
Abstract
Genetic sex determination by W and Z chromosomes has developed independently in different groups of organisms. To better understand the evolution of sex chromosomes and the plasticity of sex-determination mechanisms, we sequenced the whole genomes of a male (ZZ) and a female (ZW) half-smooth tongue sole (Cynoglossus semilaevis). In addition to insights into adaptation to a benthic lifestyle, we find that the sex chromosomes of these fish are derived from the same ancestral vertebrate protochromosome as the avian W and Z chromosomes. Notably, the same gene on the Z chromosome, dmrt1, which is the male-determining gene in birds, showed convergent evolution of features that are compatible with a similar function in tongue sole. Comparison of the relatively young tongue sole sex chromosomes with those of mammals and birds identified events that occurred during the early phase of sex-chromosome evolution. Pertinent to the current debate about heterogametic sex-chromosome decay, we find that massive gene loss occurred in the wake of sex-chromosome 'birth'.
Similar content being viewed by others


Main
The existence of males and females, which are often strikingly different in morphology, reproductive strategies and behavior, is one of the most widespread phenomena in biology. However, the genetic mechanisms that generate this ubiquitous pattern are surprisingly diverse and do not follow a phylogenetic pattern. Sex-determination mechanisms can differ between even closely related species and arise frequently and independently. Fish provide a paradigmatic example, as their sex-determination mechanisms range from environmental to different modes of genetic determination. The evolutionary meaning of this remarkable plasticity is unknown. For genetic sex determination, where the trigger for female or male development comes from the genetic constitution of the individual, the evolution of sex-determination mechanisms is connected to a very peculiar genomic process, namely the formation of sex chromosomes1,2,3,4.
To improve understanding of the function and evolution of sex chromosomes, their genetic organization must be deciphered. However, owing to their degenerate nature and high repetitive DNA content, sex chromosomes pose almost insurmountable problems in deciphering their gene content and organization. So far, only the human5, chimpanzee6 and rhesus macaque Y chromosomes7 and the male-specific region on the Y chromosome of one fish, the medaka8, have been sequenced. These analyses have nevertheless provided important insights into the evolution of Y chromosomes, their genomic organization and their degeneration processes, as well as predictions as to their likely evolutionary fate9,10,11,12.
Much less genomic information exists on W chromosomes because, as with Y chromosomes, they are predominantly highly repetitive in nature. The prevailing theory of the evolution of sex chromosomes predicts that degeneration of the heterogametic sex chromosome is a stepwise process that occurs over an extended period of time. We therefore reasoned that an evolutionarily young W chromosome should be more amenable for sequencing than the highly degenerated, up to 200-million-year-old W chromosome of birds and would also provide insights into the early steps of W-chromosome evolution. Fish have long been known to have relatively young sex chromosomes13,14,15. We selected a flatfish for our study on the basis that these fish would have young sex chromosomes and offer the advantage of a relatively small genome, one that is not much larger than that of pufferfishes. We selected the Chinese half-smooth tongue sole, C. semilaevis, because of good genomic resources, as well as an available molecular marker genetic map16,17,18,19,20. This species is also an example of a group of fish with specific adaptations to a benthic lifestyle, most strikingly an asymmetric body shape with lateralization of the eyes to the same side during metamorphosis21,22.
Results
Genome assembly and annotation
We sequenced DNA from one male (ZZ) and one female (ZW) C. semilaevis separately with the Illumina sequencing platform using a whole-genome shotgun approach with 212-fold coverage (Table 1, Supplementary Fig. 1 and Supplementary Tables 1 and 2). We first assembled the male and female genomes separately (Supplementary Tables 2 and 3 and Supplementary Note). We considered scaffolds with double coverage in males as putative Z scaffolds (a total of 23.3 Mb) (Supplementary Note). 94% of these scaffolds could be anchored in one linkage group of the male genetic map by microsatellite markers (Supplementary Table 4 and Supplementary Note). We further confirmed Z-chromosome assignment by quantitative PCR in ZZ and ZW individuals (Supplementary Table 5 and Supplementary Note). We retrieved W sequences by subtracting the male assembly from the female assembly, yielding 16.4 Mb of scaffolds (Table 1 and Supplementary Table 4), which is consistent with the different sizes of the female and male haploid genomes as estimated by k-mer calculation (Supplementary Figs. 2 and 3). Owing to the lack of marker resolution, we ordered the W scaffolds in a pseudo-W chromosome on the basis of their synteny with the Z chromosome. We derived autosome sequences from the female whole-genome sequence after subtraction of the putative W and Z chromosomes. As a result, the final genome assembly was 477 Mb with a scaffold N50 size of 867 kb (Table 1 and Supplementary Table 3). We next constructed a high-resolution genetic map with 12,142 SNPs by restriction site–associated DNA sequencing (RAD-seq), which we assigned to 22 linkage groups corresponding to the 20 autosomes and both sex chromosomes (Supplementary Table 4 and Supplementary Note). Together with the former genetic map (942 simple sequence repeat (SSR) markers), we consistently anchored 445 Mb (93.3%) of the assembly on 20 autosomes, Z and W harboring 92% of the predicted genes (Supplementary Table 4).
Transposable elements (TEs) can constitute a large portion of a eukaryotic genome; for example, the human genome is 45% TEs23. The genome of the tongue sole, however, is only 5.85% TEs (Supplementary Tables 6 and 7 and Supplementary Figs. 4, 5, 6, 7), which is in the range of the compact pufferfish genomes (365 Mb, 2.7% TEs)24. Clearly, small genomes will be poor in TEs. We found a low diversity of DNA transposons (2.45%, mainly hAT and Tc-Mariner) and retrotransposons (1.34%, mainly RTE from LINE) (Supplementary Fig. 4 and Supplementary Table 7). More than two-thirds of the copies are almost complete, suggesting recent activity (Supplementary Fig. 4 and Supplementary Tables 8 and 9). Penelope retroelements are highly repeated, but only a few copies are complete, suggesting an ancient invasion that is now extinct (Supplementary Table 8). Moreover, the genome is almost devoid of long terminal repeat retrotransposons (Supplementary Fig. 5 and Supplementary Table 8).
We generated a total of 12.38 Gb of transcriptome sequences to aid in gene annotation (Supplementary Table 10). Using homologous protein alignment, ab initio prediction and expression data prediction, we annotated 21,516 protein-coding genes (Supplementary Fig. 8 and Supplementary Tables 11 and 12). 99% of the predicted genes were supported by homologs in other organisms or in the transcriptome (Supplementary Figs. 8 and 9 and Supplementary Table 11). We also identified 1,439 expanded gene families and 2,743 contracted gene families by comparing the family size between the tongue sole and the teleost common ancestor (Supplementary Figs. 10, 11, 12, 13 and Supplementary Note). In addition, we annotated 674 tRNA genes, 104 rRNA genes, 285 microRNAs and 434 small nuclear RNAs in the assembly (Supplementary Table 13).
Adaptation to a benthic lifestyle
Flatfish are characterized by a transition from pelagic to benthic habitats, which is accompanied by a morphological transition from a symmetric to an asymmetric body shape22,25. As such, it is a natural paradigm of an adaptive response to a changed environment with concomitant changes in development and behavior. The availability of the genome sequence of the tongue sole allowed us to study, at a genome-wide level, differential gene expression patterns between pelagic and benthic stages (i.e., fish before and after metamorphosis). Gene Ontology (GO) categories for differentially expressed genes between fish before and after metamorphosis are related to general metabolism and immunity but also include those categories potentially associated with a metamorphic transition (for example, response to steroid hormones or organ regeneration) and environmental cues (for example, response to light stimulus and gravity) that complement adaptation to a benthic lifestyle (Supplementary Tables 14,15,16,17,18). When we performed likelihood ratio tests based on branch-site models26, 15 of these differentially expressed genes showed a highly significant signature for positive selection (P < 0.01) (Supplementary Table 19 and Supplementary Note). This group included genes (cp, mep1b, hnf4a, ace2 and tmem67) with a putative role in the development and function of internal organs, genes (fbn1, cdhr2, pepd and itih2) that function in skin and cartilage remodeling, which have undergone intensive alteration during metamorphosis, and genes (mgam and cpb1) that may be related to changes in diet that accompany metamorphosis (Supplementary Fig. 14).
The most conspicuous feature of the transition from symmetric to asymmetric body shape is lateralization of the eyes to the same side21. We noted that the expression of opsin genes displays significant differences between the pelagic and benthic stages. The expression levels of rod pigment (rh1) and cone pigment (lws1, also called opn1lw1) genes, which are responsible for scotopic vision and long-wavelength sensitivity27, respectively, are significantly higher in the benthic stages (P < 0.01), whereas the expression of the middle wavelength–sensitive pigment gene (rh2) was significantly higher in the pelagic stages (P < 0.01) (Supplementary Fig. 14 and Supplementary Table 20). These observations may suggest an enhanced sensitivity to longer wavelengths in the benthic phase, reflecting an adaptation of the visual system accompanying the shift to the benthic environment with weak light. Notably, by screening the genomes of six teleosts for vision-related genes, we found that two crystallin genes (crybb2 and crybb3), which encode proteins that maintain the transparency and refractive index of the lens28, have been lost specifically in tongue sole (Supplementary Table 21). We also observed that several crystallin genes (cryabb, crygs1 and crygs3) have evolved into pseudogenes, suggesting decay in the visual system (Supplementary Table 21)29. This suggestion may be explicable in terms of the emergence of dependence of the adult lateralized tongue sole on the lateral-line sense organ for prey capture, as well as papillae that are specific sense organs distributed from the tip of the head to just inside the scaly region30. The development of the strong lateral-line sense organ and species-specific sensory papillae, serving as a substitute for their decayed visual system, are likely adaptations to the benthic habitat.
Genome evolution
The tongue sole is the first flatfish to be sequenced31. To ascertain the evolutionary trajectory of the tongue sole genome, we addressed the phylogenetic branching and divergence time by comparing sets of 1:1 orthologous genes in tongue sole and other teleost fishes using human and chicken as outgroups (Supplementary Figs. 15 and 16). The phylogenetic tree based on 2,628 1:1 high-quality orthologs dates the divergence of flatfish from other bony fish to about 197 million years ago, which is consistent with previous estimates (Supplementary Fig. 16)32.
The conservation of synteny between the vertebrate genomes allowed the reconstruction of the karyotype of their last common ancestor33. We determined the orthology and paralogy relationships of the tongue sole genes and established conserved synteny with other fish genomes (Tetraodon nigroviridis, medaka and zebrafish) using human as the outgroup (Supplementary Tables 22,23,24,25 and Supplementary Note). We then clustered conserved syntenic blocks, detected rearrangements in comparison to other genomes and inferred the occurrence of such rearrangements in relation to the phylogenetic tree of the genomes being compared34. This method allowed the reconstruction of the evolutionary history of the tongue sole genome at the chromosomal level. Whole-genome duplications (WGDs) and their subsequent genomic rearrangements have important roles in genome evolution35. Two rounds of WGD have occurred at the base of vertebrate evolution. The so-called modern fish (Teleostei) underwent an additional genome duplication event (TGD) in a common ancestral lineage from which all teleost fishes evolved36. The TGD is proposed to have facilitated teleost diversification and species radiation. Previous analyses of other fish genomes have documented the TGD34,37 and indicated eight major subsequent interchromosomal rearrangements in the ancestral teleost lineage within a short time period after the TGD34. 2,733 paralogous genes in the tongue sole genome were clustered into paralogous chromosomal regions distributed over 21 tongue sole chromosomes as result of the TGD (Fig. 1, Supplementary Fig. 17 and Supplementary Table 22). The genome-wide comparison of medaka, T. nigroviridis and zebrafish with the tongue sole genome identified the orthology relationships with the protochromosomes and three major lineage-specific chromosomal fusion events, leading to the reduced chromosome number of 21 in extant tongue sole after separation from the medaka lineage, which has 24 chromosomes (Supplementary Fig. 18 and Supplementary Tables 23,24,25,26).

WGD in the tongue sole and orthology in the medaka, T. nigroviridis, zebrafish and human genomes. The arcs of concentric circles represent each tongue sole chromosome (Cse1–Cse21 and Z). A–D represent tongue sole chromosomes painted with different colors according to the location of the orthologs in the human (Hsa), zebrafish (Dre), T. nigroviridis (Tni) and medaka (Ola) genomes. A 100-kb region around a gene is painted in the same color. E represents tongue sole chromosomes painted by the corresponding ancestral chromosomes (Anc1–Anc13). In F, each line joins duplicated genes at their respective positions.
The C. semilaevis Z and W chromosomes are derived from one duplicate descendant of protochromosome A for the vertebrate ancestor and A0 for the gnathostome ancestor, which gave rise to a pair of autosomes in other teleosts (Fig. 2a and Supplementary Figs. 17 and 18). Thus, the sex chromosomes of tongue sole evolved from a pair of autosomes after separation of these lineages. Notably, these sex chromosomes share large portions a common ancestry with the chicken Z and W chromosomes (Fig. 2a and Supplementary Figs. 17 and 19). We therefore posed the question of whether this sharing is coincidence or reflects a relevant evolutionary process.

(a) Common origin of the tongue sole and chicken Z chromosomes. Ten vertebrate ancestral chromosomes are represented by differently colored bars at the top. In the genomes of tongue sole and chicken, genomic regions are assigned colors, and vertical bars that represent the correspondence of individual regions to the ancestral chromosomes in the gnathostome ancestor from which the respective regions originated. The black arrows indicate fusion, fission and duplication events. The evolutionary process of ancestral chromosomes B–J is depicted in Supplementary Figure 17. The red dashed lines indicate the tongue sole Z chromosome versus selected chicken chromosomes. The tongue sole Z chromosome is orthologous to the chicken Z chromosome (red) and autosomes 15 (purple) and 17 (red). (b) The bar representing the Z chromosome is composed of differently sized fragments assigned by four colors (blue: Z-S, Z-specific genes; gray: Z-A, orthologous genes between Z and the autosome; yellow: Z-W, homologous genes between Z and W; orange: PAR, pseudoautosomal region). The red and cyan lines above the bar indicate the 5-methylcytosine density for the female and male, respectively, in 5-kb windows throughout Z. The blue line above the bar depicts the male-to-female (M:F) expression ratio by running an average of 20 genes throughout the Z chromosome. The highly methylated region (13.6–15.6 Mb) corresponding to an expression valley is not a gene-poor region. The gray background shows the distribution of TEs across the Z chromosome using a 100-kb sliding window with a 10-kb step. The y axis on the left denotes 5-methylcytosine density, and the y axis on the right denotes the log2 M:F ratio and the repeat density in parentheses. M, million.
Genomic organization and evolution of Z and W
To find out how far beyond the recognized conservation of synteny the similarity of tongue sole and chicken sex chromosomes goes, we analyzed the genomic organization of C. semilaevis Z and C. semilaevis W. Although W is the largest chromosome in metaphases, fewer sequences are anchored on W than on Z, possibly because of its higher content of repetitive DNA and TEs (Supplementary Fig. 20 and Supplementary Table 27). Both telocentric sex chromosomes share a small region (∼640 kb) at their distal telomeric ends containing 22 protein-coding genes and 1 pseudogene with no relevant sequence divergence (Fig. 2b and Supplementary Table 28). No indication of dosage compensation for these genes was apparent. We conclude that this region represents the pseudoautosomal region (PAR), where both sex chromosomes still pair in female meiosis and crossing over occurs at a normal rate.
The remaining region of the sex chromosomes contains 297 genes, which are also present on Z and W but show some sequence divergence (Supplementary Table 29). A uniformly distributed Ks (number of synonymous substitutions per synonymous site) of around 0.15 for this region compared to 0.0115 for the autosomes and 0.0188 for the PAR (Supplementary Fig. 21) can be taken as an indication of reduced or even absent recombination38. Not surprisingly, the W chromosome displays a high content of TEs (29.94% as compared to 13.13% on Z and 4.33% on autosomes) and pseudogenes (19.74% as compared to 3.54% on Z and 2.48% on autosomes) (Supplementary Tables 27 and 30). Together with genes that are not shared with the Z chromosome, the non-PAR of the W chromosome has 317 predicted functional protein-coding genes, which is about one-third of the 904 functional genes on the Z chromosome (Supplementary Table 29). On the chimpanzee and human Y chromosomes, there are only 40–80 intact genes, as compared to 1,098 protein-coding genes on the X chromosome. 26 expressed genes have been attributed to the chicken W chromosome39, whereas the Z chromosome has about 1,000 genes40, as in tongue sole. This observation points to a relatively recent evolutionary origin for the tongue sole sex chromosome pair, where degeneration on the W chromosome has not progressed to a stage at which almost its entire original gene content has disappeared. We determined the age of the tongue sole sex chromosomes to be about 30 million years (Supplementary Table 31), which is consistent with the general view that fish have young sex chromosomes13,14,15. This age contrasts with the hundreds of millions of years for the ancestry of the mammalian Y and avian W chromosomes1,41 and could serve to explain why there are still many intact genes in the non-recombining region of the tongue sole W chromosome.
The non-PAR of the Z chromosome exhibits only one evolutionary stratum, which is evident from the almost uniform distribution of Ks values for the Z-W gene pairs (Supplementary Fig. 21). We did not find evidence for recent transposition of genes or chromosome segments from autosomes or between the sex chromosomes.
We determined whether there is dosage compensation for Z-linked genes that are present in a single dosage in females (Supplementary Tables 32,33,34,35,36,37,38,39 and Supplementary Note). For those 763 Z-linked genes (reads per kilobase of gene per million mapped reads (RPKM) >1), male expression in whole-body (without gonad) transcriptomes was on average 1.32 times higher than female expression (95% confidence interval (CI) 1.291–1.362). This result is markedly less than the expected twofold difference for a comple te absence of dosage compensation and indicates incomplete gene dosage compensation (Supplementary Fig. 22). To elucidate the underlying mechanism, we analyzed the global gene expression pattern for sex bias of autosomal as compared to Z-chromosome genes. The Z-to-A ratios in males were close to 1 (95% CI 0.962–0.967 by Wilcoxon rank-sum test). In females, however, the ratios were 0.727–0.732 (95% CI by Wilcoxon rank-sum test), which is clearly above the 0.5 Z-to-A ratio that would be expected for no compensation. This result suggests a compensatory mechanism working through upregulation of female genes (Supplementary Fig. 23), as has also been noted previously in birds and silkworm42,43.
Sex determination by the Z chromosome
We next posed the question of whether sex determination in the tongue sole operates through a Z-linked male-determining gene, a W-linked female-determining gene or a combination of both. This question is still unclear in birds44,45 but can be addressed in tongue sole because of the possibility of producing sex reversals by high-temperature treatment (Fig. 3a and Supplementary Note). Compared to a low spontaneous sex-reversal rate (14%) of ZW individuals raised at normal ambient temperature (22 °C), a rate of ∼73% sex-reversed ZW males (fertile pseudomales) appeared after high-temperature (28 °C) treatment during the sensitive developmental period (Supplementary Tables 40 and 41)46. We then crossed these pseudomales to normal ZW females and raised the offspring under non–sex reverting conditions (22 °C). Surprisingly, there was a huge male bias, with all ZZ fish being male but also 94% of the ZW fish developing as pseudomales (Fig. 3b and Supplementary Table 40). Microsatellite markers that distinguish paternal and maternal Z chromosomes revealed that all second-generation pseudomales had inherited the Z chromosome from their sex-reversed fathers (Fig. 3c and Supplementary Table 42). Moreover, Z-chromosomal genes in the offspring of sex-reverted fish retained the paternal methylation pattern, implying that transgenerational inheritance of DNA methylation status at certain loci on the Z chromosome is particularly important for the inheritance of sex reversal (Supplementary Fig. 24). In summary, these data show that in tongue sole, sex determination operates through a Z-encoded mechanism that determines male development. However, we cannot exclude the possibility that temperature-induced sex determination may operate through a different mechanism than genetic sex determination and might even involve different genes on the Z chromosome.

(a) A schematic flow chart of the production of the first (F1) and second (F2) generations of sex-reversed tongue sole with the ZW sex-determining system. The red cross indicates that the expected WW fish were not obtained because of the absence of W spermatozoa in WZ pseudomale sperm as determined by microsatellite analysis. (b) Ratio of female to male sex-reverted WZ pseudomales in families raised at normal temperature (F1, 22 °C), high temperature–treated offspring (F1, 28 °C) and WZ pseudomale offspring raised at normal temperature (F2, 22 °C). About 14% of the offspring from crossings of normal WZ females with ZZ males were sex reversed even when cultured under normal conditions (22 °C). Treatment at high temperature (28 °C) during the sex-determining stages of offspring from crosses of WZ females with ZZ males led to about 73% sex-reversed ZW male individuals. Surprisingly, there was an extremely male-skewed sex ratio (94%) in the offspring of WZ pseudomale families crossed with normal WZ females and raised under normal conditions (22 °C). (c) Paternal inheritance of the Z chromosome in three WZ pseudomale families determined by microsatellite analysis. The Z chromosome (identified by markers 1 and 2) of about 84–90% of ZW individuals is inherited from the sex-reversed father in three pseudomale families (4, 6 and 20), indicating that some genetic information on the Z chromosome is dominant in male sex determination in tongue sole.
For the purpose of identifying a putative primary sex-determining gene, we could thus exclude all W-encoded female-determining factors. On the tongue sole Z chromosome, we identified four genes that are known to be involved in sexual development in other vertebrates (Supplementary Table 43). For sf-1, ptch1 and fst, gene expression patterns at the sex-determination stage and gene methylation in normal males and females or in sex-reverted animals were incompatible with a role as a male sex-determining gene (Supplementary Figs. 25, 26, 27, 28). However, dmrt1 displayed many features that make it an outstanding candidate for a master sex-determining gene (Fig. 4 and Supplementary Figs. 26,27,28,29,30,31,32). Only the Z chromosome contains a functional copy of dmrt1, whereas we found a heavily corrupted pseudogenized copy on the W chromosome (Fig. 4a and Supplementary Fig. 29). This gene is highly expressed specifically in male germ cells and presomatic cells of the undifferentiated gonad at the sex-determination stage and persists at high levels during testis development (Fig. 4b,d and Supplementary Fig. 32). These patterns are paralleled by demethylation of the dmrt1 promoter region (Fig. 4c). The expression of dmrt1 in sex-reversed ZW males was dosage compensated by upregulation to a level that is observed in normally developing ZZ males (Supplementary Fig. 28). All of these observations are defining features for a dosage-dependent male sex-determining gene47, although functional proof cannot be obtained because tongue sole is refractory to transgenic technologies, as are all marine fish.

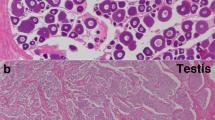
(a) dmrt1 BAC FISH analysis of tongue sole chromosomes showing a double signal in males and a single signal in females. BAC clone Hind012D10-3J, which contains the full-length dmrt1 gene, was labeled and used to probe male (ZZ) and female (ZW) chromosome spreads. Scale bars, 5 μm. (b) RT-PCR analysis of dmrt1 during developmental stages in female (black bar) and male (red bar) tongue sole. The data are shown as the mean ± s.e.m. (n = 3). (c) Methylation status across the differentially methylated region (DMR) of dmrt1 in the gonads of an adult WZ female, a ZZ male and a WZ female compared to male sex-reversed fish. The schematic diagram at the top shows the genomic structure of dmrt1 in tongue sole. Exons are depicted as blue boxes, and the 3′ and 5′ UTR regions are indicated by white boxes. The black arrow indicates the direction of the dmrt1 gene from transcriptional start site. Also shown is the methylation level of each cytosine, indicated by a green line, identified on both DNA strands throughout the dmrt1 gene in female and male fish. The gray shadow indicates the DMR. Open and filled circles represent unmethylated and methylated cytosines, respectively, validated by TA clone and Sanger sequencing. ZZ testis P, testis of the male parent; ZW testis F1, testis of a pseudomale in the first generation (temperature induced); ZW testis F2, testis of a pseudomale in the second generation (untreated); ZW ovary F1, ovary in the first-generation female; ZW ovary F2, ovary in the female offspring of a pseudomale. (d) Specific expression of dmrt1 in testis. Gonad in situ hybridization using the antisense RNA probe of dmrt1 performed in tongue sole larvae at 56 d, 83 d and 150 d during the gonad-development stage. G, gonium; OG, oogonium; OL, ovarian lamellae; OC, oocyte; SG, spermatogonia; SC, spermacyte; SE, sertoli cell; SP, spermatid; ST, spermatozoa.
Notably, we found that an E3 ubiquitin ligase gene, neurl3, is located only on the Z chromosome and is absent from W. Several E3 ubiquitin ligases have been demonstrated to be necessary for sperm development in human48 and mouse49. We assume that neurl3 may also potentially be a male-beneficial gene in tongue sole because it is highly expressed during spermatogenesis (Supplementary Figs. 33 and 34), and W-chromosome sperm that lack this gene do not develop in sex-reversed WZ pseudomales (Supplementary Fig. 35).
Discussion
Comparing the young sex chromosomes of tongue sole to those of birds and mammals allowed us to define processes that operate during the early phase of the establishment of sex chromosomes, which may also be important in stabilizing a sex-determination system10,11. The most obvious common feature between tongue sole and the much older avian and mammalian sex chromosomes is suppression of recombination. This observation is in line with the reasoning that suppression of recombination is a primary driving force of sex-chromosome evolution immediately after a genetic determinant on this chromosome has begun to operate. Notably, suppression of recombination has spread over most of the chromosome in tongue sole. The PAR of tongue sole is already very small and is comparable to the much more advanced sex chromosomes of birds and mammals. Accumulation of TEs also appears as an early event. The tongue sole Z chromosome, which is in common with the mammalian X and chicken Z chromosomes40, has a higher content of TEs. The even higher TE content of the W chromosome might explain why it, unlike the fully differentiated W chromosome, is even larger than the Z chromosome. The accumulation of sex-specific genes appears as another early event of sex-chromosome evolution. A ubiquitin E3 ligase gene, neurl3, which is clearly involved in spermatogenesis, is only present on the Z chromosome. It will be interesting to determine whether this chromosomal assignment is conserved in other flatfishes with WZ sex determination and hence might have contributed to the stabilization of the female heterogametic system in this group of fishes. Contrary to the mammalian X and chicken Z chromosomes, the tongue sole Z chromosome does not display a lower gene density as compared to the autosomes40, and we found no indication for pronounced gene traffic between autosomes and sex chromosomes.
The necessity for gene dosage compensation appears to be an early requirement during sex-chromosome evolution. Dosage compensation in tongue sole has already proceeded to the level of complexity seen in birds but differs from mammals in terms of the mechanism by which the mammalian X chromosome is compensated. The question as to whether dosage compensation in birds represents a primordial form that is common to other types of sex chromosome has not been answered because avian sex chromosomes are not at an early stage of evolution50. Our finding of the same compensation mechanisms for the relatively young sex chromosomes of tongue sole provides support for this hypothesis. However, although the dosage sensitivity of specific sets of Z-chromosome genes is evolutionarily conserved between zebrafinch and chicken, the compensated Z-chromosome genes in tongue sole are not the same as those in birds (Supplementary Tables 32,33,34,35,36,37,38,39)42.
Sex-chromosomal gene loss and degeneration2 has sparked speculation and discussion about ongoing decay and even the looming extinction of the human Y chromosome. However, it was recently shown that the process of gene loss has ceased during the past 25 million years51. Thus the majority of the Y chromosome's original gene complement must have been lost much earlier. During its 30 million years of existence, the tongue sole W chromosome has already lost about two-thirds of its original protein-coding information. Extrapolation of this rate of decay would mean that the W chromosome will arrive at the gene content that is characteristic of the mammalian and avian sex chromosomes, which are several million years old, within less than a few tens of millions of years. This observation demonstrates that gene loss on sex chromosomes is a very early and rapid feature of their evolution. If the situation in tongue sole is generalizable, then from the various theoretically possible kinetics of W or Y chromosome degradation10, an exponential decay that slows down in its final stages appears to be a reasonable scenario.
Parallel evolution of dmrt1 toward an important sex-determining gene in chicken, and probably also in tongue sole, provides support for the hypothesis that some ancestral chromosomes are more prone to evolve as sex chromosomes because of their particular gene content14,52. Genes such as dmrt1 can take over a master sex-determining role more easily than other genes, as indicated by the fact that this gene evolved to be the master male sex-determining gene independently in birds44, Xenopus laevis53, medaka54,55,56 and probably also tongue sole. It is noteworthy that of the 58 sex-related genes that we analyzed in tongue sole, we found more than twice the average number on the sex chromosome pair.
In summary, our sequencing of the genome of the half-smooth tongue sole provided insights into adaptation to its benthic life and has allowed the first comprehensive view of the structure and evolution of W chromosomes. The first full sequence of the Z chromosome outside of birds showed an unexpected parallel evolution of the same ancestral autosomal gene, dmrt1, which has been shown to be the primary male sex-determining gene in chicken. In addition to the evidence presented here, future studies in tongue sole will be necessary to provide further clues for such a function of dmrt1. Studies on the W chromosomes of other species will be informative with respect to the question of whether W chromosome evolution is just a female version of the much better studied Y chromosome evolution.
Methods
Genome sequence and assembly.
High-quality genomic DNA was extracted from one adult female and one adult male tongue sole. Then, according to the Illumina standard operating procedure, we constructed 15 paired-end libraries for the female and 11 paired-end libraries for the male (170 bp to ∼40 kb) (Supplementary Tables 1 and 2). Paired-end sequencing was performed on an Illumina HiSeq2000 for each library to produce the raw data. We then filtered artificial and low-quality reads to obtain a set of usable reads that contained 857.5 million and 730.0 million reads, representing 63.86 Gb and 46.67 Gb of data for the female and male individuals, respectively, covering in total 212 times the genome (Supplementary Fig. 1 and Supplementary Tables 1 and 2). In addition, we corrected sequencing errors for the 17-mers with a frequency lower than four as per the method described previously57. We next assembled reads to contigs and scaffolds to build the male and female genomes using SOAPdenovo (Supplementary Tables 2 and 3)58.
Sex-chromosome identification and chromosomal assignment of scaffolds.
With the same sequencing coverage, the depth of Z-linked scaffolds in the non-PAR region in the female is expected to be half that in the male Z chromosome, in female autosomes and in male autosomes (Zf = 1/2Zm = 1/2Af = 1/2Am), where f is female, m is male, Z is the Z chromosome, and A is the autosome. We thus identified 26 and 126 Z-linked scaffolds in the male and female assemblies, respectively. For the W-linked scaffolds that displayed the female assembly only, we expected that these scaffolds should not be covered by reads from the male genome, and the sequencing depth should be about half of the average value of autosomes in the female genome reads. Using this method, we identified 306 W-linked scaffolds in the non-PAR region representing a 16.4 Mb length with a scaffold N50 of 128 kb. Considering the interference of W reads and the high-quality assembly of the male genome relative to the female genome for the Z assembly (scaffold N50 of 1,305 kb as compared to 357 kb, respectively), we chose scaffolds from the male assembly as the Z-linked scaffolds in the final version. For other scaffolds representing autosomes and the W chromosome and other undetected Z-linked scaffolds (if any), we used the female version. We then used BLASTN (E value < 1 × 10−5, identity ≥95% and aligning rate >50%) to map molecular markers to scaffolds. We linked scaffolds onto chromosomes with a string of 100 'N's representing the gap between two adjacent scaffolds based on the high-resolution genetic map. In total, 944 scaffolds with 445 Mb length (93.3% of scaffolds in length) were anchored to 22 chromosomes, representing the 20 autosomes, Z and W (Supplementary Table 4).
Repeat annotation.
We used two software packages, PILER-DF59 and RepeatScout60, to construct a de novo TE library for the tongue sole genome. We ran the software with default parameters independently, filtered those that were too short (<100 bp) or had gap N > 5%, then combined the results to get a consensus library. The library contains 1,182 elements that have been classified using homologies with TEs from Repbase (Supplementary Note)61.
RNA-seq analysis.
RNA-seq reads were generated using Illumina HiSeq 2000 (Supplementary Note) and aligned to the reference genome using TopHat62 to identify exons and splice junctions ab initio. High-quality splice junctions were also predicted by the TopHat package62. High-depth regions joining known gene-coding regions directly or by high-quality junction reads were considered as UTRs. Then the gene expression was measured by reads per kilobase of gene per million mapped reads (RPKM)63 and adjusted by a scaling normalization method64. Differentially expressed genes were detected using DESeq65 and Cuffdiff66. Annotation of genes to GO categories was performed according to the orthologous relationship between the C. semilaevis gene set and the Danio rerio gene set, which had perfect GO annotation. The P value was adjusted for multiple testing by consideration of the Benjamini-Hochberg false discovery rate67. The KEGG automatic annotation server68 annotated the genes to KEGG pathways using zebrafish and human as references. Fisher's exact test and the χ2 test then identified enriched pathways69.
Gene prediction and annotation.
Genscan70 and Augustus71 were used for ab initio gene prediction. Protein sequences of Oryzias latipes, Takifugu rubripes, T. nigroviridis, Gasterosteus aculeatus, D. rerio and Homo sapiens were obtained from Ensembl database (release 57). Then the parent protein sequences were aligned to the tongue sole genome by TBLASTN at E value < 1 × 10−5. The homologous genome sequences were then aligned against the matching proteins using GeneWise72 for accurate spliced alignments. We then integrated gene models defined by three methods (de novo prediction, homology-based gene prediction and RNA-seq models) to produce the consensus gene models using GLEAN73. We obtained a final reference gene set containing 21,516 functional genes (Supplementary Table 12). For gene annotation, we detected the motifs and domains by InterProScan74 against publicly available databases, including Pfam, PRINTS, PROSITE, ProDom, SMART and PANTHER, and then retrieved GO annotation from the results of InterProScan. From the reference gene set, 17,890 and 14,935 genes could be annotated by InterPro and GO annotation, respectively. We also annotated 20,265 genes by searching the Swiss-Prot database using BLASTP at E value = 1 × 10−5.
Reconstruction of ancestral vertebrate chromosomes.
We first performed an all-against-all comparison between tongue sole protein sequences and human protein sequences using BLASTP (E value < 1 × 10−10) to identify the paralogs in the tongue sole genome (Supplementary Table 22). Then we identified reciprocal best-match orthologous genes between tongue sole and each other fish using BLASTP at E value = 1 × 10−10. 14,231, 14,310 and 13,084 orthologous genes were identified for T. nigroviridis, medaka and zebrafish, respectively (Supplementary Tables 23,24,25). We further identified doubly conserved synteny by synteny analysis and then deduced the ancestral teleost karyotype by considering the results from the human genome as an outgroup, as was performed in a previous study (Supplementary Table 26)37. The ancestral vertebrate ancestor consisting of ten protochromosomes was reconstructed, and an evolutionary hierarchy from the ancestral vertebrate ancestor to the genomes of human, chicken and medaka was assigned as reported previously (Supplementary Figs. 17 and 18)33.
Dosage compensation.
We used RNA-seq data from the whole bodies (without gonad) of male and female fish to test for dosage compensation of the Z chromosome. The male-to-female (M:F) gene expression ratio was used to measure the dosage compensation level for every gene in the female relative to the normal male calculated as the RPKM ratio of each gene between the two samples. Only genes with RPKM > 1 in both the normal male and female were considered. The Z-to-autosomes (Z:A) expression ratio for every gene in the Z chromosome was calculated by dividing the RPKM of the gene by the median RPKM of all autosomal genes. After filtering out genes with RPKM < 1, we calculated the M:F expression ratio for all Z-linked genes (Supplementary Note).
Production and genetic analyses of sex reversals.
Treatment of tongue sole with high temperature (28 °C) during the critical developmental stage directly affects the sex ratio of the progeny75. Briefly, about 3,000 larvae were collected at 25 d after hatching (dph) and allocated evenly into three tanks (3 m3) at 23 °C. The sea water was heated up gradually and maintained 28 °C until 100 dph. The genetic sex identification of larvae was performed by PCR analysis using the sex-linked SSR marker, CseF-SSR1, which produced one band of 206 bp for ZZ and two bands of 206 bp and 218 bp for ZW76. The phenotypic sex was identified by routine histology of gonads for the presence of oocytes or spermatocytes (Supplementary Note)77. Under treatment with high temperature, about 70% of ZW individuals developed as male, whereas under normal conditions (22 °C), the spontaneous sex-reversal rate was about 14%. The sex-reversed fish produced by high temperature could cross with normal females. Surprisingly, there was an extremely male-skewed sex ratio (>94%) in offspring of pseudo-male families raised under normal conditions (22 °C) (Supplementary Table 40). This was caused by an extremely high sex-reversal rate of genetic females to phenotypic males (∼94% compared to the sex-reversal rate of 14% under normal conditions). A similar phenomenon was also detected in the offspring of pseudo-male families crossings of spontaneous sex-reverted males with normal females.
FISH mapping.
Metaphase chromosome spreads were obtained from the head kidney of female and male fish and were then passed through an ethanol series and air dried. The dmrt1 BAC DNA was extracted and labeled by nick translation using a Nick Translation System (DIG-Nick Translation Mix, Roche). The BAC FISH probe contained 1 μg of labeled dmrt1 BAC DNA, 50 μg of sonicated salmon sperm DNA and 10 μg of Cot-1 DNA. After hybridization in a moist chamber at 37 °C for 24 h, chromosome slides were subjected to a series of washing steps (2× SSC (TaKaRa) for 5 min, 50% formamide for 5 min and 1× SSC for 5 min). Signal detection and amplification were performed using sheep anti-digoxigenin and FITC donkey anti-sheep (Dig-Nick Translation Mix, Roche, 11745816910). FISH staining was performed with propidium iodide. Image capture was carried out with a NIS-element fluorescence microscope (Nikon) and then analyzed by the LUCIA system and Adobe photoshop software (Supplementary Note).
RT-PCR and bisulfite PCR (BS-PCR) analysis.
Total RNA was isolated and purified from all the samples using a traditional phenol method78, and the RNA concentration was measured using Nanodrop technology. Primers for quantitative RT-PCR (qRT-PCR) analysis were designed using the Primer Premier 5 program. qRT-PCR was performed on ABI PRISM 7500 Real-Time PCR System using Hotstart Taq polymerase (Qiagen), and the β-actin gene (Actb) was used as the internal reference (Supplementary Note). BS-PCR was performed on the first exon and intron of dmrt1 to verify the authenticity of the DMR. PCR products were purified and cloned using the pMD18-T Simple Vector cloning kit following the manufacturer's protocol. For each sample, a minimum of 15 clones were sequenced. All clones were sequenced on an ABI 3730xl DNA analyzer using SP6 or T7 primers. BS-PCR together with sequencing of several clones provided allele-specific methylation profiles (Supplementary Note). Additional results are shown in Supplementary Figures 36,37,38 and Supplementary Tables 44,45,46,47,48,49,50,51,52,53,54,55.
URLs.
SOAP, http://soap.genomics.org.cn/; Ensembl, http://www.ensembl.org/index.html; KEGG, http://www.genome.jp/kegg/; Repbase, http://www.girinst.org/repbase/index.html; SOALR, http://treesoft.svn.sourceforge.net/viewvc/treesoft/; RepeatMasker, http://repeatmasker.org/; GLEAN, http://sourceforge.net/projects/glean-gene/.
Accession codes.
The tongue sole whole-genome shotgun project has been deposited in DDBJ/EMBL/GenBank under the project accession PRJNA73987 (with the associated accession code AGRG00000000). RNA-Seq and miRNA sequencing reads have been deposited in the NCBI Sequence Read Archive (SRA047922 and SRA122228), and BAC sequences have been deposited in the GSS division of GenBank (JQ003878, JQ003879, JQ003880, JQ003881).
Accession codes
Primary accessions
BioProject
NCBI Reference Sequence
Sequence Read Archive
References
Ellegren, H. Sex-chromosome evolution: recent progress and the influence of male and female heterogamety. Nat. Rev. Genet. 12, 157–166 (2011).
Charlesworth, B. & Charlesworth, D. The degeneration of Y chromosomes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 355, 1563–1572 (2000).
Mank, J.E. Sex Chromosomes and the evolution of sexual dimorphism: lessons from the Genome. Am. Nat. 173, 141–150 (2009).
Quinn, A.E., Sarre, S.D., Ezaz, T., Marshall Graves, J.A. & Georges, A. Evolutionary transitions between mechanisms of sex determination in vertebrates. Biol. Lett. 7, 443–448 (2011).
Skaletsky, H. et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423, 825–837 (2003).
Hughes, J.F. et al. Chimpanzee and human Y chromosomes are remarkably divergent in structure and gene content. Nature 463, 536–539 (2010).
Hughes, J.F., Skaletsky, H. & Page, D.C. Sequencing of rhesus macaque Y chromosome clarifies origins and evolution of the DAZ (Deleted in AZoospermia) genes. Bioessays 34, 1035–1044 (2012).
Kondo, M. et al. Genomic organization of the sex-determining and adjacent regions of the sex chromosomes of medaka. Genome Res. 16, 815–826 (2006).
Bellott, D.W. & Page, D.C. Reconstructing the evolution of vertebrate sex chromosomes. Cold Spring Harb. Symp. Quant. Biol. 74, 345–353 (2009).
Marshall Graves, J.A. Sex chromosome specialization and degeneration in mammals. Cell 124, 901–914 (2006).
Schartl, M. Sex chromosome evolution in non-mammalian vertebrates. Curr. Opin. Genet. Dev. 14, 634–641 (2004).
Charlesworth, B. Sex determination: primitive Y chromosomes in fish. Curr. Biol. 14, R745–R747 (2004).
Charlesworth, D., Charlesworth, B. & Marais, G. Steps in the evolution of heteromorphic sex chromosomes. Heredity 95, 118–128 (2005).
Marshall Graves, J.A. & Peichel, C.L. Are homologies in vertebrate sex determination due to shared ancestry or to limited options? Genome Biol. 11, 205 (2010).
Mank, J.E. & Avise, J.C. Evolutionary diversity and turn-over of sex determination in teleost fishes. Sex Dev. 3, 60–67 (2009).
Song, W. et al. Construction of a high-density microsatellite genetic linkage map and mapping of sexual and growth-related traits in half-smooth tongue sole (Cynoglossus semilaevis). PLoS ONE 7, e52097 (2012).
Sha, Z. et al. Generation and analysis of 10 000 ESTs from the half-smooth tongue sole Cynoglossus semilaevis and identification of microsatellite and SNP markers. J. Fish Biol. 76, 1190–1204 (2010).
Shao, C.W. et al. Construction of two BAC libraries from half-smooth tongue sole Cynoglossus semilaevis and identification of clones containing candidate sex-determination genes. Mar. Biotechnol. (NY) 12, 558–568 (2010).
Chen, S.L. et al. Isolation of female-specific AFLP markers and molecular identification of genetic sex in half-smooth tongue sole (Cynoglossus semilaevis). Mar. Biotechnol. (NY) 9, 273–280 (2007).
Zhou, L.Q., Yang, A.G., Liu, X.Z., Du, W. & Zhuang, Z.M. The karyotype of the tonguefish Cynoglossus semilaevis. J. Fish Sci. China 3, 417–419 (2005).
Friedman, M. The evolutionary origin of flatfish asymmetry. Nature 454, 209–212 (2008).
Schreiber, A.M. Asymmetric craniofacial remodeling and lateralized behavior in larval flatfish. J. Exp. Biol. 209, 610–621 (2006).
Lander, E.S. et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001).
Aparicio, S. et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 297, 1301–1310 (2002).
Schreiber, A.M. Flatfish: an asymmetric perspective on metamorphosis. Curr. Top. Dev. Biol. 103, 167–194 (2013).
Zhang, J., Nielsen, R. & Yang, Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 22, 2472–2479 (2005).
Meredith, R.W., Gatesy, J., Emerling, C.A., York, V.M. & Springer, M.S. Rod monochromacy and the coevolution of cetacean retinal opsins. PLoS Genet. 9, e1003432 (2013).
Graw, J. Genetics of crystallins: cataract and beyond. Exp. Eye Res. 88, 173–189 (2009).
Ma, A.J., Wang, X.A., Zhuang, Z.M., Zhang, X.M. & Zhang, L.J. Structure of retina and visual characteristics of the half-smooth tongue-sole Cynoglossus semilaevis Gunther. Acta Zool. Sin. 53, 354–363 (2007).
Ma, A.J., Wang, X.A. & Zhuang, Z.M. Lateral-line sense organs and dermal surface structures of the tongue sole Cynoglossus semilaevis. Acta Zool. Sin. 53, 1113–1120 (2007).
Bernardi, G. et al. The fishes of genome 10K. Mar. Genomics 7, 3–6 (2012).
Hurley, I.A. et al. A new time-scale for ray-finned fish evolution. Proc. Biol. Sci. 274, 489–498 (2007).
Nakatani, Y., Takeda, H., Kohara, Y. & Morishita, S. Reconstruction of the vertebrate ancestral genome reveals dynamic genome reorganization in early vertebrates. Genome Res. 17, 1254–1265 (2007).
Kasahara, M. et al. The medaka draft genome and insights into vertebrate genome evolution. Nature 447, 714–719 (2007).
Smith, J.J. et al. Sequencing of the sea lamprey (Petromyzon marinus) genome provides insights into vertebrate evolution. Nat. Genet. 45, 415–421 (2013).
Amores, A., Catchen, J., Ferrara, A., Fontenot, Q. & Postlethwait, J.H. Genome evolution and meiotic maps by massively parallel DNA sequencing: spotted gar, an outgroup for the teleost genome duplication. Genetics 188, 799–808 (2011).
Jaillon, O. et al. Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature 431, 946–957 (2004).
Wright, A.E., Moghadam, H.K. & Mank, J.E. Trade-off between selection for dosage compensation and masculinisation on the avian Z chromosome. Genetics 192, 1433–1445 (2012).
Moghadam, H.K., Pointer, M.A., Wright, A.E., Berlin, S. & Mank, J.E. W chromosome expression responds to female-specific selection. Proc. Natl. Acad. Sci. USA 109, 8207–8211 (2012).
Bellott, D.W. et al. Convergent evolution of chicken Z and human X chromosomes by expansion and gene acquisition. Nature 466, 612–616 (2010).
Marshall Graves, J.A. How to evolve new vertebrate sex determining genes. Dev. Dyn. 242, 354–359 (2013).
Itoh, Y. et al. Sex bias and dosage compensation in the zebra finch versus chicken genomes: general and specialized patterns among birds. Genome Res. 20, 512–518 (2010).
Zha, X. et al. Dosage analysis of Z chromosome genes using microarray in silkworm, Bombyx mori. Insect Biochem. Mol. Biol. 39, 315–321 (2009).
Küpper, C. et al. Triploid plover female provides support for a role of the W chromosome in avian sex determination. Biol. Lett. 8, 787–789 (2012).
Smith, C.A. et al. The avian Z-linked gene DMRT1 is required for male sex determination in the chicken. Nature 461, 267–271 (2009).
Deng, S.P., Chen, S.L., Xu, J.Y. & Liu, B.W. Molecular cloning, characterization and expression analysis of gonadal P450 aromatase in the half-smooth tongue-sole, Cynoglossus semilaevis. Aquaculture 287, 211–218 (2009).
Matson, C.K. & Zarkower, D. Sex and the singular DM domain: insights into sexual regulation, evolution and plasticity. Nat. Rev. Genet. 13, 163–174 (2012).
Liu, Y.Q. et al. Human RING finger protein ZNF645 is a novel testis-specific E3 ubiquitin ligase. Asian J. Androl. 12, 658–666 (2010).
Yin, Y. et al. The E3 ubiquitin ligase Cullin 4A regulates meiotic progression in mouse spermatogenesis. Dev. Biol. 356, 51–62 (2011).
Birchler, J.A. Dosage compensation for the birds. Heredity 102, 423–424 (2009).
Hughes, J.F. et al. Strict evolutionary conservation followed rapid gene loss on human and rhesus Y chromosomes. Nature 483, 82–86 (2012).
O'Meally, D., Ezaz, T., Georges, A., Sarre, S.D. & Marshall Graves, J.A. Are some chromosomes particularly good at sex? Insights from amniotes. Chromosome Res. 20, 7–19 (2012).
Yoshimoto, S. et al. A W-linked DM-domain gene, DM-W, participates in primary ovary development in Xenopus laevis. Proc. Natl. Acad. Sci. USA 105, 2469–2474 (2008).
Nanda, I. et al. A duplicated copy of DMRT1 in the sex-determining region of the Y chromosome of the medaka, Oryzias latipes. Proc. Natl. Acad. Sci. USA 99, 11778–11783 (2002).
Matsuda, M. et al. DMY is a Y-specific DM-domain gene required for male development in the medaka fish. Nature 417, 559–563 (2002).
Kondo, M. et al. Absence of the candidate male sex-determining gene dmrt1b(Y) of medaka from other fish species. Curr. Biol. 13, 416–420 (2003).
Li, R. et al. The sequence and de novo assembly of the giant panda genome. Nature 463, 311–317 (2010).
Li, R. et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272 (2010).
Edgar, R.C. & Myers, E.W. PILER: identification and classification of genomic repeats. Bioinformatics 21 (suppl. 1), i152–i158 (2005).
Price, A.L., Jones, N.C. & Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 21 (suppl. 1), i351–i358 (2005).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Trapnell, C., Pachter, L. & Salzberg, S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
Mortazavi, A., Williams, B.A., Mccue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628 (2008).
Robinson, M.D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010).
Roberts, A., Trapnell, C., Donaghey, J., Rinn, J.L. & Pachter, L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 12, R22 (2011).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J.R. Stat. Soc. 57, 289–300 (1995).
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A.C. & Kanehisa, M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185 (2007).
Huang, W., Sherman, B.T. & Lempicki, R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 (2009).
Salamov, A.A. & Solovyev, V.V. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 10, 516–522 (2000).
Stanke, M. & Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19 (suppl. 2), ii215–ii225 (2003).
Birney, E., Clamp, M. & Durbin, R. GeneWise and genomewise. Genome Res. 14, 988–995 (2004).
Elsik, C.G. et al. Creating a honey bee consensus gene set. Genome Biol. 8, R13 (2007).
Zdobnov, E.M. & Apweiler, R. InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001).
Deng, S.P., Chen, S.L., Tian, Y.S., Liu, B.W. & Zhuang, Z.M. Gonadal differentiation and effects of temperature on sex determination in half-smooth tongue sole, Cynoglossus semilaevis. J. Fish Sci. China 14, 714–719 (2007).
Chen, S.L. et al. Induction of mitogynogenetic diploids and identification of WW super-female using sex-specific SSR markers in half-smooth tongue sole (Cynoglossus semilaevis). Mar. Biotechnol. (NY) 14, 120–128 (2012).
Chen, S.L. et al. Selection of the families with high growth rate and high female proportion in half-smooth tongue sole (Cynoglossus semilaevis). J. Fish China 37, 481–488 (2013).
Chen, S., Hong, Y., Scherer, S.J. & Schartl, M. Lack of ultraviolet-light inducibility of the medakafish (Oryzias latipes) tumor suppressor gene p53. Gene 264, 197–203 (2001).
Acknowledgements
We thank H. Lin, F. Zhao, J. Lei, Z. Zhu and Q. Wang for their support of the tongue sole genome project. We thank Z. Liu, Z. Zhuang, R. Li, Z. Jiang, P. Ni, X. Qian and T. Xu for discussion. We thank J. Zhai, S. Liu and Z. Li for their help in the culture of tongue sole families. We thank M. Yi and L. Huang for their help with chromosome FISH. We are grateful to the faculty and staff of Yellow Sea Fisheries Research Institute, CAFS and BGI-Shenzhen, whose names were not included in the author list but who nevertheless contributed to this project. This work was supported by grants from the Special Fund for Agro-Scientific Research in the Public Interest (200903046) of China to S.C., the State 863 High Technology R&D Project of China (2012AA092203) to S.C., the State 863 High Technology R&D Project of China (2012AA10A403-2) to C.S., the National Nature Science Foundation of China (31130057 and 31072202) to S.C., the National Nature Science Foundation for Young Scholar (41006107) to C.S., the Taishan Scholar Project Fund of Shandong of China to S.C., the JSPS RONPAKU (Dissertation PhD) program to C.S., the Basic Research Program Supported by Shenzhen City (JC2010526019) to G.Z., the Key Laboratory Project Supported by Shenzhen City (CXB200903110066A) to J.W., the Shenzhen Key Laboratory of Transomics Biotechnologies (CXB201108250096A) to J.W. and the Shenzhen Key Laboratory of Gene Bank for National Life Science.
Author information
Authors and Affiliations
Contributions
S.C., G.Z., C.S., Q. Huang, G.L. and P.Z. contributed equally to this work. S.C., G.Z., J.W. and Q.T. initiated, managed and drove the tongue sole genome sequencing project. S.C., M.S., G.Z., C.S. and Q. Huang designed the analysis. S.C., J.Y., C.S., K.W., Y.T. and M.D. prepared the samples and performed the sequencing. L.Y., Q.Z., G.L., Q.Y., H. Zhou, J. Zhang, Z.S. and N.A. performed the genome assembly and annotation. J.-N.V. and D.C. performed the repeat analysis. W.S., S.C., G.M., C.S., X.L., Y. Li and Y. Zhao constructed the high-density genetic map. G.Z., Q. Huang, G.L., H. Zhou and Q.Z. performed genome evolution analysis. G.Z., Q. Huang, G.L., C.S., P.Z., N.A., Q.Y. and H. Zhou performed the sex chromosome evolution analysis. N.A. performed the gene evolution analysis. G.Z., P.Z., Q.L. and G.L. performed the dosage compensation analysis. G.Z., Q.L., P.Z., H.X., Q. Hu, L.J., J.L. and B.S. performed the methylation analysis. S.C., Y. Liu, C.S., N.W., C.Y., Q. Hu, S.L., Z.L., F.Y., L.M., Y. Zhu, H. Zhang and J. Zhang performed the gene expression and the in situ hybridization. S.C., Y.H. and M.X. performed the chromosome FISH. All authors contributed to data interpretation. M.S., C.S., G.Z., Q. Huang and S.C. wrote the manuscript, and M.S., C.S., G.Z., S.C., J.W. and Q.T. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–38, Supplementary Tables 1–13,15–43 and 45–55, and Supplementary Note. (PDF 6023 kb)
Supplementary Table 14
Differentially expressed genes between pre- and post-metamorphosis. (XLS 107 kb)
Supplementary Table 44
Differentially expressed genes between normal female ovaries and pseudomale testes. (XLS 121 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/.
About this article
Cite this article
Chen, S., Zhang, G., Shao, C. et al. Whole-genome sequence of a flatfish provides insights into ZW sex chromosome evolution and adaptation to a benthic lifestyle. Nat Genet 46, 253–260 (2014). https://doi.org/10.1038/ng.2890
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.2890
This article is cited by
-
Regulatory mechanism of LncRNAs in gonadal differentiation of hermaphroditic fish, Monopterus albus
Biology of Sex Differences (2023)
-
Gonadal transcriptomes reveal sex-biased expression genes associated with sex determination and differentiation in red-tail catfish (Hemibagrus wyckioides)
BMC Genomics (2023)
-
Massive expansion of sex-specific SNPs, transposon-related elements, and neocentromere formation shape the young W-chromosome from the mosquitofish Gambusia affinis
BMC Biology (2023)
-
Consequences of polyploidy and divergence as revealed by cytogenetic mapping of tandem repeats in African clawed frogs (Xenopus, Pipidae)
European Journal of Wildlife Research (2023)
-
SNP Panel and Genomic Sex Identification in Atlantic Halibut (Hippoglossus hippoglossus)
Marine Biotechnology (2023)
