the expansion of gene families, including those with roles in flowering time, defense response, flavor, and pigment accumulation. We identified a candidate gene, DCAR_032551, that conditions carotenoid accumulation (Y) in carrot taproot and is coexpressed with several isoprenoid biosynthetic genes. The primary mechanism regulating carotenoid accumulation in carrot taproot is not at the biosynthetic level. We hypothesize that DCAR_032551 regulates upstream photosystem development and functional processes, including photomorphogenesis and root de-etiolation.
Similar content being viewed by others

Main
Carrot (Daucus carota subsp. carota L.; 2n = 2x = 18) is a globally important root crop whose production has quadrupled between 1976 and 2013 (FAO Statistics; see URLs), outpacing the overall rate of increase in vegetable production and world population growth (FAO Statistics; see URLs) through development of high-value products for fresh consumption, juices, and natural pigments and cultivars adapted to warmer production regions1.
The first documented colors for domesticated carrot root were yellow and purple in Central Asia approximately 1,100 years ago2,3, with orange carrots not reliably reported until the sixteenth century in Europe4,5. The popularity of orange carrots is fortuitous for modern consumers because the orange pigmentation results from high quantities of alpha- and beta-carotene, making carrots the richest source of provitamin A in the US diet6. Carrot breeding has substantially increased nutritional value, with a 50% average increase in carotene content in the United States as compared to 40 years ago6. Lycopene and lutein in red and yellow carrots, respectively, are also nutritionally important carotenoids, making carrot a model system to study storage root development and carotenoid accumulation.
Carrot is the most important crop in the Apiaceae family, which includes numerous other vegetables, herbs, spices, and medicinal plants that enhance the epicurean experience7, including celery, parsnip, arracacha, parsley, fennel, coriander, and cumin. The Apiaceae family belongs to the euasterid II clade, which includes important crops such as lettuce and sunflower8. Genome sequences of euasterid I species have been reported, but only two genomes9,10 have been published among the other euasterid II species.
Here we report a high-quality genome assembly of a doubled-haploid orange carrot, characterization of the mechanism controlling carotenoid accumulation in storage roots, and the resequencing of 35 accessions spanning the genetic diversity of the Daucus genus. Our comprehensive genomic analyses provide insights into the evolution of the asterids and several gene families. These results will facilitate biological discovery and crop improvement in carrot and other crops.
Results
Genome sequencing and assembly
An orange, doubled-haploid, Nantes-type carrot (DH1) was used for genome sequencing. We used BAC end sequences and a newly developed linkage map with 2,075 markers to correct 135 scaffolds with one or more chimeric regions (Supplementary Figs. 1 and 2, and Supplementary Note).
The resulting v2.0 assembly spans 421.5 Mb and contains 4,907 scaffolds (N50 of 12.7 Mb) (Table 1), accounting for ∼90% of the estimated genome size (473 Mb; Supplementary Table 1)11. The scaftig N50 of 31.2 kb is similar to those of other high-quality genome assemblies such as potato12 and pepper13. About 86% (362 Mb) of the assembled genome is included in only 60 superscaffolds anchored to the nine pseudomolecules (Supplementary Table 2). The longest superscaffold spans 30.2 Mb, 85% of chromosome 4.
In mapping of unassembled Illumina reads against the assembled genome, 99.7% of the reads aligned (Supplementary Table 3), suggesting that the unassembled fraction of the carrot genome (∼10%) likely consists of assembled duplicated sequences. No substantial sequence contamination was detected (Supplementary Fig. 3). In mapping of carrot ESTs14, genes identified from transcriptome analysis in 20 unique DH1 tissue types, and 248 ultraconserved genes from the Core Eukaryotic Genes15 data set, ∼94%, 98%, and 99.9% aligned to the carrot genome assembly, respectively, demonstrating that the assembly covers the majority of gene space (Supplementary Tables 4–6).
Mapping of 99.9% of 454 paired-end reads and 95.6% of paired-end BAC reads, within their estimated fragment lengths (Supplementary Table 7), confirmed an accurate assembly. A linkage map including 394 markers aligned with high collinearity to 36 superscaffolds (covering 343.5 Mb) demonstrates correct ordering and orientation of these superscaffolds (Supplementary Figs. 4 and 5).
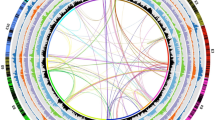
Cytological evaluations using subtelomeric BAC clones and a telomeric probe indicated that the assembly extends into telomeric and subtelomeric regions, further supporting the high physical coverage of the carrot genome assembly (Fig. 1 and Supplementary Fig. 6).

(a) The integrated linkage map for carrot is shown to the far left (the vertical bar to the left indicates genetic distance in cM). Lines connect a subset of markers to the pseudomolecule. Next, from left to right, are shown the cM/Mb ratio, predicted genes (percent of nucleotides/200-kb window), transcriptomes (percent of nucleotides/200-kb window), class I and class II repetitive sequences (percent of nucleotides/200-kb window), noncoding RNAs (percent of nucleotides/200-kb window), and SNPs (number of SNPs/100-kb window). Genes and TEs are more abundant in the distal and pericentromeric regions of the chromosomes, respectively. DNA pseudomolecules are shown in orange to the right. Gray horizontal lines indicate gaps between superscaffolds. Horizontal blue and red lines labeled on the right indicate the locations of BAC probes hybridized to pachytene chromosome 1 (see b); a horizontal yellow line indicates the location of the telomeric repeats. To the far right is a digitally straightened representation of carrot chromosome 1 probed with oligonucleotide probes to the telomeric repeats (Telo; blue) and the CL80 and Cent-Dc repeats (red) and with probes corresponding to BAC 68M03 (red) specific to chromosome 1 and BACs 20G08 and 20P12 (green) flanking the CL80 repeat. (b) FISH mapping of oligonucleotide probes to telomeric repeats (Telo; yellow) and the CL80 repeat (red) and probes corresponding to BAC clones specific to the termini of the short (1S; green) and long (1L; red) arms of carrot chromosome 1. (c–e) FISH mapping of the CL80 (red) and Cent-Dc (K11; green) repeats on the pachytene complements of DH1 (c), D. guttatus (d), and Daucus littoralis (e). Cent-Dc did not generate any detectable signals in D. guttatus or D. littoralis. Scale bars, 5 μm.
Together, the assembly statistics and corroborating evaluations demonstrate that the assembly achieved standard parameters of high quality16. On the basis of genome coverage and length of sequence contiguity, the carrot genome assembly is one of the most complete genomes reported (Supplementary Table 8).
Genome characterization
Carrot coding regions, tandem repeats, and mobile elements were characterized to evaluate the structural and functional features contributing to carrot evolution (Supplementary Note). Repetitive sequences accounted for 46% of the genome assembly (Table 1), of which 98% (193.7 Mb) were annotated as transposable elements (TEs) (Supplementary Table 9). Class II TEs accounted for 57.4 Mb—a greater amount of the genome than in similarly sized plant genomes, including rice (48 Mb)17. Given the abundance of class II TEs, we studied the evolution and distribution of insertion sites for two miniature inverted-repeat transposable element (MITE) class II families, Tourist-like Krak18 and Stowaway-like DcSto19. The expansion of DcSto elements was characterized by multiple amplification bursts (Supplementary Fig. 7). Over 50% of DcSto and Krak insertion sites were located near (<2 kb away from) or inside predicted genes. However, no evidence was found to support their preferential insertion in genic regions (Supplementary Fig. 8), supporting the hypothesis that the impact of DNA transposons on gene function and genome evolution may reflect the interplay of stochastic events and selective pressure20.
Tandem repeat–rich regions create a technical challenge to genome assembly21. By using RepeatExplorer22 and cytology, we identified four major tandem repeat families accounting for ∼7% of the DH1 genome and traced their evolutionary history in the Daucus genus (Supplementary Table 10). These tandem repeats included the carrot centromeric satellite Cent-Dc (CL1)23 and three new tandem repeats (CL8, CL80, and CL81). In DH1 and related species, 39- to 40-bp Cent-Dc monomers were organized in a higher-order repeat structure (Supplementary Fig. 9). Daucus species distantly related to carrot were enriched for the CL80 repeat, which occupied most subtelomeric and pericentromeric regions (Fig. 1 and Supplementary Fig. 10). Conversely, the carrot CL80 sequence was associated with a knob on chromosome 1. Because Cent-Dc and CL80 were detected in members of the divergent Daucus clades (Daucus I and II), we hypothesize that their origin predates the estimated divergence of the two clades ∼20 million years ago24. After Daucus radiated, these repeat families presumably underwent differential expansion and shrinkage of their repeat arrays and structural reorganization of monomers.
In assembly v1.0 gene annotation, 32,113 genes were predicted (Table 1 and Supplementary Note), of which 79% had substantial homology with known genes (Supplementary Tables 11 and 12). The majority (98.7%) of gene predictions had supporting cDNA and/or EST evidence (Supplementary Table 13), demonstrating the high accuracy of gene prediction. Relative to five other closely related genomes, carrot was enriched for genes involved in a wide range of molecular functions (Supplementary Table 14). We also identified 564 tRNAs, 31 rRNA fragments, 532 small nuclear RNA (snRNA) genes, and 248 microRNAs (miRNAs) distributed among 46 families (Fig. 1 and Supplementary Table 15).
Carrot diversity analysis
To evaluate carrot domestication patterns, we resequenced 35 carrot accessions, representative D. carota subspecies, and outgroups (Daucus syrticus, Daucus sahariensis, Daucus aureus, and Daucus guttatus) (Supplementary Table 16). After filtering, 1,393,431 high-quality SNPs (accuracy >95%; Supplementary Note) were identified, with the largest number of diverging or alternate alleles in outgroups, a signature of genome divergence (Supplementary Table 17). Phylogenetic and cluster analysis separated samples by geographical distribution relative to carrot's Central Asian center of origin (eastern or western) and cultivation status (wild, cultivated, open pollinated, or inbred) (Fig. 2a). Eastern wild accessions were most closely related to cultivated carrots, further demonstrating a primary center of carrot domestication in the Middle East and Central Asia3. Cluster analysis showed extensive allelic admixture (Fig. 2a), reflective of the outcrossing nature within carrot combined with extensive geographical overlap between wild and cultivated carrot lines4. This pattern was particularly evident in eastern wild and cultivated samples, likely caused by less intensive carrot breeding in eastern regions. Indeed, some eastern cultivated carrots still maintain primary taproot lateral branching and reduced pigmentation (Supplementary Fig. 11). In contrast, western cultivars clearly separated from wild and eastern cultivated carrots, and some inbred lines (I3 and I4) have a purified genetic pattern shared with western cultivated accessions, reflecting the intensive breeding practiced in western regions.

(a) Top, neighbor-joining phylogenetic tree of carrot and other Daucus accessions based on SNPs. Bottom, population structure of Daucus accessions. Each color represents a subpopulation, and each accession is represented by a vertical bar. The length of the colored segments in the vertical bars represents the proportion contributed by each subpopulation. (b) The 26 inner tracks depict the SNP frequency distributions for 100-kb non-overlapping windows in the 8 wild D. carota subsp. carota accessions (blue tracks), 14 open-pollinated cultivars and local land races (green tracks), and 4 inbred lines (orange tracks). The outermost track shows the positions of SNPs with the top 1% of FST values, estimated by comparing SNPs from wild and cultivated eastern accessions. The track below this shows the location of markers spanning the QTLs associated with the Y (chromosome 5) and Y2 (chromosome 7) loci40.
Nucleotide diversity (π)25 estimates showed that wild carrots have a slightly higher level of genetic diversity than cultivated carrots (Supplementary Table 18), indicating the occurrence of a limited domestication bottleneck, consistent with previous findings3,26. When D. carota subspecies, which have morphological characteristics contributing to their sexual isolation relative to carrot27, were excluded from diversity estimates, this observation was more evident from comparative analysis (wild, π = 9.5 × 10−4 versus cultivated, π = 8.6 × 10−4). In contrast, a clear reduction in genetic diversity and heterozygosity was found in inbred lines (Fig. 2b and Supplementary Table 17), likely resulting from their use in hybrid carrot breeding programs28.
To identify genomic regions associated with domestication events, we computed pairwise population differentiation (FST) levels for wild and cultivated eastern accessions29, as these samples resemble the genetic pool for primary carrot domestication. We identified local differentiation signals on chromosomes 2, 5, 6, 7, and 8. Peaks on chromosomes 5 and 7 overlap with previously mapped quantitative trait loci (QTLs) controlling carotenoid accumulation in tap root (Fig. 2b), a major domestication trait in carrot.
Genome evolution
Comparative phylogenomic analysis among 13 plant genomes (Supplementary Table 19 and Supplementary Note) indicated that carrot diverged from grape ∼113 million years ago, from kiwifruit ∼101 million years ago, and from potato and tomato ∼90.5 million years ago, confirming the previously estimated dating of the asterid crown group to the Early Cretaceous and its radiation in the Late–Early Cretaceous8 (Fig. 3a and Supplementary Fig. 12). Further divergence between carrot and lettuce, both members of the euasterid II clade, likely occurred ∼72 million years ago.

(a) Evolutionary relationships of the eudicot lineage (Supplementary Fig. 12). Circles indicate the ages of WGD (red) or WGT (blue) events. Age estimates for the A. thaliana, kiwifruit, lettuce, and Solanaceae WGD and WGT events and for the γ WGT event were obtained from the literature30,65,66. The polyploidization level of the kiwifruit WGDs (purple circles) awaits confirmation. Mya, million years ago. (b) Age distribution of fourfold-degenerate sites for genes from the D. carota, A. thaliana, and Solanum lycopersicum genomes. The x axis shows fourfold-degenerate transversion rates; the y axis shows the percentage of gene pairs in syntenic or collinear blocks. The γ peak represents the ancestral γ WGT shared by core eudicots; Dc-α and Dc-β represent carrot-specific WGD and WGT events, respectively. (c) The distribution of remaining carrot duplicated blocks derived from the seven eudicot protochromosomes. (d) Synteny of carrot protochromosome A19 with corresponding blocks on grape, coffee, tomato, and kiwifruit chromosomes. Vertical bars indicate the depth of primary correspondence to carrot protochromosome A19. Of the 110 syntenic blocks identified in comparison of carrot and grape protochromosome A19, a substantial portion (43; 39.1%) correspond to 6 grape blocks. A similar pattern was observed for the carrot–coffee, carrot–kiwifruit, and carrot–tomato comparisons, indicating that carrot has experienced either 3 × 2 or 2 × 3 WGD events. (e) Representation of carrot-specific genome duplications. The tracks, from outermost to innermost, show GC content (%), density of tandem duplications (number per 0.5-Mb window), genes retained in the carrot Dc-α (cyan) and Dc-β (blue) events, chromosomal blocks descending from the seven ancestral core eudicot protochromosomes (colored as in c), and duplicated segments derived from the Dc-α (dashed links; duplicates) and Dc-β (solid links; triplicates) events.
We identified two new whole-genome duplications (WGDs) specific to the carrot lineage, Dc-α and Dc-β, superimposed on the earlier γ paleohexaploidy event shared by all eudicots (Fig. 3a,b). These WGDs likely occurred ∼43 and ∼70 million years ago, respectively (Fig. 3a). Estimating the timing of the Dc-β WGD to around the Cretaceous–Paleogene (K–Pg) boundary further supports the hypothesis that a WGD burst occurred around that time, perhaps reflecting a selective polyploid advantage in comparison to diploid progenitors30. These results may also suggest a co-occurrence of the Dc-β WGD with Apiales–Asterales divergence. To address this possibility, we compared the carrot genome with the genome of horseweed (Conyza canadensis) (Supplementary Note), an Asteraceae with a low-pass whole-genome assembly9. Pairwise paralog and ortholog gene divergence indicated that a possible WGD occurred in the horseweed genome that does not overlap with the carrot Dc-β event, as it occurred after divergence with carrot (Supplementary Fig. 13). This WGD is likely shared with lettuce and may represent a whole-genome triplication (WGT) recently described in lettuce that is basal to Asteraceae31.
Using methods previously described32,33, we reconstructed the carrot paleopolyploidy history. Carrot chromosomal blocks descending from the seven ancestral core eudicot chromosomes were highly fragmented and dispersed along the nine carrot chromosomes (Fig. 3c). The two lineage-specific WGDs were clearly evident from the distribution of the fourfold-degenerate transversion rates of carrot paleohexaploid paralogous genes, whereas genes from the shared eudicot γ WGT were largely lost, likely owing to extensive genome fractionation (Supplementary Fig. 14). Comparative analysis with the grape, tomato, coffee, and kiwifruit genomes identified a clear pattern of multiplicons (1:5 or 1:6 ratio) (Fig. 3d). Depth analysis of duplicated blocks harboring paralogous genes under the Dc-α fourfold-degenerate transversion peak indicated over-retention of duplicated blocks. In contrast, duplicated blocks harboring paralogous genes under the Dc-β peak retained a larger number of triplicated blocks (Fig. 3e). We suggest that at least 60 chromosome fusions or translocations and a lineage-specific WGT (Dc-β) followed by a WGD (Dc-α) contributed to diversification of the 9 carrot chromosomes from the 21-chromosome intermediate ancestor.
Characterization of Dc-α and Dc-β duplicated blocks demonstrated that extensive gene fractionation has occurred during the evolutionary history of the carrot genome (Supplementary Tables 20 and 21). Dc-α ohnologs are significantly enriched (P ≤ 0.01) in protein domains involved in selective molecule interactions (binding) and protein dimerization functions (Supplementary Table 22), supporting the gene dosage hypothesis34; this observation predicts that categories of genes encoding interacting products will likely be over-retained.
Regulatory genes
Characterization of orthologous gene clusters across multiple genomes identified 26,320 carrot genes in 13,881 families, with 10,530 genes unique to carrot (Supplementary Fig. 15). Protein domains involved in regulatory functions (binding) and signaling pathways (protein kinases) were abundant among the genes unique to carrot (Supplementary Tables 23 and 24).
We identified 3,267 (10% of the total) regulatory genes in carrot, a number similar to that in tomato (3,209 regulatory genes) and rice (3,203 regulatory genes) (Supplementary Tables 25 and 26, and Supplementary Note). Overall, genomes that experienced WGDs after the γ paleohexaploidization event harbored more regulatory genes. In carrot, large-scale duplications represented the most common mode of regulatory gene expansion, with ∼33% of these genes retained after the two carrot WGDs, demonstrating the evolutionary impact of large-scale duplications on plant regulatory network diversity34 (Supplementary Table 27). Six regulatory gene families involved in lineage-specific duplications were expanded in carrot (Supplementary Table 28). The expanded families include a zinc-finger (ZF-GFR) regulatory gene family, the JmjC, TCP, and GeBP families, the B3 superfamily, and response regulators. The over-represented regulatory gene subgroups shared orthologous relationships with functionally characterized genes involved in cytokinin signaling, which can influence the circadian clock as well as plant morphology and architecture (Supplementary Figs. 16–20). For example, the expanded JmjC, response regulator, and B3-domain subgroups share ancestry with the Arabidopsis thaliana REF6; PRR5, PRR6, and PRR7; and VRN1 genes, respectively, which regulate flowering time35,36,37, a major trait in plant adaption and survival.
Pest and disease resistance genes
Using the MATRIX-R pipeline38 with additional manual data curation, we predicted 634 putative pest and disease resistance (R) genes in carrot (Supplementary Tables 29–34 and Supplementary Note). Most R gene classes were under-represented in carrot. The expanded orthologous subgroups included classes containing the NBS and LRR protein domains (NL) and coiled-coil NBS and LRR domains (CNL). Lineage-specific duplications contributed to the expansion and diversification of these R gene families in carrot and other genomes (Supplementary Fig. 21 and Supplementary Table 35). Many R genes (206) were located in clusters, and these clusters tended to harbor genes from multiple R gene classes (Supplementary Tables 36 and 37). The expansion of the NL and CNL families might reflect evolutionary events generating tandem duplications, resulting in preferential clustering on chromosomes 2 and 3–7, respectively (Supplementary Fig. 22). One cluster containing three RLK genes and one LRR gene, spanning only 50 kb, colocalized with the carrot Mj-1 region, which controls resistance to Meloidogyne javanica, a major carrot pest39 (Supplementary Fig. 22). This analysis demonstrates the important role of tandem duplications in the expansion of R genes in carrot. Additionally, R gene clusters may provide a reservoir of genetic diversity for evolving new plant–pathogen interactions.
A candidate gene controlling high carotenoid accumulation
Carotenoids were first discovered in carrot and named accordingly. The Y and Y2 gene model explains the phenotypic differences between white and orange carrots40,41, with elevated carotenoid accumulation in homozygous-recessive genotypes (yyy2y2). In spite of the striking color variation attributed to these two genes, little is known about the molecular basis of carotenoid accumulation in carrot. Although homologs of all known carotenoid biosynthesis genes have been identified in carrot, none appear to be responsible for carotenoid accumulation42,43,44,45,46. Using two mapping populations, we demonstrated that Y regulates high carotenoid accumulation in both yellow and dark orange roots (Fig. 4a, Supplementary Figs. 23 and 24, Supplementary Table 38, and Supplementary Note), a result consistent with the previously proposed model41. Fine-mapping analysis identified a 75-kb region on chromosome 5 that harbors the Y gene (Fig. 4b–e and Supplementary Fig. 25). Of the eight genes predicted in this region, none had homology with known isoprenoid biosynthesis genes (Supplementary Table 39), implying that regulation of carotenoid accumulation in carrot roots by the Y locus extends beyond the isoprenoid biosynthesis genes. Within the 75-kb region, DCAR_032551 was the only gene to have a mutation that segregated with high carotenoid pigmentation. DCAR_032551 harbors a 212-nt insertion in its second exon that creates a frameshift mutation in both yellow and dark orange carrots (Supplementary Fig. 26 and Supplementary Table 39).

(a) Phenotypes associated with the Y locus, including pale orange (pOr), dark orange (dOr), yellow (Y), and white (W) roots, from the indicated populations. (b) Previously published genetic map and location of the Y locus41. (c) Carrot chromosome 5 and the molecular markers used for fine-mapping of the Y locus. The genotypes of the 76-kb region in recombinant individuals are illustrated (Supplementary Fig. 25). Het, heterozygous. (d) The fine-mapped region controlling the Y locus. Numbers represent the eight genes predicted in this region. Gene 7, DCAR_032551, was the only gene differentially expressed (upregulated) in RNA-seq analysis of yellow versus white and dark orange versus pale orange samples. Below are all the nonsynonymous SNPs (for example, G>A) and insertions (ins) identified in the four genes located in the 65-kb haplotype block associated with the Y locus in the resequencing samples (Supplementary Table 40). The number of accessions with each haplotype block classification (I–III; Supplementary Table 17) is given. The DCAR_032551 ya variant harbors a 212-nt insertion in the second exon, and the yb variant harbors a 1-nt insertion in the second exon. Het, heterozygous. (e,f) Nucleotide diversity (π) estimated in wild (blue) and cultivated (orange) carrots (e) and the top 1% of FST values (blue) (f) in the 75-kb region (gray shading) of carrot chromosome 5. (g,h) Patterns of LD in wild (g) and cultivated (h) carrots. Red and white spots indicate regions of strong (r2 = 1) and weak (r2 = 0) LD, respectively. The gray bar indicates the position of the 75-kb fine-mapped region harboring the Y candidate gene.
Using resequencing data, a haplotype block extending for 65 kb, with 64 kb overlapping the fine-mapped region, was associated with all but two highly pigmented root samples (C1 and I2) (Supplementary Fig. 27). In contrast, within the 65-kb region, seven haplotype blocks were detected in wild accessions. Polymorphism detection within the haplotype block identified eight nonsynonymous SNPs in four genes and two indels, including the 212-nt insertion in DCAR_032551, in yellow and dark orange samples (Fig. 4f and Supplementary Table 40). No wild or cultivated white samples had the 212-nt insertion. The two highly pigmented (yy) accessions, C1 and I2, that did not share the 65-kb haplotype block were heterozygous for the insertion. However, further analysis of DCAR_032551 identified a 1-nt insertion in the second exon, 60 nt upstream of the 212-nt insertion site (Fig. 4f and Supplementary Fig. 26). The 1-nt insertion was in trans phase relative to the 212-nt insertion, indicating that these accessions harbor two frameshift mutations that likely disrupt functioning of the Y gene product. Thus, resequencing supports the central role of DCAR_032551 in conditioning high pigment accumulation in carrot roots and identifies a second, independent mutation in this same gene, which we speculate should also be recessive to the wild-type allele.
To determine whether this region was ever under selection, we scanned for differences in nucleotide diversity, differentiation, and linkage disequilibrium (LD) between wild and cultivated accessions. An FST peak on chromosome 5, located between 24.4 and 25.0 Mb, overlapped the 75-kb fine-mapped region underlying DCAR_032551 (Figs. 2c and 4g,h). In this region, LD was increased in highly pigmented cultivated materials and nucleotide diversity was drastically reduced in cultivated carrots (wild, π = 3.1 × 10−4 versus cultivated, π = 2.0 × 10−4) (Fig. 4g,h). The 50-kb window encompassing the Y candidate gene had the highest level of differentiation (FST = 1.0) and the lowest level of nucleotide diversity (π = 1.5 × 10−4) among cultivated carrots. The selective sweep in the Y region is relatively short in comparison with those for other genes controlling carotenoid accumulation, including the selective sweep for y1 in maize, which extends 200 kb upstream and 600 kb downstream of the gene47. Rather, this scenario resembles the short sweep (60–90 kb) identified in maize around teosinte branched1 (tb1), a major domestication-associated gene48. A short sweep may reflect the highly effective rates of recombination expected in an outcrossing species like carrot. Gene flow between wild and cultivated carrot followed by recurrent phenotypic selection that likely occurred throughout the history of carrot4 may have had a role in increasing the recombination rate around the Y locus.
Selection signatures, including reduction in nucleotide diversity and a decrease in the number of haplotypes, associated with the Y gene region further support the inclusion of carotenoid accumulation as a major domestication trait—a trait that contributes substantial nutritional and economic value to modern carrots. Furthermore, the identification of a second haplotype block for pigmentation surrounding the Y candidate gene suggests that this gene has been selected multiple times. These results may elucidate the timing and origin of the pigmented taproot phenotype during carrot domestication.
A model for carotenoid accumulation in carrot roots
To investigate gene expression in the region of the Y candidate, comparative transcriptome analysis was performed for white versus yellow and pale orange versus dark orange roots (Supplementary Note). DCAR_032551 was the only significantly differentially expressed (upregulated; P ≤ 0.001) gene in the yy (yellow and dark orange) relative to the Y– (white and pale orange) genotype (Supplementary Table 39), further supporting our mapping and resequencing results.
Weighted gene coexpression network analysis (WGCNA) indicated that DCAR_032551 is coordinated with a set of 925 genes (Supplementary Table 41). Gene Ontology (GO) term enrichment analysis indicated that isoprenoid pathway genes were particularly enriched (Supplementary Table 42). Among cellular components, membrane terms and molecular function terms related to oxidative reactions and biological processes in response to acids and chemicals were highly enriched (Supplementary Table 43). Assuming a conserved function of Y in yellow and dark orange roots, we annotated genes that were differentially expressed (upregulated or downregulated) in white versus yellow and pale orange versus dark orange comparisons. This analysis identified a positive relationship between high carotenoid accumulation and overexpression of several light-induced genes, including those involved in photosynthetic system activation and function, plastid biogenesis, and chlorophyll metabolism (Supplementary Tables 44 and 45), an unexpected finding in non-photosynthetic root tissue. These findings tie into the WGCNA analysis as components of photomorphogenesis are located in the thylakoid membranes and involve many oxidative processes and chemical responses, including hormonal regulation. Analysis of the 98 genes annotated in the plastidal methylerythritol phosphate (MEP) and carotenoid pathways (Supplementary Table 46 and Supplementary Note) confirmed coordinated overexpression of several genes in these pathways and carotenoid accumulation in yy plants. Furthermore, an inverse relationship was observed between the majority of differentially expressed terpene synthase genes (Supplementary Table 47) and high carotenoid accumulation, consistent with substrate flux into the carotenoid pathway. DXS1 and LCYE were the only genes in the MEP and carotenoid pathways that were differentially expressed in yy genotype samples with high carotenoid accumulation in both populations, suggesting that they possibly encode enzymes that regulate carotenoid accumulation. Although LCYE has not been reported to be a carotenoid regulatory gene target, its elevated expression may account for the relative abundance of lutein in yellow carrots and alpha-carotene in orange carrots. DXS1 is a limiting factor in upregulation of the carotenoid pathway in A. thaliana49. DXS1 expression is induced by light50,51, and it is the main DXS isoform catalyzing the biosynthesis of isoprenoid and carotenoid precursors in photosynthetic metabolism52,53. DXS1 also regulates carotenoid accumulation in A. thaliana and tomato54,55. Overall, these results indicate that DCAR_032551 is coexpressed with isoprenoid pathway genes and that overexpression of the light-induced/photosynthetic transcriptome cascades in orange and yellow carrot roots may explain elevated carotenoid accumulation.
The DCAR_032551 gene product represents a plant-specific protein of unknown function, and mutants of the A. thaliana homolog PSEUDO-ETIOLATION IN LIGHT (PEL) have an etiolated phenotype, a phenotype associated with defective responses to light56 (Supplementary Table 44). In many ways, the physiology and genetics of carotenoid accumulation in dark orange and yellow (yy) carrots are similar to the phenotypes of the A. thaliana det, cop, and fus de-etiolated mutants. These mutants lack the ability to inhibit the light-induced photosynthetic transcriptome cascade associated with de-etiolation and photomorphogenesis in non-photosynthetic tissues such as roots57. De-etiolated mutants grown in the dark have characteristics of light-grown seedlings, including carotenoid accumulation and overexpression of light-induced photosystem and plastid biogenesis genes58,59. In contrast, when exposed to light, these mutants demonstrate ectopic expression of genes involved in chloroplast formation58. Physiological studies have demonstrated that, unlike other species, carrots with carotenoid-rich roots have ectopic chloroplast accumulation when exposed to light44,60 and that highly pigmented carrot roots have upregulation of photosystem-related genes in comparison with white roots27,61. These observations when coupled with the transcriptome data presented here indicate that, similar to de-etiolated A. thaliana mutants, carrot roots with high levels of carotenoid accumulation may have lost the ability to inhibit the transcriptome cascade associated with de-etiolation and photomorphogenesis. The recessive nature of the Y gene in such roots is compatible with loss of the constitutive negative feedback function associated with the recessive det, cop, and fus mutants in A. thaliana. In addition, the A. thaliana homolog of the Y candidate produces a protein that interacts with genes such as FAR1 and COP9, involved in the light signaling pathway (Supplementary Table 48). Our hypothesis is further supported by the WGCNA analysis indicating that DCAR_032551 is coexpressed with COP1 and HY5 (Supplementary Table 41), genes both directly involved in the regulation of photomorphogenesis. Together, these findings make DCAR_032551 a plausible regulatory candidate. Considering our results coupled with previous physiological studies44, we hypothesize that carotenoid accumulation in carrot taproot results from root de-etiolation, whereby the repression of photomorphogenic development typically found in etiolated roots is lifted. The resulting overexpression of DXS1 provides precursors to the carotenoid biosynthetic pathway, which leads to an accumulation of carotenoids in orange and yellow (yy) carrot roots (Fig. 5).

Upward- and downward-pointing arrows indicate upregulated and downregulated genes, respectively, in the yellow versus white (yellow arrows) and dark orange versus pale orange (orange arrows) comparisons. The orange box delimits the isoprenoid biosynthetic branch that leads to the carotenoid pathway. As shown in the green box, the majority of the upregulated genes in yellow and dark orange roots are involved in the photosynthetic pathway (Supplementary Table 45); genes that are included are involved in the assembly and function of photosystems I and II and plastid development. We hypothesize that loss of the constitutive repression mechanisms conditioned by genes involved in de-etiolation and photomorphogenensis in non-photosynthetic tissue, such as carrot roots, induces overexpression of DXS1 and, consequently, activation of the metabolic cascade that leads to high levels of carotenoid accumulation in carrot roots.
Discussion
Vitamin A deficiency is a global health challenge62, making the development of sustainable vitamin A sources a priority for crop improvement. Its plentiful carotenoids make carrot an important source of provitamin A in the human diet6. Although carrot was a model organism to study plant development and totipotency in the 1950s63,64, the molecular basis of neither carrot growth nor phytochemical accumulation has been well described. The high-quality carrot genome sequence described here, in combination with mapping and comparative transcriptome analysis, demonstrates that carotenoid accumulation in carrot is controlled at the regulatory level and that root de-etiolation leading to overexpression of the photosynthetic transcriptome cascade may have an important role in this regulatory mechanism. These results provide the foundation for new genetic mechanisms regulating carotene accumulation in plants, with potential application to several crops.
This study included the first comparative genomic and phylogenomic analyses comprising members of the euasterid II clade and clarified the evolutionary events surrounding the radiation of the main asterid clades. The two new WGD events (Dc-α and Dc-β) identified provide a new tool to study genome polyploidization. The two WGDs specific to the carrot lineage and the new WGD identified in the horseweed genome, which is possibly shared with lettuce, prompt important evolutionary questions about the possible involvement of the latter WGD in the early radiation of the Asterales order. The carrot genome is the first chromosome-scale Apiaceae genome to be sequenced and will provide a foundation for future comparative genomic and evolutionary studies.
Resequencing diverse Daucus species emphasized a high level of variability in repetitive sequence structure and chromosomal location, demonstrated a high level of genetic diversity retained in cultivated carrots, and identified a genetic sweep associated with domestication. This information lays the groundwork for future studies on carrot domestication and chromosome evolution across the Daucus genus.
The high-quality carrot reference genome and large set of SNP markers will accelerate marker-facilitated trait mapping through genome-wide association studies and genomic selection. The carrot genome sequence will support crop improvement efforts and help identify additional candidate genes underlying isoprenoid and flavonoid accumulation, biotic and abiotic stress resistance, and regulatory pathways controlling growth, flowering, seed production, and regeneration in tissue culture—all important traits for sustained agricultural production and improved human health.
Methods
Plant materials and sequencing.
Genome assembly used doubled-haploid NCBI BioSample SAMN03216637. Sequences included 3 paired-end Illumina libraries, 5 mate-paired Illumina libraries, and 40,693 BAC end sequences (Supplementary Tables 49 and 50). The abundance of 17-nt k-mers from 170- and 800-nt libraries (Supplementary Fig. 28) was used to estimate the genome size (k_num/peak depth) (Supplementary Note).
De novo assembly.
Genome assembly used SOAPdenovo version 2.04 (ref. 67). Gaps were filled using GapCloser. This generated assembly v1.0 (Supplementary Table 51).
To guide the construction of superscaffolds and anchor the genome, an integrated linkage map was developed using JoinMap 4.0 (ref. 68). CheckMatrix (see URLs) was used to remove markers with inconsistent placement. The collinearity of common markers was inspected using MapChart 2.2 (ref. 69), and inconsistent markers were removed before merging maps. Markers in common were used as anchor points (Supplementary Tables 52 and 53). Marker order correlations between composite and component map linkage groups were calculated in SAS 9.2 using the PROC CORR Spearman function (Supplementary Table 54). Linkage groups were assigned to chromosomes, oriented, and numbered using published classification23.
To build superscaffolds and to identify chimeric scaffolds and correct them, 29,875 paired-end BACs, 20-kb and 40-kb Illumina mate-paired sequences, and 2,075 marker sequences mapped in the carrot integrated linkage map were aligned to the v1.0 assembly. For each scaffold or contig, unambiguously aligned sequences were visualized in GBrowse. Superscaffolding was initiated with scaffolds containing sequences of mapped markers. Scaffold connections supported by at least two paired-end BACs were annotated, and sequences were further connected using a custom Perl script (cp1; see URLs). The quality of each scaffold assembly and contiguity were verified by visually inspecting the coverage of large-insert libraries (20 and 40 kb) and the consistency of marker order along the linkage map.
Possible chimeric scaffolds (Supplementary Fig. 2) were identified as those containing sequences of markers mapped to different linkage groups or to distal locations of the same linkage group or those containing regions not covered by mate-paired sequences. Within each chimeric scaffold, the chimeric regions were identified as those regions not covered by mate-paired or paired-end BAC sequences and were then manually inspected. The midpoint between the closest unambiguously aligned paired-end sequences flanking the chimeric region was defined as the misassembly point. Corrected scaffolds were then used to progressively construct superscaffolds as described above. This process generated assembly v2.0 and nine carrot pseudomolecules (Supplementary Figs. 29 and 30, and Supplementary Table 55).
See the Supplementary Note for additional details.
De novo assembly of the plastid and mitochondrial genomes is described in the Supplementary Note.
Genome quality evaluation.
The presence of possible sequence contamination was evaluated using DeconSeq70 with scaftigs from the v2.0 assembly.
To evaluate the correctness of the assembled sequences, we used (i) an 8-kb 454 library of DH1 (SRA accession SRX1135252) and (ii) 4,717 paired-end BACs that were not used to join scaffolds into superscaffolds during assembly. Paired-end reads that aligned with both ends to a unique location in the carrot plastid genome or the v2.0 assembly were used to calculate the mean insert size.
A new linkage map including GBS SNP markers was developed to verify the order of the scaffolds and superscaffolds. GBS libraries were prepared as described by Elshire et al.71, with minimal modification. TASSEL version 4.3.11 (ref. 72) was used for analysis, with paired-end data preprocessed for TASSEL compatibility using a custom Perl script, bb.tassel (see URLs). SNPs were called using documented GBS pipeline procedures73. Sequences containing SNPs unambiguously aligned to the carrot genome assembly were kept (18,007 SNPs). SNPs scored as heterozygous but with an allele ratio a:b far from 1:1 were eliminated if the ratio was <0.3 or >3.0, where a and b were the two alleles for a given SNP. Mapping was carried out as described74 (Supplementary Fig. 31 and Supplementary Note).
FISH experiments were carried out to evaluate consistency and coverage of the carrot genome assembly in telomeric regions. Anther preparation and the FISH procedure were carried out according to published protocols75,76 using five types of probes: (i) BAC probes specific for subtelomeric regions on the short (1S, 2S, 4S, 5S, 6S, 8S, 9S) and long (1L, 2L, 4L, 5L, 6L, 8L, 9L) arms of each chromosome, (ii) carrot chromosome-specific BAC probes23, (iii) telomeric probe (Telo), (iv) a probe corresponding to the CL80 repetitive sequence, and (v) plasmid K11 containing the putative carrot centromere repeat (Cent-Dc)23 (Supplementary Table 56).
Gene space coverage was evaluated using carrot ESTs14, RNA-seq data from 20 different DH1 tissues (NCBI BioSamples SAMN03965304–SAMN03965323), and 258 ultraconserved genes from the Core Eukaryotic Genes data set. Previously published carrot ESTs14 were aligned to the genome using BLASTN77; RNA-seq data from 20 different DH1 tissues (NCBI BioProject PRJNA291977) were assembled with Trinity r2013_08_14 and mapped to the assembly using TopHat v2.0.11 (ref. 78). Scaftigs from the carrot assembly were aligned to the Core Eukaryotic Genes data set15 using CEGMA v2.4.
See the Supplementary Note for additional details.
Repetitive sequences, gene prediction, and genome annotation.
RepeatMasker v3.2.9 (see URLs) was applied to screen the genome assembly for low-complexity DNA sequences and interspersed repeated elements using a custom library. Ab initio prediction with RepeatModeler version 1.1.0.4 (see URLs) generated a de novo repeat library from the assembled genome. RepeatMasker and LTR_FINDER version 1.1.0.5 (ref. 79) were then used to identify and classify repeat elements in the genome (Supplementary Fig. 32 and Supplementary Table 9).
MITEs belonging to the Tourist-like Krak18 and Stowaway-like DcSto19 families were identified using TIRfinder80, including the carrot, kiwifruit, pepper, tomato, and potato genomes. MITE copies were grouped into families fulfilling the 80–80–80 criterion81 (Supplementary Fig. 33 and Supplementary Tables 57–59). Consensus sequences were used to investigate intra- and interspecific relationships among families with Circoletto82,83 (Supplementary Fig. 34). Stowaway-like elements carrying insertions >10 nt in length were removed from subsequent steps. Within-family similarity was calculated from a Kimura two-parameter pairwise distance matrix. The evolutionary history of related DcSto elements was investigated using MEGA6 (ref. 84).
Tandem repetitive sequences were analyzed with RepeatExplorer22 and SeqGrapheR85 using a subset of 1 × 107 Illumina reads from DH1 and five resequenced genotypes representative of Daucus clades I and II. To select tandem repetitive sequences, the node/edge ratio (number of nodes/number of edges) among aligned sequences in each cluster was calculated. Clusters with a ratio >0.09, representing more than 0.05% of the genome, were selected for further analysis. Tandem repeats were identified using Tandem Repeats Finder v4.07b86 (Supplementary Tables 60 and 61).
The abundance and localization of selected repetitive sequences in DH1 and other Daucus species were also investigated by FISH (Supplementary Note).
For gene model prediction, mobile element–related repeats were masked using RepeatMasker (see URLs). De novo prediction using AUGUSTUS v2.5.5 (ref. 87), GENSCAN v.1.1.0 (ref. 88), and GlimmerHMM-3.0.1 (ref. 89) was trained using model species A. thaliana and S. lycoperisum training sets. The protein sequences of S. lycoperisum, Solanum tuberosum, A. thaliana, Brassica rapa, and Oryza sativa were mapped to the carrot genome using TBLASTN77 (BLAST All 2.2.23) and analyzed with GeneWise version 2.2.0 (ref. 90). Carrot ESTs14 were aligned to the genome using BLAT91 and analyzed with PASA92 to detect spliced gene models. RNA-seq reads from 20 DH1 libraries were aligned with TopHat 2.0.9 (ref. 78). Transcripts were predicted by Cufflinks93. All gene models produced by de novo prediction, protein homology searches, and prediction and transcript-based evidence were integrated using GLEAN v1.1 (ref. 94).
Putative gene functions were assigned using the best BLASTP77 match to SwissProt and TrEMBL databases. Gene motifs and domains were determined with InterProScan version 4.7 (ref. 95) against the ProDom, PRINTS, Pfam, SMART, PANTHER, and PROSITE protein databases. GO IDs for each gene were obtained from the corresponding InterPro entries. All genes were aligned against KEGG (release 58) proteins.
miRNAs and snRNAs in the assembled genome were detected using INFERNAL96 against the Rfam database (release 9.1). tRNA loci were detected using tRNAscan-SE v1.1.23 (ref. 97). rRNA was detected by homologous BLASTN77 searches using the closest available species with complete sequences, Panax ginseng, P. quinquefolius, and Thapsia garganica (accessions KM036295.1, KM036296.1, KM036297.1, and AJ007917.1).
See the Supplementary Note for additional details.
Resequencing.
Resequencing data under NCBI BioProject PRJNA291976 (BioSamples SAMN03766317–SAMN03766351) include 18 cultivated accessions, 13 wild accessions, and 4 other Daucus species (Supplementary Table 16).
DNA from single plants was extracted as described by Murray and Thompson98. Paired-end libraries with insert sizes of 250–350 nt were sequenced using Illumina technology at BGI.
Reads were mapped with BWA-MEM version 0.7.10 (ref. 99). Alignments were filtered using SAMtools version 0.1.19 (ref. 100). Duplicate reads were marked using MarkDuplicates from Picard tools version 1.119 (see URLs). GATK version 3.3-0 (ref. 101) was used to identify SNPs for each genotype.
The accuracy of SNP calls was evaluated with 3,202 previously characterized SNPs3. A random subset of 49,365 biallelic SNPs was analyzed with STRUCTURE v2.3.4 (ref. 102), and the most accurate population structure was determined by the method discussed in Evanno et al.103.
Phylogenic analysis used PHYLIP v3.5 (ref. 104) with this same subset. Seqboot was used for bootstrapping with 1,000 replicates, and genetic distances were calculated using gendist. A neighbor-joining tree was created using the neighbor function, and a consensus tree was generated using consense.
See the Supplementary Note for additional details.
Genome evolution.
Gene clusters with 13 other species were identified using OrthoMCL v2.0.2 (ref. 105) (Supplementary Tables 19 and 62).
Peptide sequence from 312 single-copy orthologous gene clusters was used to construct phylogenetic relationships and estimate divergence time. Alignments from MUSCLE106 were converted to coding sequences. Fourfold-degenerate sites were concatenated and used to estimate the neutral substitution rate per year and divergence time. PhyML107 was used to construct the phylogenetic tree.
The Bayesian Relaxed Molecular Clock (BRMC) approach was used to estimate the species divergence time using the program MCMCTREE v4.0, which is part of the PAML package108. The 'correlated molecular clock' and 'JC69' models were used. Published times for sorghum–rice (<55 million years ago, >35 million years ago)109,110,111, tomato–potato (<4 million years ago, >2 million years ago)112, and grape–rice (<130 million years ago, >240 million years ago)113 divergence were used to calibrate divergence time.
Chromosome collinearity within carrot and between carrot and tomato, grape, and kiwifruit was evaluated with MCscan114 (Supplementary Table 63). The synonymous mutation rate (ks) and fourfold-degenerate transversion rate were calculated using the HKY model115.
The paleopolyploid history was determined as described by Salse33 (Fig. 3c and Supplementary Figs. 35 and 36). Grape–carrot syntenic blocks descending from the seven ancestral chromosomes were detected in carrot as compared with grape, kiwifruit, tomato, and coffee (Supplementary Fig. 37).
Divergence and WGD time points in the carrot and tomato genomes were estimated using a method described by Vanneste et al.30 (Supplementary Table 64).
The comparative analysis with the horseweed genome9 used the same gene prediction pipeline described earlier. In total 38,199 genes were predicted and clustered using OrthoMCL to find single-copy gene families across 14 species. A maximum-likelihood tree was reconstructed on the basis of the fourfold-degenerate sites from the 963 single-copy gene families. Reciprocal best BLASTN69 hits within horseweed or between horseweed and other species were used to calculate the paralog/ortholog gene divergence (Supplementary Fig. 13).
We collected all syntenic blocks containing genes associated with the Dc-α, Dc-β, and Dc-γ WGD events (Supplementary Table 65). FUNC116 was used to carry out a hypergeometric test to identify GO categories with over-representation or under-representation of Dc-α WGD retained and tandem duplicated genes.
See the Supplementary Note for additional details.
Regulatory and resistance genes: gene family analysis.
We used PlantTFcat117 to annotate possible candidate transcription factors, transcription regulators, and chromatin regulators, collectively referred to as regulatory genes. Eleven genomes, including D. carota, S. lycopersicum, S. tuberosum, Coffea canephora, Actinidia chinensis, A. thaliana, B. rapa, Vitis vinifera, Prunus persica, Carica papaya, and O. sativa, were screened and filtered for InterPro domains specific for each regulatory gene family.
Predicted regulatory gene classes were grouped with OrthoMCL as described. We then carried out a detailed analysis of expanded carrot regulatory gene families (Supplementary Fig. 38 and Supplementary Tables 66–79). See the Supplementary Note for the classification of duplication modes of each regulatory gene. For phylogenetic analysis, multiple-sequence alignments with complete protein sequence were conducted using Clustal W118 with default parameters. Phylogenetic trees were constructed using the neighbor-joining method, with pairwise deletion, using MEGA6 (ref. 84).
MATRIX-R38 was used to annotate and classify R genes from nine species, including D. carota, S. lycopersicum, S. tuberosum, C. canephora, Capsicum annuum, A. chinensis, A. thaliana, V. vinifera, and O. sativa. Proteins identified via hidden Markov model (HMM) profiling were further analyzed using InterProScan version 5.0 (ref. 119) for conserved domains and motifs characteristic of R proteins (NBS, LRR, TIR, kinase, serine/threonine).
See the Supplementary Note for additional details.
A candidate gene controlling carotenoid accumulation.
Mapping populations, 97837 (n = 253) and 70796 (n = 285), were used to study the Y locus that regulates carotenoid accumulation in carrot root, where 97837 was derived from an intercross between yellow- and white-rooted cultivars and 70796 was derived from a cross between a dark orange inbred carrot and a wild white-rooted carrot (Supplementary Figs. 39–41). Carotenoids were quantified as described by Simon and Wolff120 and Simon et al.121.
Analysis of marker–trait associations was carried out with molecular markers considered as fixed effects in a linear model implemented in the GLM function of TASSEL72. The primers used for fine-mapping are reported in Supplementary Table 80. Genome assembly v2.0 was used as a reference to identify marker locations (Supplementary Tables 81 and 82). The genome-wide significance threshold was determined by the Bonferroni method122. QTL analysis for population 70796 used R package qtl123 (Supplementary Table 83).
Resequencing of polymorphisms and phenotypes were used to identify the haplotype block associated with pigmented versus non-pigmented roots. SNPs covering the region associated with high carotenoid accumulation were loaded into TASSEL72 and manually inspected to identify the start and end of the haplotype block. Sequence from the haplotype block and its flanking sequences were used for haplotype network analysis with PopArt v1.7 (ref. 124).
Haploview v4.2 (ref. 125) was used to calculate and visualize LD in the candidate region. FST analysis of 1,393,431 original filtered SNPs was conducted pairwise between each of the 35 resequenced genotypes using VCFTools126 with default parameters. The top 1% of FST values were determined and visualized by a custom Perl script (cp2; see URLs). Nucleotide diversity (π) was estimated in TASSEL72 as described by Nei and Lin25.
See the Supplementary Note for additional details.
Gene expression analysis.
Root tissue was collected from population 97837 plants with yellow (yyY2Y2) and white (YYY2Y2) genotypes, with two biological replicates per genotype, 80 d after planting. Root tissue was collected from population 70796 plants with dark orange (yyy2y2) and pale orange (YYy2y2) genotypes, with three biological replicates, 100 d after planting. Total RNA was extracted from whole-root tissue using the TRIzol Plus RNA Purification kit. RNA quantity and integrity were confirmed with an Experion RNA StdSens Analysis kit. All samples had RQI values above 8.0. Paired-end libraries (insert size of 133 nt) were sequenced on Illumina HiSeq 2000 lanes (2 × 100-nt reads).
Filtered reads were aligned to the v2.0 genome assembly using TopHat v2.0.12 (ref. 78). The aligned read files were processed by Cufflinks v2.2.1 (ref. 93). Testing for differential expression was done at the level of genes, isoforms, and promoters. PCR was carried out to verify the 212-nt indel in the Y candidate gene (DCAR_032551) (Supplementary Fig. 42).
Expression values were log2 transformed, and the WGCNA package127 in R with signed correlations was used to determine gene coexpression modules with a soft threshold value β of 10 and a treecut value of 0.6. Functional annotation of genes within this module was determined by BLASTP search of protein sequences within this module against the A. thaliana TAIR10 (ref. 128) predictions, and GO enrichment analysis based on BLASTP best hits to TAIR10 was performed using AgriGO129 and PANTHER130.
Genes that were simultaneously upregulated or downregulated in both yellow and dark orange samples, relative to the white and pale orange samples, were manually annotated. GO annotations and subcellular localization are also reported.
See the Supplementary Note for additional details.
Identification of flavonoid and isoprenoid pathway genes.
The peptide sequences for carrot predicted genes were aligned against annotated flavonoid and isoprenoid pathway genes in the KEGG database (Supplementary Tables 46, 84, and 85). BLASTP69 was carried out using default parameters. Sequences with <50% identity, <50 residues were excluded. Peptide sequences from genomes having orthologous relationships with retained carrot genes were extracted from the genome evolution analysis. Genes annotated from the A. thaliana and tomato genomes were manually verified. Multiple-sequence alignments were generated with Clustal W118. Phylogenetic analyses were carried out using MEGA6 (ref. 84) (Supplementary Figs. 43 and 44). Carrot peptide sequences annotated as InterProScan IDs IPR001906 and IPR005630 and containing the N-terminal domains PF011397 and PF03936 (Supplementary Table 47) along with known terpene synthases (TPSs) from seven other species were used for analysis with MEGA. The amino acid substitution models tested were WAG, mtREV, Dayhoff, JTT, VT, Blosum62, and CpREV. The tree with the highest AICc value was obtained with the JTT+F model with estimation of the gamma distribution. The phylogenetic tree was then rooted at the split between the type I (TPS-c, TPS-e, TPS-f, and TPS-h) and type III (TPS-a, TPS-b, and TPS-g) subfamilies (Supplementary Fig. 45).
See the Supplementary Note for additional details.
Accession codes.
The genome assembly has been deposited at GenBank under accession LNRQ00000000 and at Phytozome. The version described in this paper is version LNRQ01000000. All raw reads have been deposited in the Sequence Read Archive (SRA) under umbrella project PRJNA285926, accessions SRP062070, SRP062113, and SRP062159. Further information is available through our website (see URLs).
URLs.
Food and Agriculture Organization of the United Nations (FAO) Statistics, http://faostat3.fao.org/; SOAPaligner, http://soap.genomics.org.cn/soapaligner.html; bb.tassel, https://github.com/dsenalik/bb; CheckMatrix, http://www.atgc.org/XLinkage; cp1 and cp2 scripts, http://www.ars.usda.gov/pandp/Docs.htm?docid=25732; RepeatMasker and RepeatModeler, http://www.repeatmasker.org/; Picard tools, http://broadinstitute.github.io/picard/; TargetP web-based predictor, http://www.cbs.dtu.dk/services/TargetP/; Arabidopsis database, https://www.arabidopsis.org/Blast/index.jsp. Information from this publication is available at http://www.ars.usda.gov/pandp/Docs.htm?docid=25732.
Accession codes
Primary accessions
BioProject
NCBI Reference Sequence
Sequence Read Archive
Referenced accessions
BioProject
NCBI Reference Sequence
Sequence Read Archive
References
Simon, P.W. et al. in Carrot. Handbook of Plant Breeding, Vegetables II (eds. Prohens, J. & Nuez, F.) 327–357 (Springer, 2008).
Zagorodskikh, P. New data on the origin and taxonomy of the cultivated carrot. C.R. (Doklady) Acad. Sci. USSR 25, 522–525 (1939).
Iorizzo, M. et al. Genetic structure and domestication of carrot (Daucus carota subsp. sativus) (Apiaceae). Am. J. Bot. 100, 930–938 (2013).
Simon, P.W. Domestication, historical development, and modern breeding of carrot. Plant Breed. Rev. 19, 157–190 (2000).
Arscott, S.A. & Tanumihardjo, S.A. Carrots of many colors provide basic nutrition and bioavailable phytochemicals acting as a functional food. Compr. Rev. Food Sci. Food Saf. 9, 223–239 (2010).
Simon, P.W. Plant breeding for human nutritional quality. Plant Breed. Rev. 31, 325–392 (2009).
Rubatzky, V.E., Quiros, C.F. & Simon, P.W. Carrots and Related Vegetable Umbelliferae (CABI, 1999).
Bremer, B. in Asterids.The Timetree of Life (eds. Hedges, S.B. & Kumar, S.) 177–178 (Oxford University Press, 2009).
Peng, Y. et al. De novo genome assembly of the economically important weed horseweed using integrated data from multiple sequencing platforms. Plant Physiol. 166, 1241–1254 (2014).
Scaglione, D. et al. The genome sequence of the outbreeding globe artichoke constructed de novo incorporating a phase-aware low-pass sequencing strategy of F1 progeny. Sci. Rep. 6, 19427 (2016).
Arumuganathan, K. & Earle, E.D. Nuclear DNA content of some important plant species. Plant Mol. Biol. Rep. 9, 208–218 (1991).
Potato Genome Sequencing Consortium. Genome sequence and analysis of the tuber crop potato. Nature 475, 189–195 (2011).
Kim, S. et al. Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat. Genet. 46, 270–278 (2014).
Iorizzo, M. et al. De novo assembly and characterization of the carrot transcriptome reveals novel genes, new markers, and genetic diversity. BMC Genomics 12, 389 (2011).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Chain, P.S. et al. Genome project standards in a new era of sequencing. Science 326, 236–237 (2009).
International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature 436, 793–800 (2005).
Grzebelus, D., Yau, Y.-Y. & Simon, P.W. Master: A novel family of PIF/Harbinger-like transposable elements identified in carrot (Daucus carota L.). Mol. Genet. Genomics 275, 450–459 (2006).
Macko-Podgorni, A., Nowicka, A., Grzebelus, E., Simon, P.W. & Grzebelus, D. DcSto: carrot Stowaway-like elements are abundant, diverse, and polymorphic. Genetica 141, 255–267 (2013).
Santiago, N., Herráiz, C., Goñi, J.R., Messeguer, X. & Casacuberta, J.M. Genome-wide analysis of the Emigrant family of MITEs of Arabidopsis thaliana. Mol. Biol. Evol. 19, 2285–2293 (2002).
Miga, K.H. et al. Centromere reference models for human chromosomes X and Y satellite arrays. Genome Res. 24, 697–707 (2014).
Novák, P., Neumann, P., Pech, J., Steinhaisl, J. & Macas, J. RepeatExplorer: a Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 29, 792–793 (2013).
Iovene, M. et al. Comparative FISH mapping of Daucus species (Apiaceae family). Chromosome Res. 19, 493–506 (2011).
Spalik, K. et al. Amphitropic amphiantarctic disjunctions in Apiaceae subfamily Apioideae. J. Biogeogr. 37, 1977–1994 (2010).
Nei, M. & Li, W.-H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 76, 5269–5273 (1979).
Rong, J. et al. New insights into domestication of carrot from root transcriptome analyses. BMC Genomics 15, 895 (2014).
Arbizu, C., Reitsma, K.R., Simon, P.W. & Spooner, D.M. Morphometrics of Daucus (Apiaceae): a counterpart to a phylogenomic study. Am. J. Bot. 101, 2005–2016 (2014).
Welch, J.E. & Grimball, E.L. Jr. Male sterility in the carrot. Science 106, 594 (1947).
Weir, B.S. & Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 38, 1358 (1984).
Vanneste, K., Baele, G., Maere, S. & Van de Peer, Y. Analysis of 41 plant genomes supports a wave of successful genome duplications in association with the Cretaceous–Paleogene boundary. Genome Res. 24, 1334–1347 (2014).
Truco, M.J. et al. An ultra-high-density, transcript-based, genetic map of lettuce. G3 3, 617–631 (2013).
Argout, X. et al. The genome of Theobroma cacao. Nat. Genet. 43, 101–108 (2011).
Salse, J. et al. Reconstruction of monocotelydoneous proto-chromosomes reveals faster evolution in plants than in animals. Proc. Natl. Acad. Sci. USA 106, 14908–14913 (2009).
Lang, D. et al. Genome-wide phylogenetic comparative analysis of plant transcriptional regulation: a timeline of loss, gain, expansion, and correlation with complexity. Genome Biol. Evol. 2, 488–503 (2010).
Noh, B. et al. Divergent roles of a pair of homologous jumonji/zinc-finger-class transcription factor proteins in the regulation of Arabidopsis flowering time. Plant Cell 16, 2601–2613 (2004).
Nakamichi, N. et al. Arabidopsis clock-associated pseudo-response regulators PRR9, PRR7 and PRR5 coordinately and positively regulate flowering time through the canonical CONSTANS-dependent photoperiodic pathway. Plant Cell Physiol. 48, 822–832 (2007).
Levy, Y.Y., Mesnage, S., Mylne, J.S., Gendall, A.R. & Dean, C. Multiple roles of Arabidopsis VRN1 in vernalization and flowering time control. Science 297, 243–246 (2002).
Sanseverino, W. et al. PRGdb 2.0: towards a community-based database model for the analysis of R-genes in plants. Nucleic Acids Res. 41, D1167–D1171 (2013).
Simon, P.W., Matthews, W.C. & Roberts, P.A. Evidence for simply inherited dominant resistance to Meloidogyne javanica in carrot. Theor. Appl. Genet. 100, 735–742 (2000).
Buishand, J.G. & Gabelman, W.H. Investigations on the inheritance of color and carotenoid content in phloem and xylem of carrot roots (Daucus carota L.). Euphytica 28, 611–632 (1979).
Just, B.J., Santos, C.A., Yandell, B.S. & Simon, P.W. Major QTL for carrot color are positionally associated with carotenoid biosynthetic genes and interact epistatically in a domesticated × wild carrot cross. Theor. Appl. Genet. 119, 1155–1169 (2009).
Just, B.J. et al. Carotenoid biosynthesis structural genes in carrot (Daucus carota): isolation, sequence-characterization, single nucleotide polymorphism (SNP) markers and genome mapping. Theor. Appl. Genet. 114, 693–704 (2007).
Clotault, J. et al. Expression of carotenoid biosynthesis genes during carrot root development. J. Exp. Bot. 59, 3563–3573 (2008).
Fuentes, P. et al. Light-dependent changes in plastid differentiation influence carotenoid gene expression and accumulation in carrot roots. Plant Mol. Biol. 79, 47–59 (2012).
Bowman, M.J., Willis, D.K. & Simon, P.W. Transcript abundance of phytoene synthase 1 and phytoene synthase 2 is associated with natural variation of storage root carotenoid pigmentation in carrot. J. Am. Soc. Hortic. Sci. 139, 63–68 (2014).
Wang, H., Ou, C.-G., Zhuang, F.-Y. & Ma, Z.-G. The dual role of phytoene synthase genes in carotenogenesis in carrot roots and leaves. Mol. Breed. 34, 2065–2079 (2014).
Palaisa, K., Morgante, M., Tingey, S. & Rafalski, A. Long-range patterns of diversity and linkage disequilibrium surrounding the maize Y1 gene are indicative of an asymmetric selective sweep. Proc. Natl. Acad. Sci. USA 101, 9885–9890 (2004).
Wang, R.-L., Stec, A., Hey, J., Lukens, L. & Doebley, J. The limits of selection during maize domestication. Nature 398, 236–239 (1999).
Ruiz-Sola, M.Á. & Rodríguez-Concepción, M. Carotenoid biosynthesis in Arabidopsis: a colorful pathway. Arabidopsis Book 10, e0158 (2012).
Cordoba, E. et al. Functional characterization of the three genes encoding 1-deoxy-D-xylulose 5-phosphate synthase in maize. J. Exp. Bot. 62, 2023–2038 (2011).
Kim, B.R., Kim, S.U. & Chang, Y.J. Differential expression of three 1-deoxy-D-xylulose-5-phosphate synthase genes in rice. Biotechnol. Lett. 27, 997–1001 (2005).
Saladié, M., Wright, L.P., Garcia-Mas, J., Rodriguez-Concepcion, M. & Phillips, M.A. The 2-C-methylerythritol 4-phosphate pathway in melon is regulated by specialized isoforms for the first and last steps. J. Exp. Bot. 65, 5077–5092 (2014).
Walter, M.H., Hans, J. & Strack, D. Two distantly related genes encoding 1-deoxy-D-xylulose 5-phosphate synthases: differential regulation in shoots and apocarotenoid-accumulating mycorrhizal roots. Plant J. 31, 243–254 (2002).
Estévez, J.M., Cantero, A., Reindl, A., Reichler, S. & León, P. 1-Deoxy-D-xylulose-5-phosphate synthase, a limiting enzyme for plastidic isoprenoid biosynthesis in plants. J. Biol. Chem. 276, 22901–22909 (2001).
Lois, L.M., Rodríguez-Concepción, M., Gallego, F., Campos, N. & Boronat, A. Carotenoid biosynthesis during tomato fruit development: regulatory role of 1-deoxy-D-xylulose 5-phosphate synthase. Plant J. 22, 503–513 (2000).
Ichikawa, T. et al. The FOX hunting system: an alternative gain-of-function gene hunting technique. Plant J. 48, 974–985 (2006).
Huang, X., Ouyang, X. & Deng, X.W. Beyond repression of photomorphogenesis: role switching of COP/DET/FUS in light signaling. Curr. Opin. Plant Biol. 21, 96–103 (2014).
Wei, N. & Deng, X.-W. The role of the COP/DET/FUS genes in light control of Arabidopsis seedling development. Plant Physiol. 112, 871–878 (1996).
Lau, O.S. & Deng, X.W. The photomorphogenic repressors COP1 and DET1: 20 years later. Trends Plant Sci. 17, 584–593 (2012).
Rodriguez-Concepcion, M. & Stange, C. Biosynthesis of carotenoids in carrot: an underground story comes to light. Arch. Biochem. Biophys. 539, 110–116 (2013).
Bowman, M.J. Gene Expression and Genetic Analysis of Carotenoid Pigment Accumulation in Carrot (Daucus carota L.). PhD thesis, Univ. Wisconsin–Madison, (2012).
Sherwin, J.C., Reacher, M.H., Dean, W.H. & Ngondi, J. Epidemiology of vitamin A deficiency and xerophthalmia in at-risk populations. Trans. R. Soc. Trop. Med. Hyg. 106, 205–214 (2012).
Steward, F.C. Growth and organized development of cultured cells. III. Interpretation of the growth from free cell to carrot plant. Am. J. Bot. 45, 709–713 (1958).
Vogel, G. How does a single somatic cell become a whole plant? Science 309, 86 (2005).
Huang, S. et al. Draft genome of the kiwifruit Actinidia chinensis. Nat. Commun. 4, 2640 (2013).
Jiao, Y. et al. A genome triplication associated with early diversification of the core eudicots. Genome Biol. 13, R3 (2012).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18 (2012).
Van Ooijen, J.W. JoinMap 4: Software for the Calculation of Genetic Linkage Maps in Experimental Populations (Kyazma, 2006).
Voorrips, R.E. MapChart: software for the graphical presentation of linkage maps and QTLs. J. Hered. 93, 77–78 (2002).
Schmieder, R. & Edwards, R. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS One 6, e17288 (2011).
Elshire, R.J. et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6, e19379 (2011).
Bradbury, P.J. et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635 (2007).
Glaubitz, J.C. et al. TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS One 9, e90346 (2014).
Cavagnaro, P.F. et al. A gene-derived SNP-based high resolution linkage map of carrot including the location of QTL conditioning root and leaf anthocyanin pigmentation. BMC Genomics 15, 1118 (2014).
Dong, F. et al. Development and applications of a set of chromosome-specific cytogenetic DNA markers in potato. Theor. Appl. Genet. 101, 1001–1007 (2000).
Iovene, M., Grzebelus, E., Carputo, D., Jiang, J. & Simon, P.W. Major cytogenetic landmarks and karyotype analysis inDaucus carota and other Apiaceae. Am. J. Bot. 95, 793–804 (2008).
Altschul, S.F., Gish, W., Miller, W., Myers, E.W. & Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Gambin, T. et al. TIRfinder: a web tool for mining class II transposons carrying terminal inverted repeats. Evol. Bioinform. Online 9, 17–27 (2013).
Wicker, T. et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 8, 973–982 (2007).
Darzentas, N. Circoletto: visualizing sequence similarity with Circos. Bioinformatics 26, 2620–2621 (2010).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Novák, P., Neumann, P. & Macas, J. Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinformatics 11, 378 (2010).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94 (1997).
Majoros, W.H., Pertea, M. & Salzberg, S.L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res. 14, 988–995 (2004).
Kent, W.J. BLAT—the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002).
Campbell, M.A., Haas, B.J., Hamilton, J.P., Mount, S.M. & Buell, C.R. Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genomics 7, 327 (2006).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Elsik, C.G. et al. Creating a honey bee consensus gene set. Genome Biol. 8, R13 (2007).
Zdobnov, E.M. & Apweiler, R. InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001).
Nawrocki, E.P. & Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Lowe, T.M. & Eddy, S.R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Murray, M.G. & Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 8, 4321–4325 (1980).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Pritchard, J.K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Felsenstein, J. PHYLIP—Phylogeny Inference Package (Version 3.2). Cladistics 5, 164–166 (1989).
Li, L., Stoeckert, C.J. Jr. & Roos, D.S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Edgar, R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Zhang, G. et al. Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat. Biotechnol. 30, 549–554 (2012).
Paterson, A.H., Bowers, J.E. & Chapman, B.A. Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc. Natl. Acad. Sci. USA 101, 9903–9908 (2004).
Paterson, A.H. et al. The Sorghum bicolor genome and the diversification of grasses. Nature 457, 551–556 (2009).
Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641 (2012).
Jaillon, O. et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467 (2007).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
Hasegawa, M., Kishino, H. & Yano, T. Dating of the human–ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 22, 160–174 (1985).
Prüfer, K. et al. FUNC: a package for detecting significant associations between gene sets and ontological annotations. BMC Bioinformatics 8, 41 (2007).
Dai, X., Sinharoy, S., Udvardi, M. & Zhao, P.-X. PlantTFcat: an online plant transcription factor and transcriptional regulator categorization and analysis tool. BMC Bioinformatics 14, 321 (2013).
Larkin, M.A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Simon, P.W. & Wolff, X.Y. Carotenes in typical and dark orange carrots. J. Agric. Food Chem. 35, 1017–1022 (1987).
Simon, P.W., Wolff, X.Y., Peterson, C.E. & Kammerlohr, D.S. High carotene mass carrot population. HortScience 24, 174–175 (1989).
Bland, J.M. & Altman, D.G. Multiple significance tests: the Bonferroni method. Br. Med. J. 310, 170 (1995).
Broman, K.W. & Sen, S. A Guide to QTL Mapping with R/qtl (Springer, 2009).
Leigh, J.W. & Bryant, D. POPART: full-feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116 (2015).
Barrett, J.C., Fry, B., Maller, J. & Daly, M.J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008).
Lamesch, P. et al. The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 40, D1202–D1210 (2012).
Du, Z., Zhou, X., Ling, Y., Zhang, Z. & Su, Z. agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 38, W64–W70 (2010).
Mi, H., Muruganujan, A. & Thomas, P.D. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 41, D377–D386 (2013).
Acknowledgements
The authors appreciate the financial support of the carrot industry and the following vegetable seed companies—Bejo, Carosem, Monsanto, Nunhems, Rijk Zwaan (RZ), Sumika, Takii, and Vilmorin—with additional thanks to RZ for providing DH1, BAC libraries, and BES. S.E. was supported by the National Science Foundation under grant 1202666. M. Iovene thanks the projects RGV-FAO (D.M.3824) for partial financial support and PONa3_00025-BIOforIU for funding the acquisition of a fluorescence microscope. A.M.-P., E.M., E.G., and D.G. were supported by the Polish National Science Center, project 2012/05/B/NZ9/03401, and the statutory funds for science granted by the Polish Ministry of Science and Higher Education to the Faculty of Biotechnology and Horticulture, University of Agriculture in Krakow. The authors thank H. Ruess for assembly and annotation of the plastid genome and R. Kane for support in the development of plant materials.
Author information
Authors and Affiliations
Contributions
M. Iorizzo, S.E., D. Senalik, H.A., A.V.D., and P. Simon conceived the project. M. Iorizzo, A.V.D., and P. Simon jointly supervised the research. M. Iorizzo, S.E., D. Senalik, M. Iovene, W.S., E.G., D.G., S.C., D. Spooner, A.V.D., and P. Simon conceived and designed the experiments. M. Iorizzo, S.E., D. Senalik, M. Iovene, P.Z., W.S., A.M.-P., and S.C. managed several components of the project. M. Iorizzo, S.E., D. Senalik, M. Iovene, W.S., P.C., M.Y., E.G., D.G., and P. Simon performed material preparation and genetic mapping. M. Iorizzo, D. Senalik, P.Z., Z.Z., S.C., and J.H. performed sequencing and assembly. M. Iorizzo, D. Senalik, M. Iovene, A.M.-P., E.M., E.G., and D.G. performed evaluation and analysis of repetitive elements and in situ hybridization. M. Iorizzo, D. Senalik, P.Z., and S.C. performed evolution analysis. M. Iorizzo and W.S. performed the resistance gene analysis. M. Iorizzo and D. Senalik performed the transcription factor analysis and the isoprenoid and flavonoid biosynthetic pathway analysis. M. Iorizzo, S.E., D. Senalik, M.B., and D. Spooner performed the resequencing analysis. M. Iorizzo, S.E., P. Satapoomin, and P. Simon performed the carotenoid accumulation analysis. M. Iorizzo, S.E., D. Senalik, P.Z., P. Satapoomin, M.B., M. Iovene, A.M.-P., E.G., D.G., D. Spooner, A.V.D., and P. Simon wrote the paper. M. Iorizzo and P. Simon organized the manuscript. A.V.D. and P. Simon coordinated the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Note. (PDF 3541 kb)
Supplementary Figures 1–9
Supplementary Figures 1–9. (PDF 12165 kb)
Supplementary Figure 10
Supplementary Figure 10. (PDF 26072 kb)
Supplementary Figures 11–34
Supplementary Figures 11–34. (PDF 30884 kb)
Supplementary Figures 35–45
Supplementary Figures 35–45. (PDF 22675 kb)
Supplementary Tables 1–85
Supplementary Tables 1–85. (XLSX 4063 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International licence. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Iorizzo, M., Ellison, S., Senalik, D. et al. A high-quality carrot genome assembly provides new insights into carotenoid accumulation and asterid genome evolution. Nat Genet 48, 657–666 (2016). https://doi.org/10.1038/ng.3565
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3565
This article is cited by
-
Population genomics identifies genetic signatures of carrot domestication and improvement and uncovers the origin of high-carotenoid orange carrots
Nature Plants (2023)
-
The changing colour of carrot
Nature Plants (2023)
-
A comprehensive and conceptual overview of omics-based approaches for enhancing the resilience of vegetable crops against abiotic stresses
Planta (2023)
-
Functional characterization of DcMYB11, an R2R3 MYB associated with the purple pigmentation of carrot petiole
Planta (2023)
-
Assessment of genetic homogeneity of somatic embryo-derived plants and seed-derived plants of a robusta coffee cultivar using molecular markers and functional genes sequencing
Plant Cell, Tissue and Organ Culture (PCTOC) (2023)
