
Abstract
The sensation of pain protects the body from serious injury1,2,3. Using exome sequencing, we identified a specific de novo missense mutation in SCN11A in individuals with the congenital inability to experience pain who suffer from recurrent tissue damage and severe mutilations. Heterozygous knock-in mice carrying the orthologous mutation showed reduced sensitivity to pain and self-inflicted tissue lesions, recapitulating aspects of the human phenotype. SCN11A encodes Nav1.9, a voltage-gated sodium ion channel that is primarily expressed in nociceptors, which function as key relay stations for the electrical transmission of pain signals from the periphery to the central nervous system4,5. Mutant Nav1.9 channels displayed excessive activity at resting voltages, causing sustained depolarization of nociceptors, impaired generation of action potentials and aberrant synaptic transmission. The gain-of-function mechanism that underlies this channelopathy suggests an alternative way to modulate pain perception.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others
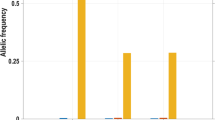
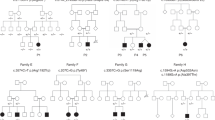
Accession codes
Primary accessions
NCBI Reference Sequence
Referenced accessions
NCBI Reference Sequence
References
Basbaum, A.I., Bautista, D.M., Scherrer, G. & Julius, D. Cellular and molecular mechanisms of pain. Cell 139, 267–284 (2009).
Williams, F.M. et al. Genes contributing to pain sensitivity in the normal population: an exome sequencing study. PLoS Genet. 8, e1003095 (2012).
Woolf, C.J. & Salter, M.W. Neuronal plasticity: increasing the gain in pain. Science 288, 1765–1769 (2000).
Dib-Hajj, S.D., Cummins, T.R., Black, J.A. & Waxman, S.G. Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 33, 325–347 (2010).
Eijkelkamp, N. et al. Neurological perspectives on voltage-gated sodium channels. Brain 135, 2585–2612 (2012).
Gold, M.S. & Gebhart, G.F. Nociceptor sensitization in pain pathogenesis. Nat. Med. 16, 1248–1257 (2010).
Kurth, I. et al. Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat. Genet. 41, 1179–1181 (2009).
Rotthier, A., Baets, J., Timmerman, V. & Janssens, K. Mechanisms of disease in hereditary sensory and autonomic neuropathies. Nat. Rev. Neurol. 8, 73–85 (2012).
Cox, J.J. et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 444, 894–898 (2006).
Yuan, J. et al. Hereditary sensory and autonomic neuropathy type IID caused by an SCN9A mutation. Neurology 80, 1641–1649 (2013).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010).
Conrad, D.F. et al. Variation in genome-wide mutation rates within and between human families. Nat. Genet. 43, 712–714 (2011).
Abecasis, G.R. et al. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010).
Veltman, J.A. & Brunner, H.G. De novo mutations in human genetic disease. Nat. Rev. Genet. 13, 565–575 (2012).
Cummins, T.R. et al. A novel persistent tetrodotoxin-resistant sodium current in SNS-null and wild-type small primary sensory neurons. J. Neurosci. 19, RC43 (1999).
Amaya, F. et al. The voltage-gated sodium channel Nav1.9 is an effector of peripheral inflammatory pain hypersensitivity. J. Neurosci. 26, 12852–12860 (2006).
Leo, S., D'Hooge, R. & Meert, T. Exploring the role of nociceptor-specific sodium channels in pain transmission using Nav1.8 and Nav1.9 knockout mice. Behav. Brain Res. 208, 149–157 (2010).
Lolignier, S. et al. Nav1.9 channel contributes to mechanical and heat pain hypersensitivity induced by subacute and chronic inflammation. PLoS ONE 6, e23083 (2011).
Maingret, F. et al. Inflammatory mediators increase Nav1.9 current and excitability in nociceptors through a coincident detection mechanism. J. Gen. Physiol. 131, 211–225 (2008).
Östman, J.A., Nassar, M.A., Wood, J.N. & Baker, M.D. GTP up-regulated persistent Na+ current and enhanced nociceptor excitability require Nav1.9. J. Physiol. (Lond.) 586, 1077–1087 (2008).
Priest, B.T. et al. Contribution of the tetrodotoxin-resistant voltage-gated sodium channel Nav1.9 to sensory transmission and nociceptive behavior. Proc. Natl. Acad. Sci. USA 102, 9382–9387 (2005).
Herzog, R.I., Cummins, T.R. & Waxman, S.G. Persistent TTX-resistant Na+ current affects resting potential and response to depolarization in simulated spinal sensory neurons. J. Neurophysiol. 86, 1351–1364 (2001).
Vanoye, C.G., Kunic, J.D., Ehring, G.R. & George, A.L. Jr. Mechanism of sodium channel Nav1.9 potentiation by G-protein signaling. J. Gen. Physiol. 141, 193–202 (2013).
Catterall, W.A. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26, 13–25 (2000).
Payandeh, J., Scheuer, T., Zheng, N. & Catterall, W.A. The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358 (2011).
Huang, Z. et al. Presynaptic HCN1 channels regulate Cav3.2 activity and neurotransmission at select cortical synapses. Nat. Neurosci. 14, 478–486 (2011).
Jacus, M.O., Uebele, V.N., Renger, J.J. & Todorovic, S.M. Presynaptic Cav3.2 channels regulate excitatory neurotransmission in nociceptive dorsal horn neurons. J. Neurosci. 32, 9374–9382 (2012).
Cheng, X. et al. Deletion mutation of sodium channel Nav1.7 in inherited erythromelalgia: enhanced slow inactivation modulates dorsal root ganglion neuron hyperexcitability. Brain 134, 1972–1986 (2011).
Dib-Hajj, S.D., Yang, Y., Black, J.A. & Waxman, S.G. The Nav1.7 sodium channel: from molecule to man. Nat. Rev. Neurosci. 14, 49–62 (2013).
Fertleman, C.R. et al. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron 52, 767–774 (2006).
Faber, C.G. et al. Gain of function Nav1.7 mutations in idiopathic small fiber neuropathy. Ann. Neurol. 71, 26–39 (2012).
Faber, C.G. et al. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc. Natl. Acad. Sci. USA 109, 19444–19449 (2012).
Subramanian, N. et al. Role of Nav1.9 in activity-dependent axon growth in motoneurons. Hum. Mol. Genet. 21, 3655–3667 (2012).
Wetzel, A., Jablonka, S. & Blum, R. Cell-autonomous axon growth of young motoneurons is triggered by a voltage-gated sodium channel. Channels (Austin) 7, 51–56 (2013).
Copel, C., Clerc, N., Osorio, N., Delmas, P. & Mazet, B. The Nav1.9 channel regulates colonic motility in mice. Front. Neurosci. 7, 58 (2013).
Rugiero, F. et al. Selective expression of a persistent tetrodotoxin-resistant Na+ current and Nav1.9 subunit in myenteric sensory neurons. J. Neurosci. 23, 2715–2725 (2003).
Copel, C. et al. Activation of neurokinin 3 receptor increases Nav1.9 current in enteric neurons. J. Physiol. (Lond.) 587, 1461–1479 (2009).
Schröder, J.M., Hoheneck, M., Weis, J. & Deist, H. Ethylene oxide polyneuropathy: clinical follow-up study with morphometric and electron microscopic findings in a sural nerve biopsy. J. Neurol. 232, 83–90 (1985).
Schwenk, F., Baron, U. & Rajewsky, K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 23, 5080–5081 (1995).
Ebbinghaus, M. et al. The anti-inflammatory effects of sympathectomy in murine antigen-induced arthritis are associated with a reduction of Th1 and Th17 responses. Ann. Rheum. Dis. 71, 253–261 (2012).
Richter, F. et al. Interleukin-17 sensitizes joint nociceptors to mechanical stimuli and contributes to arthritic pain through neuronal interleukin-17 receptors in rodents. Arthritis Rheum. 64, 4125–4134 (2012).
Blum, R., Kafitz, K.W. & Konnerth, A. Neurotrophin-evoked depolarization requires the sodium channel Nav1.9. Nature 419, 687–693 (2002).
Dib-Hajj, S.D. et al. Transfection of rat or mouse neurons by biolistics or electroporation. Nat. Protoc. 4, 1118–1126 (2009).
Sinning, A. et al. Synaptic glutamate release is modulated by the Na+-driven Cl−/HCO3− exchanger Slc4a8. J. Neurosci. 31, 7300–7311 (2011).
Acknowledgements
We are grateful to the families participating in the study. Excellent technical assistance was provided by K. Schorr, A. Roßner, P. Schroth and the team from the Jena University Hospital animal facility. Scn11a−/− mice were generously provided by J.N. Wood (Wolfson Institute for Biomedical Research, University College London). ND7/23 cells were generously provided by C. Nau (University Hospital Erlangen). We thank D.G.G. McMillan for critical reading of the manuscript. This work was supported by grants from the DFG (Deutsche Forschungsgemeinschaft) to I.K. (KU 1587/2-1) and C.A.H. (HU 800/5-1, RTG 1715, HU 800/6-1 and HU 800/7-1). Funding to J.B., P.D.J. and V.T. was provided by the University of Antwerp, Fund for Scientific Research (FWO-Flanders), Association Belge contre les Maladies neuro-Musculaires (ABMM) and Medical Foundation Queen Elisabeth (GSKE). Funding to R.B. was provided by the DFG (BL567/3-1). Funding to J.W. was provided by the DFG (WE 1406/13-1) and IZKF (Interdisziplinäres Zentrum für Klinische Forschung) Aachen (N5-3).
Author information
Authors and Affiliations
Contributions
I.K., C.A.H., E.L. and S.H.H. designed this study. G.C.K., J.B., V.T., P.D.J. and T.S. assessed the phenotypes of the affected individuals. M.B. and J.W. performed neuropathological analysis. J.A., H.T. and P.N. performed exome sequencing. Additional experiments were performed by I.K. (genetics, generation of knock-in mice and molecular biology), E.L., R.O.G. and L.L. (electrophysiology), S.G. (molecular biology and histology), J.C.H., A.W. and R.B. (molecular biology) and T.H. (tail-flick assay and histology). M.E. and H.-G.S. performed behavioral analysis and evaluation. I.K., L.L., E.L., S.H.H. and C.A.H. analyzed the data and wrote the manuscript with input from the coauthors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–4 and Supplementary Table 1 (PDF 1231 kb)
Rights and permissions
About this article
Cite this article
Leipold, E., Liebmann, L., Korenke, G. et al. A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nat Genet 45, 1399–1404 (2013). https://doi.org/10.1038/ng.2767
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.2767
This article is cited by
-
Migraine, chronic kidney disease and kidney function: observational and genetic analyses
Human Genetics (2023)
-
Pathological changes of the sural nerve in patients with familial episodic pain syndrome
Neurological Sciences (2022)
-
Single cell transcriptomics of primate sensory neurons identifies cell types associated with chronic pain
Nature Communications (2021)
-
The physiological function of different voltage-gated sodium channels in pain
Nature Reviews Neuroscience (2021)
-
Mechanical allodynia triggered by cold exposure in mice with the Scn11a p.R222S mutation: a novel model of drug therapy for neuropathic pain related to NaV1.9
Naunyn-Schmiedeberg's Archives of Pharmacology (2021)
